")
Back to Journals » OncoTargets and Therapy » Volume 9
Mutational analysis of primary and metastatic colorectal cancer samples underlying the resistance to cetuximab-based therapy
Authors Nemecek R, Berkovcova J, Radova L, Kazda T , Mlcochova J, Vychytilova-Faltejskova P, Slaby O , Svoboda M
Received 21 December 2015
Accepted for publication 12 May 2016
Published 28 July 2016 Volume 2016:9 Pages 4695—4703
DOI https://doi.org/10.2147/OTT.S102891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Min Li
Radim Nemecek,1 Jitka Berkovcova,2 Lenka Radova,3 Tomas Kazda,4 Jitka Mlcochova,3 Petra Vychytilova-Faltejskova,1,3 Ondrej Slaby,1,3 Marek Svoboda1
1Department of Comprehensive Cancer Care, Masaryk Memorial Cancer Institute, Masaryk University, Brno, Czech Republic; 2Department of Oncological and Experimental Pathology, Masaryk Memorial Cancer Institute, Brno, Czech Republic; 3Central European Institute of Technology, Masaryk University, Brno, Czech Republic; 4Department of Radiation Oncology, Masaryk Memorial Cancer Institute, Masaryk University, Brno, Czech Republic
Purpose: Although several molecular markers predicting resistance to cetuximab- or panitumumab-based therapy of metastatic colorectal cancer were described, mutations in RAS proto-oncogenes remain the only predictors being used in daily clinical practice. However, 35%–45% of wild-type RAS patients still do not respond to this anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibody-based therapy, and therefore the definition of other predictors forms an important clinical need. The aim of the present retrospective single-institutional study was to evaluate potential genes responsible for resistance to anti-EGFR therapy in relation to mutational analysis of primary versus metastatic lesions.
Patients and methods: Twenty-four paired primary and corresponding metastatic tissue samples from eight nonresponding and four responding metastatic colorectal cancer patients treated with cetuximab-based therapy were sequenced using a next-generation sequencing panel of 26 genes involved in EGFR signaling pathway and colorectal carcinogenesis.
Results: Mutational status of primary tumors and metastatic lesions was highly concordant in TP53, APC, CTNNB1, KRAS, PIK3CA, PTEN, and FBXW7 genes. Metastatic samples harbor significantly more mutations than primary tumors. Potentially negative predictive value of FBXW7 mutations in relationship to anti-EGFR treatment outcomes was confirmed. Finally, new occurrences of activating KRAS mutations were identified in a group of patients initially determined as wild-type RAS by routinely used qPCR-based RAS mutational tests. All newly detected activating KRAS mutations most likely led to cetuximab treatment failure.
Conclusion: The results of the present study suggest a need of careful consideration of previously published results of anti-EGFR-targeted therapy with regard to potentially inaccurate diagnostic tools used in the past. Based on our findings, we recommend more extensive use of next-generation sequencing testing in daily clinical practice, as it brings a significant added value in terms of validity of the diagnostic procedure.
Keywords: colorectal cancer, cetuximab, resistance to anti-EGFR therapy, next-generation sequencing, FBXW7, KRAS
Introduction
Colorectal cancer (CRC) is one of the most frequent cancers worldwide, with ~1.36 million newly diagnosed cases and >694,000 fatalities each year.1 It is also the second leading cause of cancer-related death in the western world. The metastatic stage of disease is confirmed in ~25% of patients already at the time of diagnosis. Additional 25%–40% of patients develop metastases after the primary surgical and oncological treatment.2 Most of the metastatic patients are treated with systemic palliative chemotherapy in combination with monoclonal antibodies (moAbs) against the epidermal growth factor receptor (EGFR) or vascular endothelial growth factor. All these treatment modalities lead to overall survival extension.3 Chemotherapy doublets in combination with anti-epidermal growth factor receptor (anti-EGFR) moAbs show response rate of 55%–65% and are therefore successfully used, especially in order to achieve downstaging and conversion of the borderline resectable disease to the resectable one.4 Still, there is a large number of patients who exhibit resistance and will not respond to such treatment.
Resistance to EGFR blockade is quite frequent and is caused by several factors.5,6 Primary, intrinsic resistance arises within the process of colorectal carcinogenesis and is usually related to constitutive activation of signal pathways downstream of EGFR. Secondary, acquired resistance occurs during the EGFR blockade or other anticancer treatments. It is caused by mutations that disrupt binding of cetuximab or panitumumab to EGFR, by pathway-bypass mutations (such as KRAS and BRAF alterations), or by activation of parallel pathways driven by receptor tyrosine kinases such as MET and HER2. According to recent studies, epigenetic factors such as microRNAs (miRNAs) play an important role in the response to anti-EGFR moAbs, as they are involved in signaling pathways and regulate gene expression at the posttranscriptional level.7,8
Mutations in oncogenes, KRAS or NRAS (overall called “RAS”), occur in ~50% of patients with metastatic colorectal cancer (mCRC) and represent a routinely used negative predictive indicator in clinical practice.9 However, 35%–45% of wild-type RAS mCRC patients still do not respond to this treatment and therefore other molecular predictors have been intensively investigated.10 There are some data about the negative predictive value of BRAF, PIK3CA, and PTEN mutations, HER2 gene copy number variations, and miRNAs miR-31-5p/3p.6,7,11,12 On the other hand, high EGFR gene copy number and high level of EGFR ligands, epiregulin and amphiregulin, could increase the probability of response to anti-EGFR treatment.11,13 Nevertheless, most of these tests have been currently studied only within the nonrandomized retrospective clinical trials and are not used in daily clinical practice. A combination of molecular markers could increase the sensitivity of predictive diagnostic tools and therefore a growing need to analyze several genes in parallel is apparent.11,14 High-throughput sequencing methods represent a helpful support in detecting numerous genetic changes implicated in anti-EGFR moAbs resistance. Recently, the clinical use of these methods has been rapidly expanding.
The aim of the present study was to clarify the mechanisms of resistance to anti-EGFR therapy in patients with mCRC treated with cetuximab-based therapy who did not respond to it. The next-generation sequencing (NGS) panel of 26 genes involved in colorectal carcinogenesis and EGFR signaling pathway was employed.5,15 The metastatic tissue samples were examined, as they best describe the advanced metastatic disease phenotype. Simultaneously, primary tumor tissue samples of the same patients were analyzed in order to distinguish whether discovered mutations were more likely related to progression from localized to metastatic disease, or if they had arisen already under the early selection pressure changes during the process of carcinogenesis. These findings could play an important role in the mechanisms of primary and secondary resistance to anti-EGFR therapy.
Patients and methods
Patient selection
Tissue samples were retrieved from patients with histologically confirmed colon or rectal adenocarcinoma treated with cetuximab-based therapy at Masaryk Memorial Cancer Institute (MMCI, Brno, Czech Republic) between August 2005 and August 2014. At the beginning of cetuximab treatment, all patients were at metastatic stage of the disease and had confirmed wild-type KRAS CRC according to KRAS mutational test required at a given time. Subsequently, they were regularly followed up for progression of disease and death. The informed consent form was signed by all patients and the study protocol was approved by the Institutional Review Board of MMCI.
From the initial number of 160 evaluable patients with the abovementioned characteristics, a subgroup of patients was selected fulfilling two selection criteria: 1) proven resistance to anti-EGFR moAb-based treatment (defined as volume enlargement of metastatic lesions at the time of the evaluation of treatment response, which is usually provided after 2–4 months) and 2) availability of tissue from both primary tumor and metastatic lesion in sufficient amount. Out of 40 patients with progressive disease, 16 patients met the abovementioned selection criteria. In this subgroup, we additionally completed the entire mutation status of RAS (KRAS and NRAS in exons 1, 2, 3, and 4) and BRAF (V600E) according to the latest guidelines. The Cobas® KRAS Mutation Test, the CRC RAScan™ Combination Test, and the Cobas® 4800 BRAF V600 Mutation Test were used. We found KRAS mutation (codon 117N) in two patients, NRAS mutation (codon Q61) in three patients, and BRAF mutation (V600E) in one patient. Two other patients had to be excluded from the analysis due to an insufficient DNA quality necessary for NGS sequencing. Finally, eight patients met all the required criteria.
As a control group, we selected patients with wild-type mCRC, who reached complete response (CR) or strong partial response (PR) according to Response Evaluation Criteria In Solid Tumors criteria during the anti-EGFR therapy. Similar to the first group, availability of primary and metastatic tumor tissue in sufficient amount and quality was required. From the total number of 56 patients with CR or PR, we enrolled four patients to the final NGS analysis. Altogether, the multiparallel sequencing was performed on 24 samples from 12 patients. Clinicopathological features of enrolled patients are summarized in Table 1.
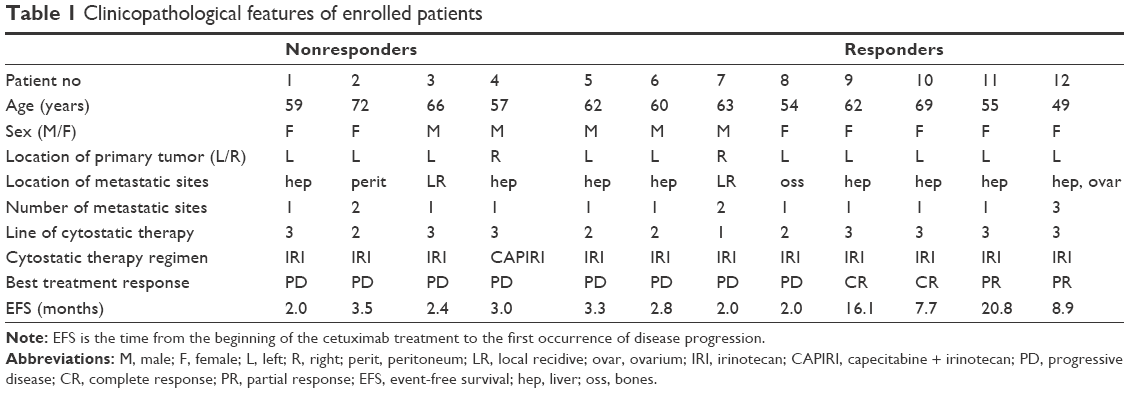 | Table 1 Clinicopathological features of enrolled patients |
Gene selection, isolation of DNA, and library preparation
The TruSight™ Tumor (Illumina, Inc., San Diego, CA, USA) gene panel was assembled to provide deep coverage of 26 genes for low mutant allele detection within hot spot regions involved in solid tumors. Gene regions are listed in Table S1. DNA was isolated from formalin-fixed, paraffin-embedded (FFPE) samples using the Cobas® DNA Sample Preparation kit (Hoffman-La Roche Ltd., Basel, Switzerland) according to the manufacturer’s protocol. The average percentage of tumor cells in tissue sections was 52%. DNA library preparation was performed according to TruSight™ Tumor Protocol (Illumina, Inc.). A volume of 100 μL of DNA was obtained from each sample and subsequently concentrated to 25 μL using the DNA Clean & Concentrator™-5 (Zymo Research Corp., Irvine, CA, USA). The concentration and purity of all samples were measured again. The ability of DNA amplification was tested using the FFPE QC Kit (Illumina, Inc.). Using the standardized DNA, ΔCT of each sample was assessed. DNAs were diluted according to this value into the hybridization reactions with a mixture of FPA (forward primer A) oligo or FPB (forward primer B) oligo. After the primary denaturation at a temperature of 95°C, hybridization was conducted at a temperature of 40°C. Afterward, nonhybridized oligos were washed out, and extension and ligation were performed.
Denatured DNA fragments were used as templates for PCR, where they were amplified and indexed. Subsequently, DNA libraries tied to magnetic beads got rid of short fragments of DNA. To check the DNA libraries, Agilent DNA 1000 chips were used, and concentration of every single library was measured using the Qubit fluorometer. All libraries were diluted to 4 nM and subsequently denatured and pooled equimolarly. The resulting pooled library was diluted into 10 pM concentration and 1% 20 pM PhiX library was added. Final library sequencing was conducted using the 300v2 sequencing kit and the standardized flow cell. Sequencing data analysis was conducted using the Illumina Variant Studio software and R/Bioconductor package. Alterations found at a frequency of <10% were excluded, based on the hypothesis that mutations at lower frequencies could marginally affect tumor behavior. This filtering step allowed removal of most of the variations derived from formalin-fixed artifacts, as well.16 The remaining mutations were compared with data presented in public databases (PolyPhen).17 Annotated nonpathogenic variations were excluded from results, whereas the remaining potentially pathogenic variations and mutations of unknown significance were retained.
Statistical analysis
Basic descriptive statistics were employed: absolute and relative frequencies for categorical variables and median for continuous variables. Robust experienced biomedical statistics were applied for analysis of raw NGS data. Statistical analysis was performed using statistical computing software “R”, Version 3.1.3.
Results
We investigated DNA of primary (P) and corresponding metastatic (M) samples from 12 RAS and BRAF wild-type patients (five men and seven women) with a median age of 61 years (Table 1). Eight patients were nonresponsive to therapy, whereas four patients displayed CR or PR of disease. Nine out of 12 patients had single metastatic location, predominantly in the liver. One patient was treated with cetuximab within the first line, four patients were treated within the second line, and seven patients within the third line of systemic anticancer therapy. All patients were treated with cetuximab + irinotecan. Capecitabine was added to the combination in one case. Later, this combination was no longer used as the highlights of the toxicity of capecitabine in combination with cetuximab were published.18 Median time to progression was 2.6 months in nonresponders and 12.5 months in responders.
In NGS analysis, at least one mutation in each sample was detected. The most frequently mutated genes were KRAS, TP53, and APC, which were mutated in nine (38%), eight (33%), and seven (29%) samples, respectively. These results are in agreement with the published literature.15 Mutation rates for the remaining genes were as follows: 25% for FBXW7 and PIK3CA; 13% for MET and PTEN; 8% for KIT, STK11, AKT1, FOXL2, and CTNNB1; and less for the other genes. No mutation was detected in MAP2K1 and CDH1. Mutation rates for every single gene in the responder and nonresponder groups are shown in Figure 1.
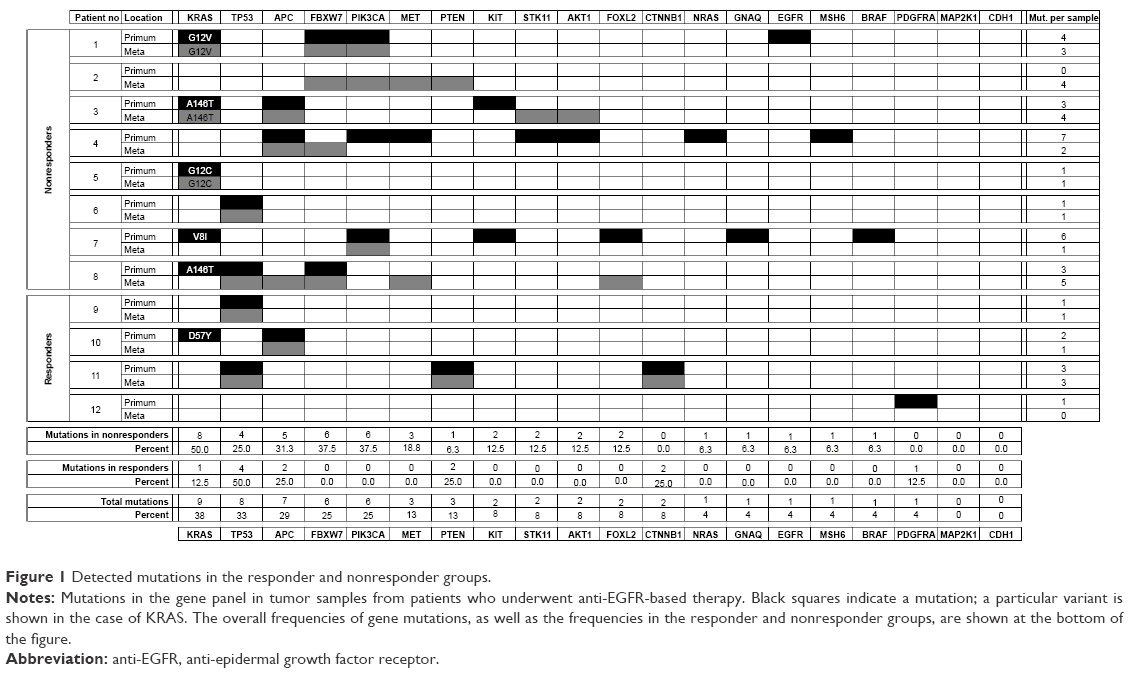 | Figure 1 Detected mutations in the responder and nonresponder groups. |
KRAS, the gene with the highest mutation frequency, displayed mutations almost exclusively in the nonresponder group, the only exception being the primary tumor sample from patient no 10, where mutation D57Y in KRAS was detected. According to PolyPhen, this mutation is described as “probably damaging”, being found in HRAS and causing CDC25 independency.19 Despite initial analyses based on the abovementioned diagnostic tests (Thera Screen® K-RAS Mutation Kit, Cobas® KRAS Mutation Test Kit, and CRC RAScan™ Combination Kit) indicating that all tumors carried a wild-type RAS gene, the subsequent NGS analysis revealed that six primary tumors and three metastases harbored a mutation in this gene. In particular, we found the following variations in KRAS alterations: G12V, G12C, A146T, V8I, and D57Y. Mutations G12V, G12C, and A146T were previously described as pathogenic for the KRAS gene and recorded in the COSMIC and PolyPhen databases, and mutation V8I was described as “possibly damaging”, according to PolyPhen.17 All of these mutations were found in patients resistant to anti-EGFR treatment. The presence of D57Y, the only one KRAS mutation discovered in the responder group, was discussed earlier.
Besides KRAS, the mutation status of BRAF, NRAS, PIK3CA, and PTEN was already shown to correlate with cetuximab resistance.20–25 In our study, we detected BRAF mutation (G596D) in P of patient no 7, NRAS mutation (P110L) in P of patient no 4, PIK3CA alterations (E542K, L456M, G106D, E545K, and E547K) even in six samples (in P of patient no 1, 4, and 7 and in M of patient no 1, 2, and 7), and PTEN alterations in three samples (in P of patient no 11 and in M of patients no 2 and 11). All but two of these alterations belonged to tumors that did not respond to anti-EGFR therapy. The only exception was the P and M of patient no 11 (from the responder group), where PTEN alteration R130* with unknown clinical significance was detected. According to the published literature, known activating mutations in KRAS, BRAF, and NRAS appeared to be mutually exclusive (Figure 1). Because all of these genes are downstream effectors of the EGFR-induced pathways, they appear to have a potentially significant impact on cetuximab resistance.
Except for genes involved in the EGFR signaling pathway, the other genes often mutated in CRC included FBXW7 (25%), MET (13%), KIT (8%), STK11 (8%), AKT1 (8%), FOXL2 (8%), and CTNNB1 (8%), whereas CTNNB1 mutation G34E was found only in samples of patient no 11 with a very good response to EGFR blockade, all other mentioned alterations were detected in patients who did not respond to this therapy. Most frequent were the mutations in FBXW7 gene, particularly R609W, W366L, W485I, T504I, F441S, I528F, and G517R. These alterations deserve a special interest, because according to the published literature some of them may predict cetuximab resistance.26 Other discovered mutations are described in Figure 1, and the relevance of these mutations in colorectal cancerogenesis is summed up in Table 2.27–30
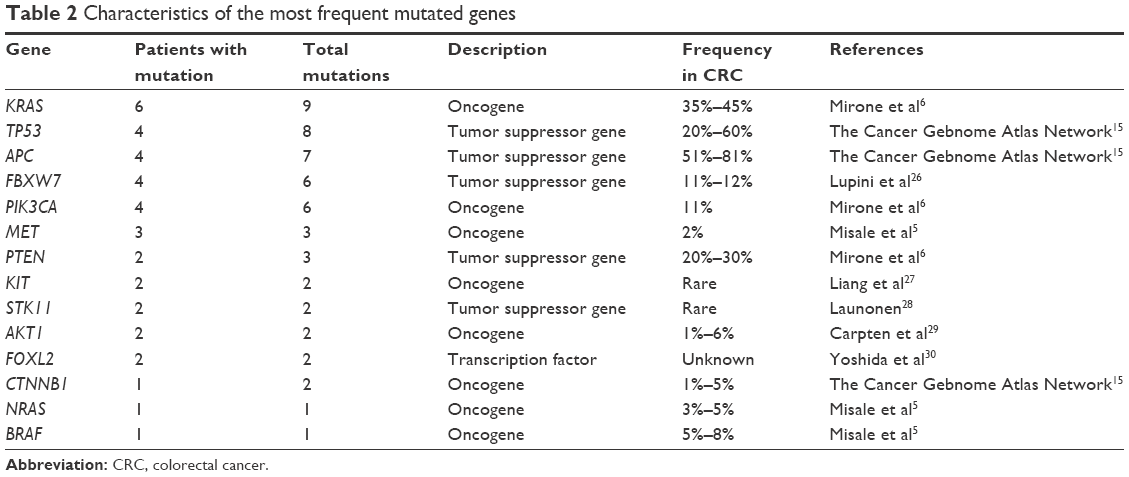 | Table 2 Characteristics of the most frequent mutated genes |
Discussion
Anti-EGFR therapy is frequently used for the treatment of patients with wild-type RAS mCRC. At the beginning of this targeted therapy era in 2004, no predictive marker was known and the moAbs were prescribed to all patients. Investigations into the molecular basis of response to EGFR blockade began in 2005. Since then, rapidly accumulating evidence indicated that the resistance to EGFR blockade in mCRC is related to constitutive activation of signaling pathways downstream of EGFR.31–33 Initially, KRAS exon 2 (codons 12 and 13) mutations were discovered to be responsible for the treatment failure. As a consequence, the use of cetuximab and panitumumab was restricted by the authorities only for the patients without these mutations (first by European Medicine Agency [EMA] in 2008 and later by the US Food and Drug Administration [FDA] in 2009). Because not all KRAS wild-type patients benefit from anti-EGFR treatment, research has been increasingly focused on the validation of additional biomarkers of resistance. It was discovered that although 80% of KRAS variants occur in exon 2 (codons 12 and 13), oncogenic mutations also affect KRAS codons 59 and 61 (in exon 3) and 117 and 146 (in exon 4) and the same codons in NRAS gene.34 Multiple studies have shown that mutations in KRAS exon 3 and 4 or NRAS exons 2–4 also predict the lack of clinical benefit to anti-EGFR-targeted therapy. Therefore, in 2013 EMA and FDA restricted the further use of cetuximab and panitumumab only to patients with wild-type RAS mCRC.9,35,36 Since 2013, no other biomarker reached the point of being widely used in the daily clinical practice. A number of scientific articles about the BRAF V600E mutation were published, especially in pretreated patients with mCRC but none of these studies convincingly confirmed its negative predictive value.37–39 Several other markers or their combinations had been intensively studied, such as PIK3CA and PTEN mutations, HER2 gene copy number variations, EGFR gene copy number, or level of EGFR ligands, epiregulin and amphiregulin, and miRNAs miR-31-5p/3p.7,8,11–14 However, none of these markers entered into the daily clinical practice.
In the present study, we performed a targeted sequencing of a group of 26 genes involved in colorectal carcinogenesis and EGFR signaling pathway. Our aim is to describe their involvement in the mechanisms of resistance to EGFR blockade. For this purpose, we sequenced the paired primary and metastatic lesions obtained from patients who did or did not respond to cetuximab-based therapy. A relatively low number of sequenced patients is a limitation that must be taken into account when interpreting the results of this study. Still, there are some interesting findings to be discussed.
The mutational status of primary versus metastatic lesions was highly concordant in TP53, APC, and CTNNB1 genes. Partial (but still high) concordance was found in KRAS (mutation in both P and M in three patients, mutation only in P in three patients), FBXW7 (mutation in both P and M in two patients, mutation only in P in one patient, and only in M in one patient), PIK3CA (mutation in both P and M in two patients and mutation only in P in other two patients), and PTEN (mutation in both P and M in one patient and only in M in one patient). Other genes were mutated either in P or in M (Figure 1). KRAS mutations in the corresponding P and M in three patients were detected (variants G12V, G12C, and A146T) after excluding samples with mutations V8I and D57Y with unclear clinical significance. Mutation (A146T) in P only was detected only in one case (patient no 8). In accordance with the published literature, the mutational status of the abovementioned genes was highly concordant in primary and metastatic tumors.40 Our results suggest that the biopsy of primary tumor could be a sufficient alternative to the biopsy of metastatic lesion in most cases in order to carry out a mutational analysis of mCRC.
In comparison with the responder group, significantly more alterations in the nonresponder group were discovered (12 alterations with median one mutation per sample and 46 detected mutations with median three mutations per sample, respectively). In the responder group, TP53 was the most frequently affected gene with mutation in four out of eight samples, followed by APC, CTNNB1, and PTEN with mutation in two samples in every single gene. Additional two mutations were in KRAS (the KRAS D57Y mutation discussed earlier with unclear clinical significance) and in PDGFRA (mutation E644Q). Both mutations were present only in the primary tumor and not in the metastatic lesion. While TP53, APC, and CTNNB1 mutations are quite common events during the colorectal carcinogenesis, inactivation of tumor suppressor PTEN could be potentially related to cetuximab resistance. However, clinical significance of PTEN mutation R130* detected in our study remains unclear. In the nonresponder group, the most frequently mutated genes included KRAS (eight out of 16 possible alterations), FBXW7 and PIK3CA (six alterations both), APC (five alterations), and TP53 (four alterations). As stated earlier, while APC and TP53 are related mainly to colorectal carcinogenesis, the activating mutations in KRAS are furthermore clearly related to cetuximab resistance. In the case of PIK3CA and FBXW7 mutations, this relationship is quite presumable as well. In the case of FBXW7, alterations in six samples were identified (particularly two in P and four M). All of them were found in the nonresponder group. No FBXW7 mutation was detected in the responder group. In three out of six cases, there was a coincidence with parallel KRAS mutation, and in other three cases a PIK3CA mutation was simultaneously detected. The involvement of FBXW7 in resistance to cytostatic chemotherapy has been previously reported, as well as the potential relationship to cetuximab or panitumumab resistance.26,41 F-box proteins constitute one of the subunits of the ubiquitin protein ligase complex, which is responsible for the degradation of several cellular proteins. FBXW7 is the component for substrate recognition and is able to mediate the degradation of some oncogenes including CYCLIN E, MYC, JUN, MTOR, and others. The exact mechanism of cetuximab or panitumumab resistance through the mutations in FBXW7 is unknown and requires further studies. Our hypothesis is that the inactivating mutations in FBXW7 could affect the functionality of the ubiquitin proteasome complex and thus impair the degradation of several proteins, including some downstream effectors of EGFR.
Using a highly precise NGS, we identified several new occurrences of activating mutations in KRAS, which had been missed by standard molecular diagnostic tools. All patients in our study were initially tested for complete mutational status of RAS and confirmed as wild-type RAS. Looking into the history of KRAS molecular testing in our institution, the Thera Screen® K-RAS Mutation Kit was in use initially in 2008 with a sensitivity of up to 1% mutant in a background of wild-type genomic DNA, analyzing the codons 12 and 13 of KRAS only. Later, since 2012, the Cobas® KRAS Mutation Test Kit was employed with a sensitivity of ~5% in a mixed sample and for analysis of codons 12, 13, and 61 of KRAS. Finally, to cover newly described clinically relevant codons 59, 117, and 146 of KRAS and all required codons of NRAS, the CRC RAScan™ Combination Kit (with a sensitivity of 2%–5%) was added to the Cobas® test in September 2013. With NGS, we were able to detect activating mutations in KRAS in eight samples (belonging to five patients with mutations in P and three patients with mutations in M), although all our patients were previously confirmed to have wild-type RAS by qPCR methods before entering the study. Particularly in patient no 1 and 5, the omission (not finding) of mutation G12V and G12C of KRAS was a failure of the Thera Screen mutational test. We could speculate that if more sensitive Cobas test had been used, KRAS mutations would have been detected. The Cobas test failed to detect mutation A146T in samples of patients no 3 and 8, as well. Nevertheless, all newly detected KRAS mutations led to a failure of cetuximab treatment. Thus, the discrepancy between the outcomes of tests routinely used in daily clinical practice and NGS is of a significant clinical relevance.
The size of the study group limits the clinical interpretation and applicability of the data due to the lack of statistical significance. Still, the research presented in this article identifies several mutations with potentially important involvement in cetuximab resistance of CRCs, which could be confirmed with broader studies in the future.
Conclusion
With the utilization of NGS panel of 26 genes involved in the EGFR pathway and colorectal carcinogenesis, we sequenced primary and corresponding metastatic FFPE tissue samples from responding and nonresponding mCRC patients treated with cetuximab. The presented results confirmed previously published evidence that the mutational status of primary tumors and metastatic lesions is highly concordant in TP53, APC, CTNNB1, KRAS, PIK3CA, PTEN, and FBXW7 genes. A number of mutations were significantly higher in the metastatic samples than in the primary tumors. We confirmed a potentially negative predictive value of FBXW7 mutations in relationship to anti-EGFR treatment outcomes. Finally, eight new occurrences of activating mutations in KRAS were identified in a group of patients with upfront-verified wild-type RAS mCRC compared with the routinely used qPCR RAS mutational tests. All newly detected KRAS mutations most likely led to a cetuximab treatment failure.
The results of the present study suggest a need of careful consideration of the previously published results of anti-EGFR-targeted therapy with regard to potentially inaccurate diagnostic tools used in the past. Based on our findings, we recommend more extensive use of NGS testing in daily clinical practice, as it brings a significant added value in terms of validity of the diagnostic procedure.
Acknowledgments
This work was supported by MH CZ – DRO (MMCI, 00209805), grant IGA NT/13860 – 4/2012 of the Czech Ministry of Health, and by the project CEITEC 2020 (LQ1601) supported by the Ministry of Education, Youth and Sports of the Czech Republic. We are grateful to Pavel Fabian, participating investigator, for assistance with sample preparation.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Van Cutsem E, Cervantes A, Nordlinger B, Arnold D; ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(suppl 3):iii1–iii9. | ||
Tay RY, Wong R, Hawkes EA. Treatment of metastatic colorectal cancer: focus on panitumumab. Cancer Manag Res. 2015;7:189–198. | ||
Khattak MA, Martin H, Davidson A, Phillips M. Role of first-line anti-epidermal growth factor receptor therapy compared with anti-vascular endothelial growth factor therapy in advanced colorectal cancer: a meta-analysis of randomized clinical trials. Clin Colorectal Cancer. 2015;14(2):81–90. | ||
Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 2014;4(11):1269–1280. | ||
Mirone G, Shukla A, Marfe G. Signaling mechanisms of resistance to EGFR- and anti-angiogenic inhibitors cancer. Crit Rev Oncol Hematol. 2016;97:85–95. | ||
Mlcochova J, Faltejskova P, Nemecek R, Svoboda M, Slaby O. MicroRNAs targeting EGFR signalling pathway in colorectal cancer. J Cancer Res Clin Oncol. 2013;139(10):1615–1624. | ||
Mlcochova J, Faltejskova-Vychytilova P, Ferracin M, et al. MicroRNA expression profiling identifies miR-31-5p/3p as associated with time to progression in wild-type RAS metastatic colorectal cancer treated with cetuximab. Oncotarget. 2015;6(36):38695–38704. | ||
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–1034. | ||
Lee MS, Kopetz S. Current and future approaches to target the epidermal growth factor receptor and its downstream signaling in metastatic colorectal cancer. Clin Colorectal Cancer. 2015;14(4):203–218. | ||
Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–5930. | ||
Martin V, Landi L, Molinari F, et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer. 2013;108(3):668–675. | ||
Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–5074. | ||
Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One. 2009;4(10):e7287. | ||
The Cancer Gebnome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. | ||
Wong SQ, Li J, Tan AY, et al; CANCER 2015 Cohort. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 2014;7:23. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Maughan TS, Adams RA, Smith CG, et al; MRC COIN Trial Investigators. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377(9783):2103–2114. | ||
Modzelewska K, Elgort MG, Huang J, et al. An activating mutation in sos-1 identifies its Dbl domain as a critical inhibitor of the epidermal growth factor receptor pathway during Caenorhabditis elegans vulval development. Mol Cell Biol. 2007;27(10):3695–3707. | ||
Oliner K, Douillard JY, Sienna S, et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first-line treatment for metastatic colorectal cancer (mCRC). J Clin Oncol. 2013;31(15_suppl):3511. | ||
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. | ||
Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27(35):5931–5937. | ||
Eklöf V, Wikberg ML, Edin S, et al. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer. 2013;108(10):2153–2163. | ||
Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–1857. | ||
Loupakis F, Pollina L, Stasi I, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–2629. | ||
Lupini L, Bassi C, Mlcochova J, et al. Prediction of response to anti-EGFR antibody-based therapies by multigene sequencing in colorectal cancer patients. BMC Cancer. 2015;15:808. | ||
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9(5):435–443. | ||
Launonen V. Mutations in the human LKB1/STK 11 gene. Hum Mutat. 2005;26(4):291–297. | ||
Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. | ||
Yoshida K, Sanada M, Ogawa S. Deep sequencing in cancer research. Jpn J Clin Oncol. 2013;43(2):110–115. | ||
Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. | ||
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. | ||
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. | ||
Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50(5):307–312. | ||
Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32(21):2240–2247. | ||
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. | ||
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. | ||
Souglakos J, Philips J, Wang R, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer. 2009;101(3):465–472. | ||
Rowland A, Dias MM, Wiese MD, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112(12):1888–1894. | ||
Vakiani E, Janakiraman M, Shen R, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012;30(24):2956–2962. | ||
Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and 658 FBW7. Nature. 2011;471(7336):110–114. |
Supplementary material
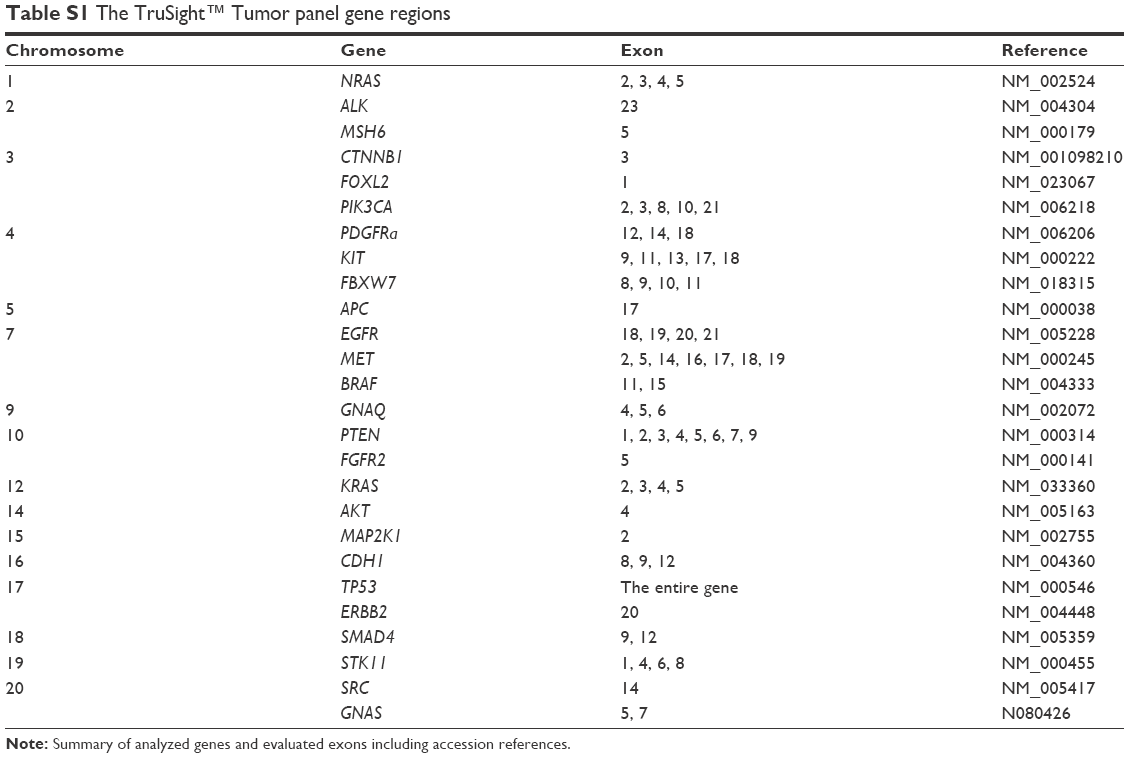 | Table S1 The TruSight™ Tumor panel gene regions |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.