")
Back to Journals » Clinical Interventions in Aging » Volume 10
Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders
Received 31 March 2015
Accepted for publication 22 April 2015
Published 14 July 2015 Volume 2015:10 Pages 1163—1172
DOI https://doi.org/10.2147/CIA.S85808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Walker
Yan Cai,1 Seong Soo A An,1 SangYun Kim2
1Department of Bionano Technology, Gachon Medical Research Institute, Gachon University, 2Department of Neurology, Seoul National University College of Medicine, Seoul National University Bundang Hospital, Seongnam-si, Gyeonggi-do, South Korea
Abstract: Alzheimer’s disease (AD) is the most common form of dementia. Mutations in the genes encoding presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein have been identified as the main genetic causes of familial AD. To date, more than 200 mutations have been described worldwide in PSEN1, which is highly homologous with PSEN2, while mutations in PSEN2 have been rarely reported. We performed a systematic review of studies describing the mutations identified in PSEN2. Most PSEN2 mutations were detected in European and in African populations. Only two were found in Korean populations. Interestingly, PSEN2 mutations appeared not only in AD patients but also in patients with other disorders, including frontotemporal dementia, dementia with Lewy bodies, breast cancer, dilated cardiomyopathy, and Parkinson’s disease with dementia. Here, we have summarized the PSEN2 mutations and the potential implications of these mutations in dementia-associated disorders.
Keywords: mutations in presenilin 2, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is the most common form of neurodegenerative disease of the brain. Pathological hallmarks of AD include intraneuronal accumulation of paired helical filaments composed of abnormal tau proteins and extracellular deposits of β-amyloid peptide (Aβ) in neuritic plaques.1 Clinically, AD can be categorized into two phenotypes based on the ages of onset: early-onset AD (EOAD; <65 years) and late-onset AD (LOAD; >65 years), of which LOAD is the more common form worldwide. The proportion of EOAD in all AD cases is between 5% and 10%.2 Presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) are mostly associated with autosomal dominant forms of EOAD.3 Apart from genetic factors, mutations are environmentally related. Genetic–environmental interactions may be caused by variation in the age of onset, neuropathological patterns, and disease duration.4 To date, more than 200 mutations have been described in PSEN1 throughout the world, but mutations in PSEN2 are extremely rare. Less than 40 mutations in PSEN2 have been identified.5 From those, two PSEN2 mutations were detected in Korean patients. Unlike PSEN1, AD patients with PSEN2 mutations have a wide range in the age of onset, from 40 to 80 years.6 Interestingly, some reports have suggested that the inherited mode of AD was autosomal inheritance with variable penetrance, which suggests that other environmental factors might also be significant for AD pathogenesis.7 In addition, mutations in PSEN2 are also closely involved in other diseases, including EOAD, LOAD, frontotemporal dementia (FTD), dementia with Lewy bodies (DLBs), breast cancer, dilated cardiomyopathy (DCM). In this review, we studied and summarized PSEN2, in particular, the known PSEN2 mutations and the potential implications of PSEN2 in AD and in other disorders.
PSEN2 gene
In 1995, PSEN2 was initially reported as a causative gene for AD, following the identification of APP and PSEN1.8 The gene was localized to chromosome lq42.13. It consists of 12 exons, of which exon 1 and exon 2 contain the untranslated regions.
PSEN2 transcription
Transcriptional regulation
PSEN2 is driven by two separate promoter elements, P1 and P2, which are located in exon 1 and exon 2, respectively. The upstream P1 is a housekeeping promoter. PSEN2-P1 activity depends on a stimulating protein 1 binding site at the most 5′ initiation site. The downstream P2 is induced by Egr-1, which represses PSEN2-P1 activity.9 Interestingly, a study showed that Egr-1 cannot regulate the PSEN2 promoter in mouse.10 APP influences the expression of Egr-1 by enhancing histone H4 acetylation of the Egr-1 promoter.11
Splice variant
The two isoforms of PSEN2 protein are produced by alternative splicing. An aberrant splice variant of PSEN2 lacks exon 5, which results in the insertion of five amino acids, SSMAG, into the protein variant, and which introduces a premature stop codon in exon 6.12 Aggregation of the PSEN2 variant protein was detected in the hippocampus and cerebral cortex of patients with sporadic AD.13 The protein variant also was detected in sporadic AD patients, in the frontal lobe of patients with bipolar disorder, and in patients with schizophrenia.14,15 The PSEN2 variant is upregulated under hypoxic conditions in cell culture, and a study has shown that the PSEN2 variant influences the conformation of tau protein in human neuroblastoma cells.12,16
PSEN2 protein
Structure
PSEN2 is located on chromosome 1, and it encodes the PSEN2 protein. PSEN2 is a transmembrane protein with 448 amino acids and a molecular weight of 55 Da.17,18 It is predicted to span the lipid bilayer nine times.19 PSEN2 and PSEN1 are homologous, with a similarity of 67%.20 The two proteins differ at the N-terminus and at the hydrophilic loop, while the hydrophobic region is highly conserved. PSEN2 is an unstable holoprotein. It undergoes autocatalytic endoproteolysis within the large cytoplasmic loop domain, to form a stable and biologically active heterodimer. In PSEN2, two aspartyl residues–D263 and D366 found in the adjacent transmembrane regions Transmembrane domain (TM)-VI and TM-VII–are the active sites of the γ-secretase complex.
Location
PSEN2 has two isoforms. Isoform 1 is found in the placenta, skeletal muscle and heart, while isoform 2, which lacks amino acids 263–296, is found in the brain, heart, placenta, liver, skeletal muscle, and kidney. Presenilin proteins that are localized in neurons reside in the endoplasmic reticulum and Golgi.21
Function
Presenilin, an aspartyl protease, is a subunit of γ-secretase. γ-Secretase participates in the cleavage of APP, which can produce different lengths of β-amyloid peptide (Aβ). The Aβ42 form aggregates easier than the Aβ40 form. The accumulation of Aβ in the brain is a pathological characteristic of AD.22 The process of Aβ aggregation is shown in Figure 1. PSEN2 mutation might increase γ-secretase activity. Cell-based studies and mouse models have shown that some PSEN2 mutations cause an increased production of Aβ42, which is a major hallmark in the brains of patients with AD. Presenilin mutations are a major risk factor for AD.23 Several studies have indicated that AD-related presenilin mutations can alter intracellular calcium signaling, which leads to Aβ aggregation to form brain plaques and neuronal cell death.24,25
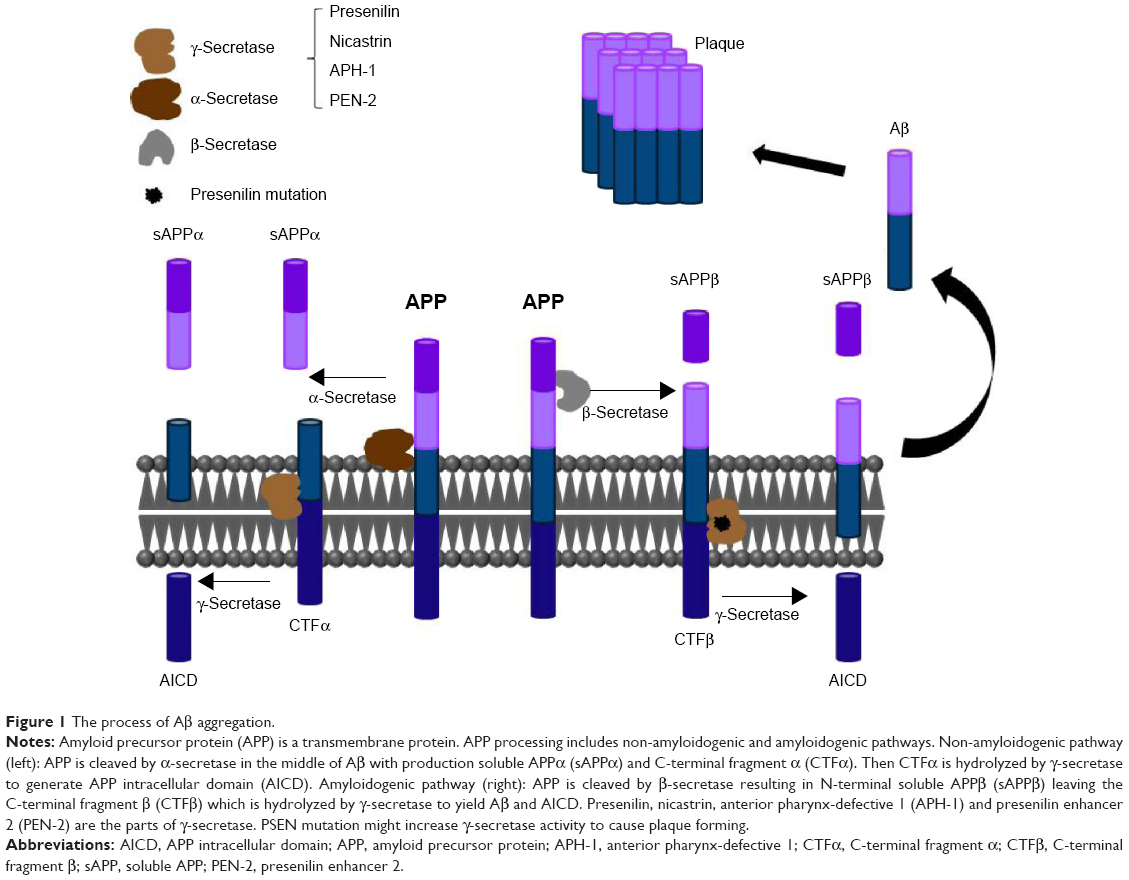 | Figure 1 The process of Aβ aggregation. |
γ-Secretase catalyzes the intramembrane cleavage of integral membrane proteins. It plays an important role in intracellular signaling, including Notch signaling and APP processing. In separate studies published in 1996, Vito et al26 and Wolozin et al27 proposed that PSEN2 is involved in apoptosis. A study demonstrated that wild-type and mutant N141I-PSEN2 trigger p53-dependent apoptosis in HEK293 human cells and in murine neurons.28 In primary rat cortical neurons, PSEN2, overexpression significantly increased susceptibility to staurosporine-induced apoptosis. PSEN2 mutations can promote apoptosis. Bcl-2 can down regulate pro-apoptotic activities, which are induced by PSEN2.29 A recent study suggested that overexpression of human mutant PSEN2 induces changes in glucose metabolism, which is accompanied by a decrease in insulin levels.30
PSEN2 mutations
Mutations in the presenilin genes are the main causes of familial EOAD. Similar to APP, mutant presenilins can enhance Aβ production and contribute to AD development, whereas PSEN2 plays less of a role than PSEN1. An extensive literature search for mutations in PSEN2 was conducted. As of date, 38 mutations have been reported. The number of mutations identified in PSEN1 is greater than five times this number.31 Two PSEN2 mutations, Glu126fs and Lys306fs, are frameshift mutations, and the others are nonsynonymous substitutions (Table 1). PSEN2 mutations are associated with variable penetrance and a wide range in the age of disease onset, from 45 to 88.32,33 PSEN2 mutations are associated with both EOAD and LOAD. Only 17 of the 38 are predicted to be disease-causing mutations (Figure 2). Ten of the mutations are not pathogenic and the others are still unclear. Sixteen mutations are located within transmembrane domains. Cell-based studies suggest that four of these mutations, T122P, N141I, M239I, and M239V, cause an increase in the amount of Aβ peptide.34 The mutations T122R, S130L, and M239I were found to alter calcium signaling.35–37 Most of these mutations were discovered in European and African populations. Until now, only four missense mutations were described in Asian populations: Asn141Tyr was associated with EOAD in a Chinese Han family;37 Gly34Ser was found in a Japanese patient;39 and Arg62Cys and Val214Leu were described in the Korean patients.6
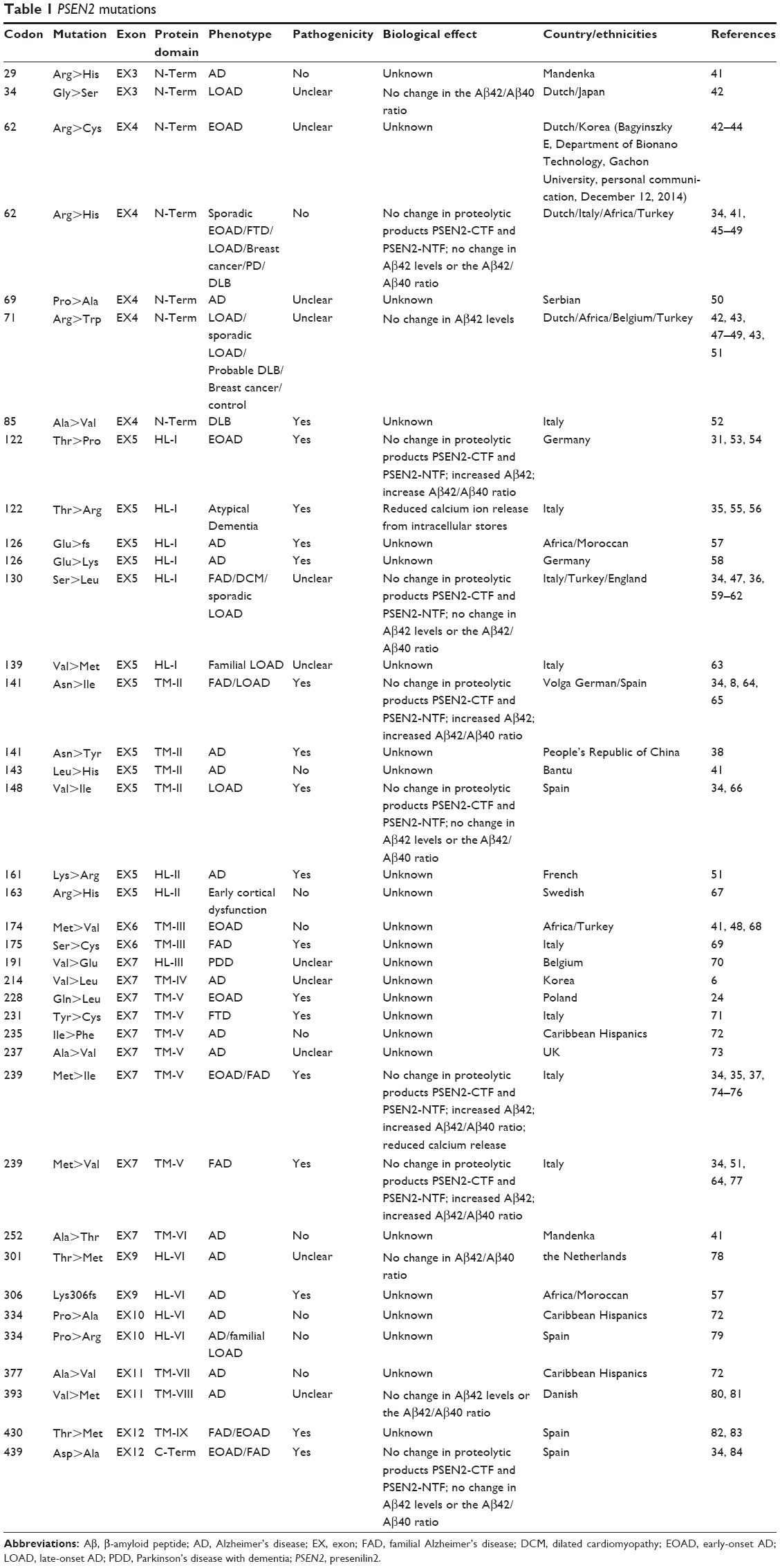 | Table 1 PSEN2 mutations |
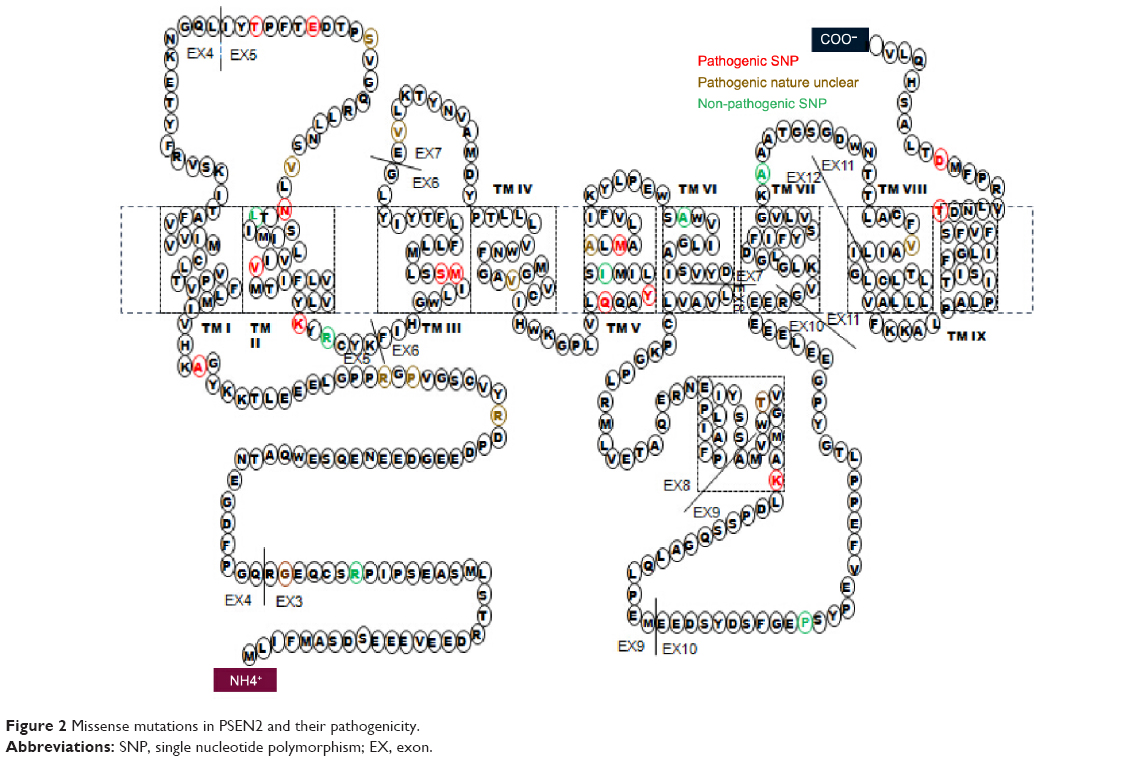 | Figure 2 Missense mutations in PSEN2 and their pathogenicity. |
Related diseases
It was well-known that some mutations in PSEN2 cause familial AD, while some PSEN2 mutations are associated with other disorders, including DLB, FTD, breast cancer, DCM, and Parkinson’s disease with dementia (PDD).
Dementia with Lewy body
DLB is a progressive degenerative disease, accounting for 10%–20% of all dementias. The core clinical features of DLB are fluctuating cognition, recurrent visual hallucinations, and motor features of Parkinson’s disease.40 Lewy bodies, an abnormal aggregation of protein, are found throughout the brain of DLB patients and in patients with other brain disorders, including AD and PDD. In 2008, a PSEN2 missense mutation, a C-to-T substitution at the second position of codon 85 leading to an alanine to valine substitution in the transcribed protein, was found in a proband with the clinical phenotype of Lewy body dementia. Neuropathological examination of the proband showed a mass of cortical Lewy bodies and hallmark lesions of AD. In his family, this mutation was identified in six carriers across two generations, with variable clinical presentation. Except for a young family member that was still asymptomatic, all carriers of the A85V mutation developed AD, DLB, or both. None of the patients carried other mutations in AD-related genes. The pathological PSEN1 mutation, A79V, is homologous to the A85V mutation in PSEN2.85 Sequence phylogenetic analysis suggested that the A85 residue is highly conserved. The mutation is located on the N-terminal, cytoplasmic side, adjacent to the TM-I domain that might be critical for the protein function. Overall, it was predicted that the A85V mutation is pathogenic. In all family members with PSEN2 A85V, the genotype of apolipoprotein E (ApoE) was ε3/ε3, which suggests that α-synuclein pathological structures are linked to PSEN2 A85V without affecting the ApoE ε4 allele.52 A PSEN2 mutation, R71W, was reported in a 73-year-old European patient with cognitive impairment and extrapyramidal symptoms, which was likely undiagnosed for DLB. One of the proband’s brothers also carried the R71W mutation and suffered an unspecified type of dementia. The other brother was healthy and did not have a PSEN2 mutation. The R71W mutation was previously identified in AD patients predicted to be possible pathogenic.70 A PSEN2 mutation, R62H, presented in a DLB patient, with no history of neurological diseases, who showed extrapyramidal signs was characterized by a slight left arm rest tremor, bilateral upper limb postural tremor, and bradykinesia on the left side.86 This mutation, located in the N-terminal of PSEN2, is conserved between PSEN1 and PSEN2. Walker et al showed that the R62H mutation did not affect Aβ42 levels or the Aβ42/Aβ40 ratio.34 Guerreiro et al used PolyPhen-2 to show that the R62H variant is likely benign.41 Based on these data, it is highly probably that PSEN2 R62H can be characterized as “not pathogenic”. Since the age of onset in carriers of the R62H mutation is significantly earlier than in affected non-carriers even after correcting for ApoE genotype, the R62H mutation may function as a disease modifier.48
Breast cancer
Breast cancer is the most common malignancy among women in Europe and the US. Two PSEN2 mutations, R62H and R71W, have been identified in patients with breast cancer. The mutations are located in the hydrophilic, N-terminal domain. In HEK293 cells, the R62H and R71W mutations did not affect the levels of the PSEN2-CTF and PSEN2-NTF proteolytic products or the Aβ(42)/Aβ(40) ratio, but did influence PSEN2 stability. Full-length PSEN2 degenerated rapidly. In a study using transgenic Caenorhabditis elegans, the R62H and R71W mutations compromised PSEN2 function in Notch signaling.49 PSEN2 has several potential roles in cancer. Deng et al and Wolozin et al reported that PSEN2 has pro-apoptotic activity.27,87 A study also has shown that PSEN2 can also adjust β-catenin levels and act in a p53-dependent mechanism to regulate cell growth.49 In 2013, a study suggested a significant role for γ-secretase in breast cancer.88
Frontotemporal dementia
FTD, a clinical phenotype of frontotemporal lobar degeneration, is the second most common form of early-onset (<65 years) neurodegenerative disease after AD.89 It is mainly characterized by deterioration of behavior, personality, and language abilities.89,90 The prevalence of FTD is between 10% and 30% of all presenile dementia.91–96 FTD has a number of clinical phenotypes and pathological subtypes.3,97,98 Clinical and molecular overlaps between AD and FTD or FTD-like phenotypes have been reported.99 To date, at least four PSEN2 mutations have been found in FTD patients. In 2010, PSEN2 R62H was found in a 31-year-old patient. The patient’s healthy mother also carried this mutation. The interaction of the H1 MAPT haplotype and the ApoE ε2 allele might function as a protective modifier against FTD, while the H1 MAPT haplotype unaccompanied by the ApoE ε2 might be a risk enhancer for FTD.43,100 These possibilities imply that modifier, suppressor, and enhancer effects of multiple genes may be crucial for genetic analysis.
Dilated cardiomyopathy
DCM is a heart muscle disease in which the heart becomes enlarged and cannot pump blood efficiently. DCM usually leads to heart failure. The causative factor for DCM has not been determined, but DCM in families is genetically linked. In 2006, the PSEN2 S130L mutation was identified in two Caucasian families. It is highly conserved. Several family members with this mutation suffered DCM and heart failure.36 Presenilin is expressed in multiple tissues, including in the heart, and it is required for cardiac development.101–104 Calcium signaling was altered in cultured skin fibroblasts from carriers of the mutation. The PSEN1 D333G also was identified in a DCM patient. Compared to the phenotypes seen in carriers of PSEN1 D333G, the phenotypes are milder in carriers of PSEN2 S130L, and PSEN2 S130L is not associated with heart failure as often. Currently, it is not clear whether γ-secretase activity is related to DCM. The Notch family of proteins is one of the major transcriptional regulators of cardiac growth and development.105 Disordered Notch signaling is associated with valvular abnormalities, syndromic cardiovascular disease, congenital heart disease, and myocyte dysfunction.106 PSEN2 knockout (PS2KO) mice grow normally without cardiac hypertrophy and fibrosis, while cardiac contractility improved.107 PSEN2 plays an important role in cardiac systolic function by modulating Ca2+ signaling.
Parkinson disease with dementia
Parkinson’s disease (PD) was first described by James Parkinson in 1817. PD is a chronic, progressive, neurological disease that results from the destruction of nerve cells in the basal ganglia. The disease mainly affects movement, but as the neurological damage progresses, the disease often affects mental functions. PDD is an impairment in thinking and reasoning that eventually affects many people with PD. A 77-year-old carrier of PSEN2 V191E showed the PDD phenotype characterized by cognitive decline, visual hallucinations, and confusion during the final years of the PD. This PSEN2 mutation is located at a highly conserved amino acid residue in the protein. In a study by Bram Meeus, the V191E mutation did not exist in more than 1,200 control individuals, so he predicted that V191E is a damaging mutation.70 A PSEN2 R163H variant has been reported in a Swedish PD family in who were also found a de novo α-synuclein A53T mutation. The proband’s mother also carried the mutation PSEN2 R163H, but she was healthy. Nevertheless, this mutation cannot be excluded with certainty as a cause of PD when in combination with α-synuclein.67 PSEN2 S130L was identified in a patient with of LOAD, and his two siblings were diagnosed with PD. Unfortunately, the genetic results from the siblings are not available. The S130L mutation was also detected in the proband’s two unaffected children, but the segregation of the disease could not be determined. The correlation between PD and AD is not clear.
Conclusion
This review described mutations in PSEN2 from diverse disorders. Mutations in PSEN2 were shown to be a rare cause of familial AD. Pathogenic mutations in the PSEN1, PSEN2, and APP gene account for 18%–50% of familial EOAD cases with autosomal dominant pattern of inheritance.108 PSEN genetic testing results could provide genetic counseling for patient’s family members. There is a considerable interest in the application of this genetic information in medical practice through genetic testing and counseling. PSEN2 mutations are involved in not only AD but also in other disorders, including FTD, DLB, PDD, breast cancer, and DCM. Why are PSEN2 mutations found in multiple diseases? Are these diseases related? Until now, the answer to this question has been unclear. There are several possible reasons that PSEN is associated with multiple diseases. PSEN2 is a transmembrane protein that is a component of γ-secretase intramembrane protease. γ-Secretase is required to process several types of integral membrane proteins, and is involved in different signaling pathways. Mutations in PSEN2 may disrupt the normal pathways and lead to different disorders. Thus, it can be hypothesized that these disorders might share underlying genetic factors. On the other hand, different neurodegenerative diseases show slightly different behavioral, language, and motor symptoms. Sometimes it is difficult to distinguish them clearly by clinical diagnosis. Many patients with both PDD and DLB have hallmark changes in the brain, including plaques and tangles that are associated with AD. These observations suggest that there may be a common pathogenetic mechanism in the formation of aggregated proteins. Therefore, mutations in PSEN2 might play a role in Aβ, α-synuclein, and tau aggregation.
Overall, genetic studies have already indicated that PSEN2 may affect people with FTD, PDD, LBD, breast cancer, and DCM. How presenilin 2 is implicated in the pathogenesis of these diseases is still unclear. This question needs to be further explored.
Acknowledgment
This manuscript was supported by grants from the Gachon University Gil Medical Center (grant number 2013-30) and the Korea Health Technology R&D Project (HI14C3331) through the Korea Health Industry Development Institute (KHIDI), Ministry of Health & Welfare, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52(1):3–13. | ||
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–227. | ||
Żekanowski C, Styczyńska M, Pepłońska B, et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol. 2003;184(2):991–996. | ||
Bagyinszky E, Youn YC, An SS, Kim SY. The genetics of Alzheimer’s disease. Clin Interv Aging. 2014;9:535–551. | ||
Alzheimer Disease and Frontotemporal Dementia Mutation Database [homepage on the Internet]. Available from: http://www.molgen.ua.ac.be/admutations. Accessed February 16, 2015. | ||
Youn YC, Bagyinszky E, Kim H, Choi BO, An SS, Kim S. Probable novel PSEN2 Val214Leu mutation in Alzheimer’s disease supported by structural prediction. BMC Neurol. 2014;14(1):105. | ||
Ertekin-Taner N. Genetics of Alzheimer’s disease: a centennial review. Neurol Clin. 2007;25(3):611–667. | ||
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | ||
Renbaum P, Beeri R, Gabai E, et al. Egr-1 upregulates the Alzheimer’s disease presenilin-2 gene in neuronal cells. Gene. 2003;318:113–124. | ||
Ounallah-Saad H, Beeri R, Goshen I, Yirmiya R, Renbaum P, Levy-Lahad. Transcriptional regulation of the murine Presenilin-2 gene reveals similarities and differences to its human orthologue. Gene. 2009;446(2):81–89. | ||
Hendrickx A, Pierrot N, Tasiaux B, et al. Epigenetic induction of EGR-1 expression by the amyloid precursor protein during exposure to novelty. PLoS One. 2013;8(9):e74305. | ||
Sato N, Hori O, Yamaguchi A, et al. A novel presenilin-2 splice variant in human Alzheimer’s disease brain tissue. J Neurochem. 1999;72(6):2498–2505. | ||
Sato N, Imaizumi K, Manabe T, et al. Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J Biol Chem. 2001;276(3): 2108–2114. | ||
Smith MJ, Sharples RA, Evin G, et al. Expression of truncated presenilin 2 splice variant in Alzheimer’s disease, bipolar disorder, and schizophrenia brain cortex. Mol Brain Res. 2004;127(1):128–135. | ||
Sato H, Takeuchi T, Sakai KL. Temporal cortex activation during speech recognition: an optical topography study. Cognition. 1999;73(3):B55–B66. | ||
Nishikawa A, Manabe T, Katayama T, et al. Novel function of PS2V: change in conformation of tau proteins. Biochem Biophys Res Commun. 2004;318(2):435–438. | ||
De Strooper B, Beullens M, Contreras B, et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J Biol Chem. 1997;272(6):3590–3598. | ||
Li J, Xu M, Zhou H, et al. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90(5):917–927. | ||
Li X, Dang S, Yan C, et al. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493(7430):56–61. | ||
Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. Sci World J. 2003;3:497–519. | ||
Annaert WG, Levesque L, Craessaerts K, et al. Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J Cell Biol. 1999;147(2):277–294. | ||
De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(1):a006304. | ||
Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature Med. 1997;3(1):67–72. | ||
Leissring MA, Yamasaki TR, Wasco W, et al. Calsenilin reverses presenilin-mediated enhancement of calcium signaling. Proc Natl Acad Sci U S A. 2000;97(15):8590–8593. | ||
Leissring MA, LaFerla FM, Callamaras N, et al. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis. 2001;8(3):469–478. | ||
Vito P, Wolozin B, Ganjei JK, et al. Requirement of the Familial Alzheimer’s Disease Gene PS2 for Apoptosis OPPOSING EFFECT OF ALG-3. Journal of Biological Chemistry. 1996;271(49):31025–31028. | ||
Wolozin B, Iwasaki K, Vito P, et al. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274(5293):1710–1713. | ||
Da Costa CA, Paitel E, Mattson MP, et al. Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc Natl Acad Sci U S A. 2002;99(6):4043–4048. | ||
Araki W, Yuasa K, Takeda S, et al. Pro-apoptotic effect of presenilin 2(PS2) overexpression is associated with down-regulation of Bcl-2 in cultured neurons. J Neurochem. 2001;79(6):1161–1168. | ||
Lee YJ, Kim JE, Hwang IS, et al. Alzheimer’s phenotypes induced by overexpression of human presenilin 2 mutant proteins stimulate significant changes in key factors of glucose metabolism[J]. Mol Med Rep. 2013;7(5):1571–1578. | ||
Larner AJ. Presenilin-1 mutation Alzheimer’s disease: a genetic epilepsy syndrome? Epilepsy & Behavior. 2011;21(1):20–22. | ||
Bird TD, Levy-Lahad E, Poorkaj P, et al. Wide range in age of onset for chromosome 1-related familial Alzheimer’s disease. Ann Neurol. 1996;40(6):932–936. | ||
Sherrington R, Froelich S, Sorbi S, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Gen. 1996;5(7):985–988. | ||
Walker ES, Martinez M, Brunkan AL, et al. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Aβ 42/40 ratios. J Neurochem. 2005;92(2):294–301. | ||
Zatti G, Burgo A, Giacomello M, et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium. 2006;39(6):539–550. | ||
Li D, Parks SB, Kushner JD, et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79(6):1030–1039. | ||
Zatti G, Ghidoni R, Barbiero L, et al. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol Dis. 2004;15(2):269–278. | ||
Niu F, Yu S, Zhang Z, et al. A novel mutation in the PSEN2 gene (N141Y) associated with early-onset autosomal dominant Alzheimer’s disease in a Chinese Han family. Neurobiol Aging. 2014;35(10):2420.e1–2420.e5. | ||
Bai YF, Tian J, Quan WX, Maeda K. Association of mutations of presenilin-2 gene and sporadic Alzheimer’s disease. J Chin Med Univ. 2011;40:357–363. | ||
Kim TH. Diagnosis and management of dementia with Lewy bodies. J Korean Geriatr Psychiatry. 2012;16(2):75–81. | ||
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. | ||
Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127(7):1641–1649. | ||
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40(8):562–583. | ||
Ertekin-Taner N, Younkin LH, Yager DM, et al. Plasma amyloid β protein is elevated in late-onset Alzheimer disease families. Neurology. 2008;70(8):596–606. | ||
Cruts M, Van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7(1):43–51. | ||
Gallo M, Tomaino C, Puccio G, et al. Novel MAPT Val75Ala mutation and PSEN2 Arg62Hys in two siblings with frontotemporal dementia. Neurol Sci. 2010;31(1):65–70. | ||
Lohmann E, Guerreiro RJ, Erginel-Unaltuna N, et al. Identification of PSEN1 and PSEN2 gene mutations and variants in Turkish dementia patients. Neurobiol Aging. 2012;33(8):1850.e17–1850.e27. | ||
Cruchaga C, Chakraverty S, Mayo K, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One. 2012;7(2):e31039. | ||
To MD, Gokgoz N, Doyle TG, et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25(25):3557–3564. | ||
Dobricic V, Stefanova E, Jankovic M, et al. Genetic testing in familial and young-onset Alzheimer’s disease: mutation spectrum in a Serbian cohort. Neurobiol Aging. 2012;33(7):1481.e7–1481.e12. | ||
Wallon D, Rousseau S, Rovelet-Lecrux A, et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J Alzheimers Dis. 2012;30(4):847–856. | ||
Piscopo P, Marcon G, Piras MR, et al. A novel PSEN2 mutation associated with a peculiar phenotype. Neurology. 2008;70(17):1549–1554. | ||
Finckh U, Müller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66(1):110–117. | ||
Finckh U, Kuschel C, Anagnostouli M, et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6(2):85–89. | ||
Binetti G, Signorini S, Squitti R, et al. Atypical dementia associated with a novel presenilin-2 mutation. Ann Neurol. 2003;54(6):832–836. | ||
Giacomello M, Barbiero L, Zatti G, et al. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol Dis. 2005;18(3):638–648. | ||
El Kadmiri N, Zaid N, Zaid Y, et al. Novel presenilin mutations within Moroccan patients with early-onset Alzheimer’s disease. Neuroscience. 2014;269:215–222. | ||
Müller U, Winter P, Bolender C, et al. Previously unrecognized missense mutation E126K of PSEN2 segregates with early onset Alzheimer’s disease in a family. J Alzheimers Dis. 2014;42(1):109–113. | ||
Tedde A, Nacmias B, Ciantelli M, et al. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch Neurol. 2003;60(11):1541–1544. | ||
Sorbi S, Tedde A, Nacmias B, et al. Novel presenilin 1 and presenilin 2 mutations in early-onset Alzheimer’s disease families. Neurobiol Aging. 2002;23(1S):S312. | ||
Tomaino C, Bernardi L, Anfossi M, et al. Presenilin 2 Ser130Leu mutation in a case of late-onset “sporadic” Alzheimer’s disease. J Neurol. 2007;254(3):391–393. | ||
Sassi C, Guerreiro R, Gibbs R, et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p. L166V and p. S230R) in British early-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(10):2422.e13–2422.e16. | ||
Bernardi L, Tomaino C, Anfossi M, et al. Late onset familial Alzheimer’s disease: novel presenilin 2 mutation and PS1 E318G polymorphism. J Neurol. 2008;255(4):604–606. | ||
Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–778. | ||
Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(4):1143–1154. | ||
Lao JI, Beyer K, Fernández-Novoa L, et al. A novel mutation in the predicted TM2 domain of the presenilin 2 gene in a Spanish patient with late-onset Alzheimer’s disease. Neurogenetics. 1998;1(4):293–296. | ||
Puschmann A, Ross OA, Vilariño-Güell C, et al. A Swedish family with de novo α-synuclein A53T mutation: Evidence for early cortical dysfunction. Parkinsonism Relat Disord. 2009;15(9):627–632. | ||
Clarimón J, Guerreiro R, Lleó A, et al. Genetic screening in a large cohort of early-onset Alzheimer’s disease patients from Spain: novel mutations in the amyloid precursor protein and presenilines. Alzheimers Dement. 2008;4(4):T583. | ||
Piscopo P, Talarico G, Crestini A, et al. A novel mutation in the predicted TMIII domain of the PSEN2 gene in an Italian pedigree with atypical Alzheimer’s disease. J Alzheimers Dis. 2010;20(1):43–47. | ||
Meeus B, Verstraeten A, Crosiers D, et al. DLB and PDD: a role for mutations in dementia and Parkinson disease genes? Neurobiol Aging. 2012;33(3):629.e5–629.e18. | ||
Marcon G, Di Fede G, Giaccone G, et al. A novel Italian presenilin 2 gene mutation with prevalent behavioral phenotype. J Alzheimers Dis. 2009;16(3):509–511. | ||
Lee JH, Kahn A, Cheng R, et al. Disease-related mutations among Caribbean Hispanics with familial dementia. Molecular Genet Genomic Med. 2014;2(5):430–437. | ||
Sassi C, Guerreiro R, Gibbs R, et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(12):2881.e1–2881.e6. | ||
Finckh U, Alberici A, Antoniazzi M, et al. Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology. 2000;54(10):2006–2008. | ||
Testi S, Fabrizi GM, Pompanin S, et al. Autosomal dominant Alzheimer’s disease with early frontal lobe involvement associated with the Met239Ile mutation of Presenilin 2 gene. J Alzheimers Dis. 2012;31(1):7–11. | ||
Tremolizzo L, Susani E, Mapelli C, et al. First report of PSEN2 mutation presenting as posterior cortical atrophy. Alzheimer Dis Assoc Disord. Epub 2014 Jul 9. | ||
Marcon G, Giaccone G, Cupidi C, et al. Neuropathological and clinical phenotype of an Italian Alzheimer family with M239V mutation of presenilin 2 gene. J Neuropath Exp Neurol. 2004;63(3):199–209. | ||
Croes EA, Theuns J, Houwing-Duistermaat JJ, et al. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J Neurol Neurosurg Psychiatry. 2004;75(8):1166–1170. | ||
Lleó A, Castellví M, Blesa R, et al. Uncommon polymorphism in the presenilin genes in human familial Alzheimer’s disease: not to be mistaken with a pathogenic mutation. Neurosci Lett. 2002;318(3):166–168. | ||
Lindquist SG, Hasholt L, Bahl J MC, et al. A novel presenilin 2 mutation (V393M) in early-onset dementia with profound language impairment. Eur J Neurol. 2008;15(10):1135–1139. | ||
Lindquist SG, Schwartz M, Batbayli M, et al. Genetic testing in familial AD and FTD: mutation and phenotype spectrum in a Danish cohort. Clin Genet. 2009;76(2):205–209. | ||
Lleó A, Blesa R, Queralt R, et al. Frequency of mutations in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch Neurol. 2002;59(11):1759–1763. | ||
Ezquerra M, Lleó A, Castellví M, et al. A novel mutation in the PSEN2 gene (T430M) associated with variable expression in a family with early-onset Alzheimer disease. Arch Neurol. 2003;60(8):1149–1151. | ||
Lleo A, Blesa R, Gendre J, et al. A novel presenilin 2 gene mutation (D439A) in a patient with early-onset Alzheimer’s disease. Neurology. 2001;57(10):1926–1928. | ||
Kauwe JSK, Jacquart S, Chakraverty S, et al. Extreme cerebrospinal fluid amyloid β levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann Neurol. 2007;61(5):446–453. | ||
Raciti L, Nicoletti A, Le Pira F, et al. Presenilin-2 gene mutation presenting as Lewy body dementia? Neurol Sci. 2011;32(3):533–534. | ||
Deng G, Pike CJ, Cotman CW. Alzheimer-associated presenilin-2 confers increased sensitivity to poptosis in PC12 cells. FEBS Lett. 1996;397(1):50–54. | ||
Peltonen HM, Haapasalo A, Hiltunen M, Kataja V, Kosma VM, Mannermaa A. γ-Secretase components as predictors of breast cancer outcome. PLoS One. 2013;8(11):e79249. | ||
Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. | ||
Hodges JR. Frontotemporal dementia (Pick’s disease): clinical features and assessment. Neurology. 2001;56(Suppl 4):S6–S10. | ||
Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology. 2002;58(11):1615–1621. | ||
Cairns NJ. Neuronal intermediate filament inclusion disease. Handb Clin Neurol. 2008;89:443–448. | ||
Snowden JS. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. | ||
Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs. 2010;24(5):375–398. | ||
Borroni B, Alberici A, Archetti S, et al. New insights into biological markers of frontotemporal lobar degeneration spectrum. Curr Med Chem. 2010;17(10):1002–1009. | ||
Rosso SM, Roks G, Stevens M, et al. Complex compulsive behaviour in the temporal variant of frontotemporal dementia. J Neurol. 2001;248(11):965–970. | ||
Hodges JR, Miller B. The neuropsychology of frontal variant frontotemporal dementia and semantic dementia. Introduction to the special topic papers: part II. Neurocase. 2001;7(2):113–121. | ||
Mackenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. | ||
Lindquist SG, Brændgaard H, Svenstrup K, et al. Frontotemporal dementia linked to chromosome 3 (FTD-3) – current concepts and the detection of a previously unknown branch of the Danish FTD-3 family. Eur J Neurol. 2008;15(7):667–670. | ||
Bernardi L, Maletta RG, Tomaino C, et al. The effects of APOE and tau gene variability on risk of frontotemporal dementia. Neurobiol Aging. 2006;27(5):702–709. | ||
Levy-Lahad E, Poorkaj P, Wang K, et al. Genomic structure and expression of STM2, the chromosome 1 familial Alzheimer disease gene. Genomics. 1996;34(2):198–204. | ||
Hébert SS, Serneels L, Dejaegere T, et al. Coordinated and widespread expression of γ-secretase in vivo: evidence for size and molecular heterogeneity. Neurobiol Dis. 2004;17(2):260–272. | ||
Nakajima M, Moriizumi E, Koseki H, et al. Presenilin 1 is essential for cardiac morphogenesis. Dev Dyn. 2004;230(4):795–799. | ||
Donoviel DB, Hadjantonakis AK, Ikeda M, et al. Mice lacking both presenilin genes exhibitearlyembryonic patterning defects. Genes Dev. 1999;13(21):2801–2810. | ||
Hansson EM, Lendahl U, Chapman G. Notch signaling in development and disease. Semin Cancer Biol. 2004;14(5):320–328. | ||
Noseda M, McLean G, Niessen K, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. 2004;94(7):910–917. | ||
Takeda T, Asahi M, Yamaguchi O, et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005;19(14):2069–2071. | ||
Kowalska A. Genetic counseling and testing for families with Alzheimer’s disease. Neurol Neurochir Pol. 2004;38(6):495–501. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.