Back to Journals » Journal of Multidisciplinary Healthcare » Volume 16
Mutational Analysis of Circulating Omicron SARS-CoV-2 Lineages in the Al-Baha Region of Saudi Arabia
Authors Almalki SSR ![]() , Izhari MA, Alyahyawi HE
, Izhari MA, Alyahyawi HE ![]() , Alatawi SK
, Alatawi SK ![]() , Klufah F
, Klufah F ![]() , Ahmed WAM
, Ahmed WAM ![]() , Alharbi R
, Alharbi R
Received 31 May 2023
Accepted for publication 20 July 2023
Published 27 July 2023 Volume 2023:16 Pages 2117—2136
DOI https://doi.org/10.2147/JMDH.S419859
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Shaia SR Almalki,1 Mohammad Asrar Izhari,1 Hanan E Alyahyawi,1 Saleha Keder Alatawi,2 Faisal Klufah,1 Waled AM Ahmed,3 Raed Alharbi1
1Department of Laboratory Medicine, Faculty of Applied Medical Sciences, Al-Baha University, Al-Baha, Saudi Arabia; 2Department of Optometry, Faculty of Applied Medical Sciences, Al-Baha University, Al-Baha, Saudi Arabia; 3Department of Nursing, Faculty of Applied Medical Sciences, Al-Baha University, Al-Baha, Saudi Arabia
Correspondence: Shaia SR Almalki, Department of Laboratory Medicine, Faculty of Applied Medical Sciences, Al-Baha University, Al-Baha, Saudi Arabia, Tel +966-556556714, Email [email protected]
Purpose: Omicron (B.1.1.529) is one of the highly mutated variants of concern of SARS-CoV-2. Lineages of Omicron bear a remarkable degree of mutations leading to enhanced pathogenicity and upward transmission trajectory. Mutating Omicron lineages may trigger a fresh COVID-19 wave at any time in any region. We aimed at the whole-genome sequencing of SARS-CoV-2 to determine variants/subvariants and significant mutations which can foster virus evolution, monitoring of disease spread, and outbreak management.
Methods: We used Illumina-NovaSeq 6000 for SARS-CoV-2 genome sequencing, MEGA 10.2 and nextstrain tools for phylogeny; CD-HIT program (version 4.8.1) and MUSCLE program for clustering and alignment. At the same time, UCSF Chimera was employed for protein visualization.
Results: Predominant Omicron pango lineages in Al-Baha were BA.5.2/B22 (n=4, 57%), and other lineages were BA.2.12/21L (n=1, 14.28%), BV.1/22B (n=1, 14.28%) and BA.5.2.18/22B (n=1, 14.28%). 22B nextstrain clade was predominant, while only one lineage showed 21L. BA.5.2/22B, BA.5.2/22B harbored a maximum of n=24 mutations in the spike region. Twelve crucial RBD mutations: D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, and Y505H were identified except the lineage BA.5.2/22B in which F486V mutation was not observed. Critical deletions S106 in membrane protein NSP6, E31in nucleocapsid, and L24 in spike region were observed in all the lineages. Furthermore, we identified common mutations of Omicron variants of SARS-CoV-2 in therapeutic hot spot spike region: T19I, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, A653V, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K, D1146D, L452R, F486V, N679K and D796Y. The effect of RBD-targeted mutations on neutralizing (NAbs) binding was considerable.
Conclusion: The outcome of this first report on SARS-CoV-2 variants identification and mutation in the Al-Baha region could be used to lay down the policies to manage and impede the regional outbreak of COVID-19 effectively.
Keywords: omicron, SARS-CoV-2, nextstrain clade, phylogeny, mutation, spike, RBD
Introduction
SARS-CoV-2 is the most lethal virus among all human coronavirus (HCoV) and is responsible for causing severe respiratory tract infections.1,2 It was first reported in Wuhan, China, at the end of December 200193–5 and found to be highly transmissible (Basic reproduction number ranging from 0.9 to 6.53).4,6 The genome of SARS-CoV-2 is plus (+) sense single-stranded (ss) RNA of size approximately 30 kb that encodes 16 non-structural proteins (nsp1-nsp16), the open reading frames (ORFs) accessory proteins 3a, 6, 7a, 7b, 8, 10. It also encodes four structural proteins viz., the spike protein (S) that forms trimeric structure, matrix/membrane-protein (M), nucleocapsid-protein (N), and an envelope-protein (E) most likely forms ion channel.7–9 The SARS-CoV-2 exudes spherical morphology and unsegmented RNA genome, with 30 poly-A tail and 50 cap-structure.10 Error-prone replication is exhibited by RNA-dependent-RNA polymerase enzyme of RNA viruses which results in an increased mutation rate.11 Though, a high mutation rate (nucleotide-nt substitution per-site/cell infection) is shown by ssRNA viruses, however, in the case of SARS-CoV-2, fixed mutations in the genome have been observed which suggests that SARS-CoV-2 changes slowly (estimated mutation rate/site × year = 1.12×10−3 nt−1 year−1) as compared to other ssRNA viruses.12,13
Adaptation of SARS-CoV-2 in the population and its global transmission has led to the accumulation of a large number of mutations, especially in spike protein; consequently, various dominant variants and lineages have been formed in year years.14 Lineage switching, generation of SARS-CoV-2 variants, and their continued global spread have aroused serious health concerns.15 The World Health Organization (WHO) has given nomenclature to these variants as variants of interest (VOI) and variants of concern (VOC).16 Major VOCs which had overwhelmed countries around the world encompass; alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Delta (B1.617.2).17
A highly transmissible variant of SARS-CoV-2 designated as Omicron was first reported in Botswana (in early November) and in South Africa dated 24-Nov-2021.18 Omicron showed many mutations (n=32), especially in spike protein alone as compared to mutation (n=16) in the tremendously infectious delta variant. The emergence of the Omicron variant (B.1.1.529) led to another fresh wave of the COVID-19 pandemic. This new variant was considered to be the most mutated SARS-CoV-2 variant.18,19 VOC are lethal variants capable of bringing about detrimental alterations in COVID-19 epidemiological parameters. A few variants, especially VOCs have raised grave epidemiological concern.20 Compared to the early-mutants or wild-type (WT) strains, these newly emerged SARS-CoV-2 variants have triggered successive COVID-19 waves with varying epidemiological features.21,22 Wild-type (WT) strain was potentially fatal among aged people with comorbid conditions; however, recent pre-clinical, clinical, and epidemiological data suggest that a vast number of emerged variants, VOCs in particular, are capable of breaching epidemiological barriers and have the potential to cause considerable fatality across the various demographic groups in different geographical locations including Al-Baha, Kingdom of Saudi Arabia.21–24 The evolution of VOCs impacts increased human-to-human transmissibility, virulence, pathogenesis, clinical manifestations, and debilitated effectiveness of the public health-safety and social measures.17 It could also influence diagnostic and therapeutic countermeasures at a greater length that compels the viral evolution research group to focus on SARS-CoV-2 variants, phenotypic characteristics, and their significant impacts on various countermeasures. The latest recovered Omicron variants were found to have a large number of mutations (>50 mutations on the entire genome and 32 mutations in the most crucial viral spike protein alone)25,26 which renders them higher infectivity (10-folds) and milder clinical manifestations26 as compared to the original virus (WT-strain). This variant has got higher potential to spread faster27 and evade the host-immune responses as a result of the accumulation of a vast number of mutations in the spike region.26
Since the classification of variants and most of the vaccine production strategies has been based on the mutations accumulated in the gene encoding spike protein,28 therefore, analyses of the SARS-CoV-2 genome sequence, especially the sequence of the spike region to characterize viral lineages and variants gain the paramount importance.29 Identification of SARS-CoV-2 variants/subvariants become crucial as many studies have demonstrated the dependency of the magnitude of neutralizing efficacy of vaccine-induced immunoglobulins on a wide range of SARS-CoV-2 variants/subvariants, especially Omicron.30,31 Many studies have explained the potential of new SARS-CoV-2 variants in scaping the immune response generated by earlier infection and/or the first dose of vaccination31–33 which necessitate the continuous tracking of the mutations in hot spot genomic region to reformulate the vaccine and therapeutic candidate.
Although, over three years of the COVID-19 pandemic a huge number of variants have been observed and sequenced worldwide,15 however, to the best of our knowledge, there is no record of SARS-CoV-2 genome sequencing in the Al-Baha region of Saudi Arabia available. The current study was aimed mainly at accomplishing whole-genome sequencing (WGS) and analyses of regional variants of SARS-CoV-2 from a phylogenetic viewpoint to characterize lineages, sub-lineages, and mutation variants. The sequence analyses enabled us to comprehend novel genomic variations that are significant indicators of vaccine development and viral evolution. The outcome of the present study could be useful in multivalent vaccine formulation and effective outbreak management.
Materials and Methods
Ethical Approval
The present study was commenced after receiving approval from the Ethical Committee of Scientific Research (ECSR) (Application number: 43121258) at Al-Baha University, Al-Baha, Kingdom of Saudi Arabia. As per the policy of ECSR, at Al-Baha University, approval is issued only to a research proposal that follows the guidelines of the Declaration of Helsinki. The study was accomplished according to the guidelines of the Declaration of Helsinki. Informed consent of all the participants was obtained and the identity of the participants was well protected.
Collection and Processing of Clinical Specimens
Nasopharyngeal (NP) swabs (n=42) collected from the patients suspected of COVID-19 infection were received from several hospitals and healthcare providers of the region and processed for diagnosis at the Regional Laboratory and Central Blood Bank, Al-Baha, Kingdom of Saudi Arabia. Viral nucleic acid (RNA) extraction from decontaminated COVID-19-positive NP swabs was carried out using the QIAamp Viral RNA Mini Kit (Qiagen, Maryland, USA) and following the manufacturer’s recommended protocol. Viral detection was carried out by Real-time Reverse Transcriptase Polymerase Chain reaction (RT-qPCR).34
SARS-CoV-2 Whole Genome Sequencing Workflow
Out of a total of 42 clinical specimens processed for viral detection, only 7 COVID-19-positive samples confirmed by RT-qPCR were further processed for accomplishing SARS-CoV-2 whole-genome sequencing.
Complementary DNA (cDNA) Synthesis
cDNA was generated from extracted viral nucleic acid (RNA) from 7 COVID-19-positive NP swabs by Reverse Transcriptase (RT) with random hexamers.
Target Amplification
The SARS-CoV-2 viral genome available in the sample was then PCR amplified using a set of primers to get overlapping segments of the entire genome and by employing two separate PCR reactions, which later on pooled together.
Library Preparation
The amplified, pooled fragments were further fragmented (On-Bead Tagmentation) and amplicons were tagged with adapter sequences. A second round of PCR amplification was performed on the adapter-tagged amplicons by applying a PCR master mix and unique index adapters. Following amplification, indexed libraries were pooled together and cleaned with the application of purification beads. Pooled purified libraries were quantified using fluorescent dye.
Execution of High-Throughput Sequencing
Pooled purified libraries were clustered onto a flow cell to perform whole-genome next-generation sequencing (NGS) applying sequencing by synthesis (SBS) chemistry using Illumina-NovaSeq 6000 Sequencing System (NovaSeq Xp S4 flow cell workflow). Sequencing of all the samples and generation of consensus sequences in FASTA format using Illumina DRAGEN™ COVIDSeq Test Pipeline was accomplished at BandrGen, Laboratory Madina, KSA. All the sequences were submitted to NCBI Sequence Read Archive (SRA) under Bio-project accession number PRJNA947168 (SAMN33843589, SAMN33843590, SAMN33843591, SAMN33843592, SAMN33843593, SAMN33843594, and SAMN33843595).
Data Analysis
Raw sequence data of all the samples were processed for quality check and report generation using PipeCoV (FastQC v0.11.9). The high-quality data were mapped to the Wuhan reference genome sequence (NC_045512.2) by applying Bowtie2 v2.3.5.1.35 Alignment of the reads was executed by applying Samtools and Bowtie2.35 All the mapped reads were assembled de novo using assembler SPAdes v3.15.0.36 The final consensus sequence obtained was annotated by Prokka v1.14.5.37 Pangolin v2.3.8 was employed to identify the lineages of SARS-CoV-2.38
Phylogenetic and Nextstrain Clade Analyses
A phylogenetic analysis based on spike sequence was undertaken by applying Molecular Evolutionary Genetics Analysis version 10.2 (MEGA 10.2 software) to determine the similarities and dissimilarities between seven SARS-CoV-2 genome sequences from the Al-Baha region and other genome sequences. The maximum likelihood method at bootstrap value 1000 was used. Nextstrain clade identification tools were used to construct a time-scaled phylogenetic tree based on genome sequences. The location and frequency of mutations were determined by employing clustering and alignment methods. CD-HIT program (version 4.8.1) was employed for clustering the sequences, while multiple sequence alignment was accomplished by the MUSCLE program.39,40 UCSF Chimera was employed for modeling and protein visualization.41
Results
COVID-19 pandemic waves are still being reported from different parts of the world due to emerging new variants of SARS-CoV-2 that exhibit high contagiousness as compared to the wild-type strain. Out of 42 Nasopharyngeal swabs processed, seven (S1, S2, S5, S6, S7, S8, S9) COVID-19-positive clinical specimens were successfully processed for whole-genome sequencing, and the sequence was analyzed to understand the phylogenetic relations and detection of mutations especially in spike region which is the critical component of the viral gene and play a significant role in disease initiation and transmission. The sequence of clinical specimen S2 showed maximum similarity (88.68%) with the reference genome sequence (NC_045512.2), while that of clinical specimen S7 exhibited minimum similarity (72.91%) (Table 1).
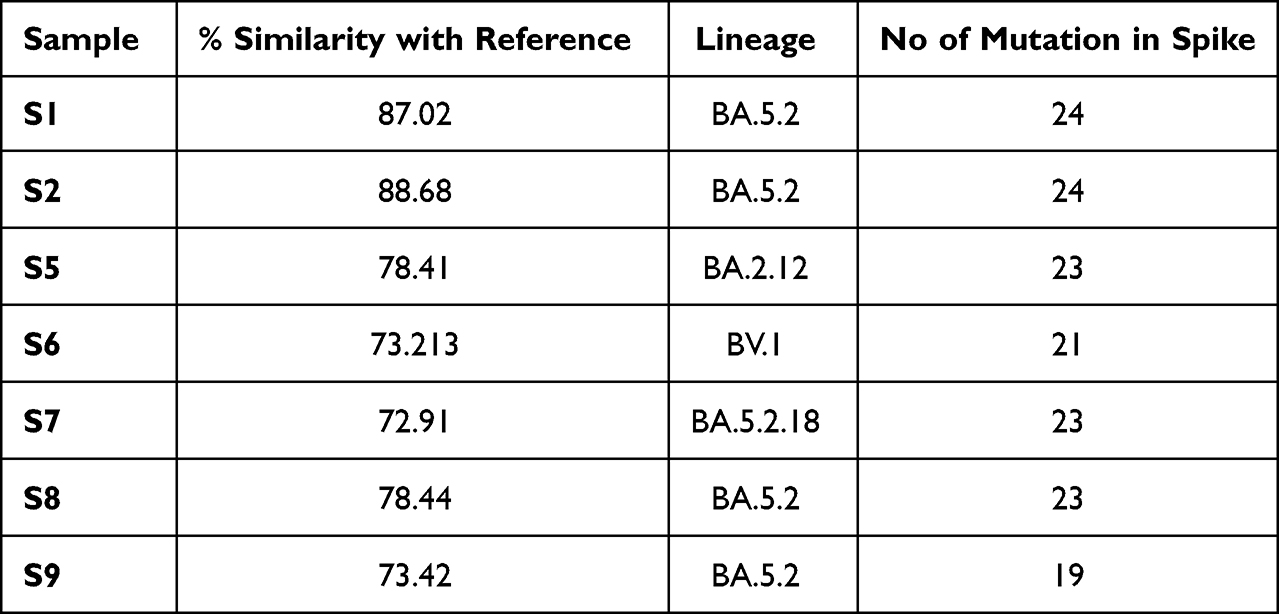 |
Table 1 Details of the Lineages and Quantum of Mutations in Spike Region of the SARS-CoV-2 |
Phylogenetic trees built based on spike sequences retrieved from all seven clinical samples unraveled the close relatedness of all the n=7 sequences with the previously sequenced omicron variants of SARS-COV-2 (Figure 1). Sequences S5 and S8 were found to be the most closely related to each other. S2 and S1 were distantly related to the other five sequences (Figure 1). The highest number of mutations detected in the spike region of SARS-CoV-2 was n=24 in the sequences obtained from clinical specimens S1 and S2, while the minimum number of spike mutations (n=19) was observed in that of clinical specimen S9 (Table 1). Lineage analysis of the sequences retrieved from S1, S2, S8, and S9 was found to share the same Pango lineage (designated as S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, and S9/BA.5.2). Three other lineages determined by analyzing the sequences from S5, S6, and S7 were BA.2.12, BV.1 and BA.5.2.18, respectively (designated as S5/BA.2.12, S6/BV.1 and S7/BA.5.2.18) (Table 1).
 |
Figure 1 Phylogenetic tree constructed based on spike sequences retrieved from all the samples by applying Molecular Evolutionary Genetics Analysis version 10.2 (MEGA 10.2 software). Maximum likelihood method at bootstrap value 1000 was used. |
Mutations and their locations in all the significant genomic regions of SARS-C0V-2, such as envelope (E), transmembrane protein (NSP4 and NSP6), membrane (M), and spike (S) were determined. Mutations in other genomic regions SARS-C0V-2, for instance, leader protein (NSP1), non-Structural protein-2 (NSP2), predicted phosphoesterase and protein-like-proteinase (NSP3), 3C-like proteinase (NSP5), non-structural protein (NSP8), ssRNA-binding protein (NSP9), RNA-dependent RNA polymerase (NSP12b), helicase (NSP13), 3’-to-5’ exonuclease (NSP14), endoRNAse (NSP15), ORF3a protein (ORF3a), ORF7a protein (ORF7a), ORF7b protein (ORF7b) and nucleocapsid protein (N) were also identified. Various mutations in these genomic regions, their locations, and annotations have been tabulated in Table 2 and Table 3. All seven sequences were compared to understand the variation of mutations across all the identified pango lineages. S5/BA.2.12 and S8/BA.5.2 showed high genetic relatedness, while S2/BA.5.2 was divergent from the other six lineages (Figure 1). The rectangular evolutionary tree exhibited the clustering of all seven lineages together with one Chinese omicron variant (Figure 1). S5/BA.2.12 was identified as 21L, whereas S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, S9/BA.5.2, S6/BV.1, and S7/BA.5.2.18 were determined to be 22B nextstrain clades (Figure 2b).
 |
Table 2 Tabulation of Mutations in an Envelope, Transmembrane Protein, Membrane, and Spike Genomic Regions of SARS-C0V-2 |
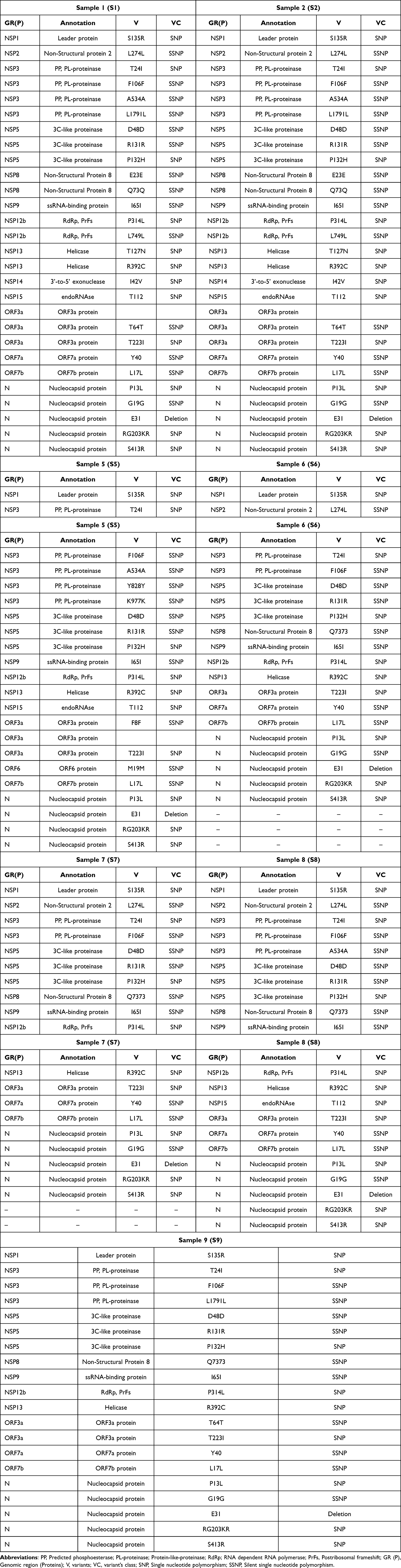 |
Table 3 Tabulation of Mutations in Genomic Regions Other Than an Envelope, Transmembrane Protein, Membr, Ane and Spike of SARS-C0V-2 |
 |
Figure 2 Nextstrian time-scaled phylogeny showing evolutionary relationships of 7 omicron lineages of SARS-CoV-2 viruses: (a) depicting spike mutations clade wise, (b) shows nextstrain clade of all the seven omicron variants identified in this study. |
Most Omicron mutations remain situated on RBD/spike that could effectively alter the epitopes (binding site) to various immunoglobulins. A trimer structural model of the spike protein of SASRS-CoV-2 was created to demonstrate most of the mutations and their locations in the genomic region using sequences obtained from S2 clinical samples (Figure 3).
 |
Figure 3 Side view of omicron’s trimeric spike protein showing mutations and their location in the spike region of SARS-CoV-2. Most frequent mutations in the spike (red color). A high frequency of mutations was detected in RBD and NTD of S1 subunits. |
Any insertion sequence in the spike region of SARS-CoV-2 was not found in any of the identified omicron lineages, namely, S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, S9/BA.5.2, S5/BA.2.12, S6/BV.1 and S7/BA.5.2.18. However, many crucial deletions were identified in spike and transmembrane proteins. A deletion S106 in transmembrane protein (NSP6) was identified in all the identified pango lineages. A deletion L24 in the spike region was also observed in all these lineages; however, one unique deletion (V47) in lineage BA.5.2 of the S2 sample (S2/BA.5.2) was observed (Table 2). E31 deletion in nucleocapsid protein was exhibited by all these omicron lineages (Table 3).
D614G mutation was observed in the spike of only S1/BA.5.2 lineage. A653V and D1146D mutations in spikes were observed in both S1/BA.5.2 and S2/BA.5.2. L5F mutation in the spike was found in only S7/BA.5.2.18. In contrast, spike Q493 mutation was observed only in S5/BA.2.12. Spike mutations V123G and G142D were commonly observed in S5/BA.2.12 and S8/BA.5.2 Pango lineages. Various other common spike mutations were observed across all the seven identified lineages such as T19I, L24, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, A653V, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K and D1146D except L452R in S9/BA.5.2 and S2/BA.5.2, F486V in S2/BA.5.2, N679K in S9/BA.5.2 and S6/BV.1, D796Y in S9/BA.5.2 (Table 2, Figure 2a and Figure 3). Omicron is the variant of SARS-CoV-2 having 37 mutations in total, 15, and 11 mutation sites remains located on the receptor-binding domain (RBD) and N-terminal domain (NTD), respectively, while the other 11 are located around the furin cleavage site as well as on the S2 subunit of the SARS-CoV-2 spike. RBD with 15 mutation sites of Spike glycoproteins of the Omicron variant is the most crucial region and plays a key role to mediate the entry of the virus into host cells. Therefore, glycoprotein especially RBD has been considered as the target for therapeutic designing and vaccine development.
Mutation sites in the RBD region of S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, S9/BA.5.2, S5/BA.2.12, S6/BV.1 and S7/BA.5.2.18 pango lineages were characterized. Total number of 12 mutations in RBD region: D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y and Y505H were identified in all the lineages except the lineage S2/BA.5.2 in which F486V mutation was not observed. All the mutations and their locations in the spike region of SARS-CoV-2 have been depicted by a ribbon diagram (Figure 3). Mutations and their locations have also been identified in other genomic regions of all the variants that have been tabulated in Table 3. Mutation T112I in endoRNAse (NSP15) was identified only in S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, S5/BA.2.12 Pango lineage. M19M mutation was observed in ORF6 protein (ORF6) of only S5/BA.2.12 Pango lineage. I42V in 3’-to-5’ exonuclease (NSP14), T127N in Helicase (NSP13), and L749L in RdRp, PrFs (NSP12b) were identified only in S1/BA.5.2 and S2/BA.5.2 Pango lineage (Table 3).
Comparative analysis of physiochemical properties (hydrophobicity and net charge) of wild and mutant RBD of the spike protein of SARS-CoV-2 was evaluated to understand the functional feature (impact of specific mutation). The effect of hydrophobicity and net charge on protein plays a vital role in deterring protein–protein interactions. Hydrophobicity and net charge of 12-targeted RBD mutations in the spike protein of wild type and mutants were compared and found to be considerably altered (Table 4).
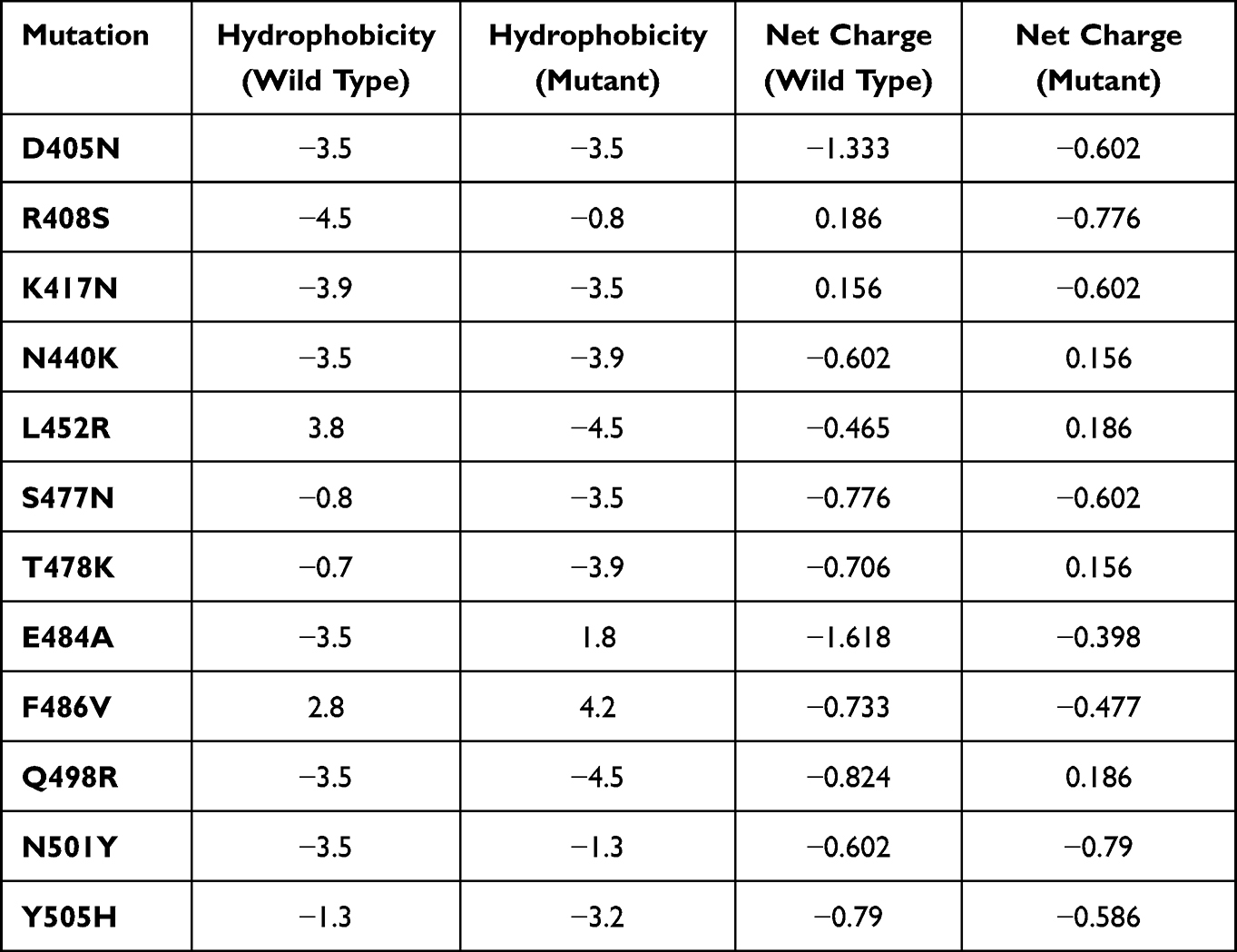 |
Table 4 Comparative Hydrophobicity and Net Charge of the Wild and Mutated Residues of RBD of S Protein Have Been Represented |
RBD interactions with distinct classes of human-neutralizing antibodies (NAbs) were evaluated to understand the effect of RBD substitutions (n=12 targeted RBD mutations) in the identified pango lineages on binding of NAbs such as Class 1(C102 Fab), Class 2 (C002 Fab), Class 3 (C135 Fab), Class 4 (CR3022) and human anti-SARS-CoV-2 spike-NTD Ab (4A8 Fab) (Figure 4). C102 Fab gains access to RBD epitope only in up RBD conformation that could be affected by potential mutations K417N, R408S, and F486V (Figure 4). C002 Fab gains access to RBD epitope up/down RBD conformations that could be potentially altered by L452R, K417N, and E484A RBD targeted mutations (Figure 4). C135 Fab is the most significant class 3 neutralizing Ab that overlaps with the ACE-2 binding site and gains access to RBD epitope in up/down RBD conformations that could be affected by the N440K and Q498R RBD mutations. In contrast, CR3022 does not overlap with ACE-2 binding site but gain access of RBD epitope only in up RBD conformation that could be impacted by R408S, N440K and D405N RBD substitutions (Figure 4). 4A8 Fab does not interfere directly with the interaction between RBD and ACE-2 receptor; however, it has a very high neutralizing ability, which could be altered by T19I mutations and L24 deletion in spike regions of SARS-CoV-2 (Figures 3 and 4).
 |
Figure 4 The model represents the interaction of RBD-specific classes of human neutralizing antibodies with RBD and NTD of the spike protein of SARS-CoV-2 and the depiction of the effect of RBD substitutions on binding to up, up/down conformation RBD. Mutation sites have been shown in red-colored spheres. |
Discussion
The emergence of most of the variants and sub-variants of SARS-CoV-2 due to mutations in spike and non-spike regions42 has rendered the ongoing pandemic. The Omicron variant dominated the COVID-19 pandemic trajectory with 37 mutations as last fall.43 COVID-19 waves are still being reported from various regions of the world and therefore, it is imperative to undertake genome sequencing at extreme lengths in different geographical regions to gain insights into mutations and lineages of SARS-CoV-2 to develop policies to monitor and curb the regional SARS-CoV-2 outbreaks in real-time. Omicron sub-variants (BA.5.2 and BF.7) were identified based on analysis of sequence retrieved from COVID-19-positive specimens in the most recent (late 2022) outbreak in China.44
Phylogeny, Evolutionary Relationship and Pango Lineage Diversity of SARS-CoV-2 in Al-Baha Region
All seven sequences were Omicron variants of SARS-CoV-2 according to the spike sequence-based phylogeny (Figure 1). The Omicron variant (B.1.1.529) was first identified in Botswana and designated as a variant of concern (VOC).45,46 Omicron is the most mutated variants of the SARS-CoV-2 showing remarkable degree of mutation, as compared to the other variants of SARS-CoV-2: Alpha variant (B.1.1.7), Beta variant (B.1.351), Gamma variant (P.1), Delta variant (B.1.617.2), Mu variant (B.1.621), Epsilon variant (B.1.427 and B.1.429), and Lambda variant (C.37).18,47 Each variant including Omicron has a diverse set of mutations that render varying degrees of transmissibility, immune evasion capability, and pathogenicity.47 Although transmission dynamics, evolutionary link, and diversity of SARS-CoV-2 were studied by sequencing various genomes of SARS-CoV-2 retrieved from NP swabs collected from patients of four metropolitan cities of Saudi Arabia: Riyadh, Madina, Jeddah, and Makkah,48 however, any identified specific variant of SARS-CoV-2 was not reported in the study. In an in silico genomic analysis of n=1270 Saudi Arabian genomes, n=30 Omicron variants of SARS-CoV-2 were reported.49
There are no previous reports of sequencing SARS-CoV-2 genomes to monitor circulating variants in the Al-Baha region. Being a highly transmissible circulating Omicron variant of SARS-CoV-2, it poses a potential public health threat in the region. Lineage analysis of the sequences retrieved from S1, S2, S8, and S9 unraveled that they had shared the same Pango lineage BA.5.2, BA.5.2, BA.5.2 and BA.5.2 (designated as S1/BA.5.2, S2/BA.5.2, S8/BA.5.2, S9/BA.5.2). Three other lineages determined by analyzing the sequences retrieved from S5, S6 and S7 were BA.2.12, BV.1 and BA.5.2.18, respectively (designated as S5/BA.2.12, S6/BV.1 and S7/BA.5.2.18) (Table 1). At first, there were three sister lineages of Omicron sub-variants: BA.1, BA.2, and BA.3. BA.1 brought about the fourth wave in South Africa but the BA.2 lineage turned out to be comparatively more lethal during a wave of Omicron.50,51 Two more predominant Omicron lineages BA.4 and BA.5 were later identified which were genetically distinct from other lineages.51,52
The most predominant Omicron sub-variant identified was BA.5.2 (n=4, 57%) which has been documented in Table 1. It was designated as Omicron/VOC that shows comparatively high replicative kinetics.53,54 BA.1, BA.2, and BA.3 sister lineages exhibited considerable resistance to neutralizing potential (artificial active and natural active immunity) of serum.51 Leu452 amino acid substitution renders high infectiousness to omicron sub-variants: BA.2.12.1, BA.2.13, BA.4, and BA.5 as compared to BA.2.55 BA.2.12 also designated as B.1.1.529.2.12 and BA.2.12.1 or B.1.1.529.2.12.1 were identified in New York, USA, as sub-variants of BA.2, which showed a considerable multiplicative advantage over parent BA.2 and thus, acquired rising infection trajectory.56 For viral entry, the dependency of BA.2 on TMPRSS2 is lesser as compared to BA.1, which enables it to replicate with high efficiency. Lineage B.V formerly BA.5.2.17 first originated in July 2022 and spread in the USA, Germany, and Denmark in particular, while BA.5.2.18 defined by mutation K444R on RF1b that emerged in Nov 2021 and caused infection in USA, Russia, UK, and Scandinavia (Cov-Lineages) (GitHub - cov-lineages/pango-designation: Repository for suggesting new lineages that should be added to the current scheme) that implies an exogenous source of the variants in Al-Baha region. Mourier et al48 reported circulating nextstrain clades in different cities of KSA: 19A in Makkah, 19B in Madinah, 20A in Jeddah, 20B in Riyadh, and 20C in the Eastern region. However, time-scaled nextstrain clades identified in our study were S5/BA.2.12/21L, S1/BA.5.2/22B, S2/BA.5.2/22B, S8/BA.5.2/22B, S9/BA.5.2/22B, S6/BV.1/22B and S7/BA.5.2.18/22B, (Figure 1 and Figure 2b) gives insights about the continuous emergence of variants across Saudi Arabia. Thus, necessitating the regular monitoring of the variants to curb the regional outbreak.
Mutations and Their Impact on the Pathogenesis of Omicron Sub-Variants of SARS-CoV-2
S1/BA.5.2/22B, S2/BA.5.2/22B harbored a maximum of n=24 mutations in the spike region, while S9/BA.5.2/22B variant carried (n=19) spike mutations (Table 1) with 12 crucial RBD mutations. Although, to date, 37 mutations in the spike region of the Omicron variant (15 on RBD, 11 on NTD, 11 on furin cleavage site, and on S2 subunit) have been reported.57 Spike glycoprotein is the most crucial region and plays a key role in mediating the entry of viruses into host cells. Therefore, glycoprotein spike especially RBD has been considered as the target for therapeutic designing and vaccine development.58 Furthermore, Omicron variants bear the most remarkable number of novel mutations among all other variants of SARS-CoV-2 leading to the genuine concern over immune evasion and vaccine failure.59 The slightly enhanced affinity of RBD with the hACE2 receptor in the case of Omicron as compared to the prototype has been reported by Cui et al.60
The biological role of spike or RBD mutations, mechanisms leading to increased transmissibility, and immune escape in highly mutating Omicron variant needs to be addressed as an imperative for the management of Omicron outbreak. Analysis of sequences unraveled the mutations and their locations in all the significant genomic regions of SARS-C0V-2, such as E, NSP4, NSP6, MS, NSP1, NSP2, NSP3, NSP5, NSP8, NSP9, NSP12b, NSP13, NSP14, NSP15, ORF3a, ORF7a, ORF7b and N (Tables 2, 3 and Figure 3). Most Omicron mutations remain situated on the spike that could effectively alter the epitopes (binding site) to various immunoglobulins. In our study, no insertion sequence was determined in any of the identified lineages. However, deletions S106 in NSP6, E31in N, and L24 in the spike region were also observed in all these lineages. Deletions S106 along with other two amino acid deletions (L105 and G107) in NSP6 have been reported in most of the omicron variants61 that are situated polar region of a transmembrane protein, therefore the absence of the amino acid may influence the protein interaction and interfere with autophagy.62 Deletion E31 in nucleocapsid region impact the detection of Omicron during molecular diagnosis as N is one of the diagnostic molecular targets.63 Omicron variants harbor various other deletions including L24 in the spike region, which may alter infectivity.64 In addition to that, one unique deletion (V47) in the spike in lineage S2/BA.5.2 was identified. Total number of 12 mutations in RBD region: D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y and Y505H were identified in all the lineages except the lineage S2/BA.5.2 in which F486V mutation was not observed. N440K, Q498R, N501Y, T478K, and S477N mutations aggrandize the binding interaction of spike to ACE2 receptor.65 K417N, E484A, N440K, S477N, T478K and N501Y mutations of RBD have been reported to be associated with decreased binding of RBD-specific nAbs.
Spike mutations may alter the significant structural epitopes, which are targets for therapeutic mAbs in clinical applications. Apart from Omicron variants, all other previously identified VOCs harbored the key substitutions: K417N, S477N, E484K, and N501Y associated with receptor binding and immune evasion.55 We determined various other common mutations: T19I, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, A653V, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K, D1146D, L452R, F486V, N679K and D796Y. D614G mutation was observed in the spike of only S1/BA.5.2 lineage. D614G is a point mutation, which fosters higher transmissibility and pathogenicity as compared to the original SARS-CoV-2 strain.66 Huang et al described the association of D614G mutation with COVID-19 severity and infectivity using machine learning mole.67 Most of the mutations in the Omicron variants/subvariants remains accumulated in the spike region which is the principal target for vaccines in use: Pfizer-BioNTech, Janssen, AstraZeneca, and Moderna.68 And therefore, mutations in the spike region bring about a decrease in the efficacy of the current vaccines leading to a higher probability of reinfection in immunized individuals. Tian et al26 reported a higher ACE2 receptor binding affinity of N501Y in comparison to the wild-type. Wise et al69 reported the possible resistance to the vaccine due to E484K substitution. L452R substitution first identified in Denmark was associated with enhanced infectivity and poor antibody neutralization.70 Substitution E484Q enhanced the ACE2 receptor affinity.71 S477G/N mutation (S477G and S477N) was found engaged in enhancing the hACE2 receptor binding.72 The effect of hydrophobicity and net charge on protein plays a vital role in deterring protein–protein interactions.
Hydrophobicity and net charge of 12-targeted RBD mutations in the spike protein of wild type and mutants were compared and found to be considerably altered (Table 4). Class 1 (C102 Fab) gains access to RBD epitope only in up RBD conformation that could be affected by potential mutations K417N, R408S, and F486V (Figure 4).73 C002 Fab gains access to RBD epitope up/down RBD conformations that could be potentially altered by L452R, K417N, and E484A RBD targeted mutations (Figure 4). C135 Fab is the most significant class 3 neutralizing Ab that overlaps with the ACE-2 binding site and gains access to RBD epitope in up/down RBD conformations that could be affected by the N440K and Q498R RBD mutations, while CR3022 does not overlap with ACE-2 binding site but gain access of RBD epitope only in up RBD conformation that could be impacted by R408S, N440K and D405N RBD substitutions (Figure 3).58 4A8 Fab does not interfere directly with the interaction between RBD and ACE-2 receptor; however, it has a very high neutralizing ability, which could be altered by T19I mutations and L24 deletion in spike regions of SARS-CoV-2 (Figures 3 and 4). Detailed structural analysis of RBD and its interactions with neutralizing antibodies along with RBD mutations in Omicron variants could be insightful about the clinical use of RBD-targeting immunoglobulins and immune response against Omicron variants of SARS-CoV-2. These findings highlight the need to identify emerging variants/subvariants to address the reduced efficacy/effectiveness of vaccines against them.74 Given the potential impact of mutations in the genomic region of SARS-CoV-2 variants on the vaccine and therapeutic options,31,32 whole-genome SARS-CoV-2 sequencing, mutation tracking, and identification of emerging variants could be part of mitigating strategies such as vaccine formulation (multivalent), delivery mechanism and modulation of the vaccine according to potential mutations.30
Conclusion
A remarkable number of mutations especially in the spike/RBD region of Omicron variants fosters the significant alteration in key biological functions and pathogenicity of Omicron variants of SARS-CoV-2. In this study, following genome sequence analyses, phylogeny, and time-scaled nextstrain clade analysis, we identified four circulating BA.5.2/22B Omicron variants. In addition to that, BA.2.12/21L, BV.1/22B, and BA.5.2.18/22B circulating variants were also identified in the Al-Baha region of the Kingdom of Saudi Arabia which is indicative of the continuous emergence of Omicron variants in the region that might trigger fresh regional SARS-CoV-2 waves. Furthermore, all the critical mutations in the RBD of spikes, such as D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, and Y505H were identified together with detection of various other common Omicron mutations, which are associated with enhanced transmissibility, infectivity, and pathogenicity of Omicron variants. We have also demonstrated the RBD targeted mutations, their locations in the trimeric spike region of SARS-CoV-2, and their possible effect on binding of NAbs such as Class 1(C102 Fab), Class 2 (C002 Fab), Class 3 (C135 Fab), Class 4 (CR3022) and human anti-SARS-CoV-2 spike-NTD Ab to get the insight into immune protection against Omicron variants. It is imperative to note that Omicron variants even with lesser virulence could pose a potential health threat, especially in case of immunosuppression and co-morbidities as they bear a significant number of spike/RBD mutations and deletions, which are associated with increased transmissibility and infectivity. Impact of identified potential mutations on disease transmissibility, immune escape and severity in vivo is recommended to be carried out to gain further insight into it. To the best of our knowledge, SARS-CoV-2 variants identification and mutation analyses have been undertaken for the first time in the Al-Baha region. Moreover, the outcome of our study would be used to lay down the policies by health authorities to effectively manage and impede the regional outbreak of COVID-19.
Funding
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number MOE-BU-1-2020.
Disclosure
The authors declare that there is no conflicts of interest.
References
1. Chen B, Tian E-K, He B, et al. Overview of lethal human coronaviruses. Signal Transduction Targeted Therapy. 2020;5(1):89.
2. Tabibzadeh A, Zamani F, Laali A, et al. SARS-CoV-2 molecular and phylogenetic analysis in COVID-19 patients: a preliminary report from Iran. Infect Genetics Evolution. 2020;84:104387.
3. Hui DS, Azhar EI, Madani TA, et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int j Infectious Dis. 2020;91:264–266.
4. Tan W, Zhao X, Ma X, et al. A novel coronavirus genome identified in a cluster of pneumonia cases—Wuhan, China 2019− 2020. China CDC Weekly. 2020;2(4):61–62.
5. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Eng j Med. 2020;2:643.
6. Linka K, Peirlinck M, Kuhl E. The reproduction number of COVID-19 and its correlation with public health interventions. Computational Mechanics. 2020;66:1035–1050.
7. Brant AC, Tian W, Majerciak V, Yang W, Zheng Z-M. SARS-CoV-2: from its discovery to genome structure, transcription, and replication. Cell Biosci. 2021;11(1):1–17.
8. Chan JF-W, Kok K-H, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Em Microbes Infections. 2020;9(1):221–236.
9. Rohaim MA, El Naggar RF, Clayton E, Munir M. Structural and functional insights into non-structural proteins of coronaviruses. Microb Pathog. 2021;150:104641.
10. Atzrodt CL, Maknojia I, McCarthy RD, et al. A Guide to COVID‐19: a global pandemic caused by the novel coronavirus SARS‐CoV‐2. FEBS J. 2020;287(17):3633–3650.
11. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502.
12. Amicone M, Borges V, Alves MJ, et al. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evolution Med Public Health. 2022;10(1):142–155.
13. Callaway E. The Coronavirus is mutating—does It Matter? Nature Publishing Group; 2020.
14. Telenti A, Arvin A, Corey L, et al. After the pandemic: perspectives on the future trajectory of COVID-19. Nature. 2021;596(7873):495–504.
15. Harvey WT, Carabelli AM, Jackson B, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409–424.
16. World Health Organization. Tracking SARS-Cov-2 Variants. Available from: Https://Www.Who.Int/En/Activities/Tracking-SARS-Cov-2-Variants/.
17. Zhang Y, Zhang H, Zhang W. SARS-CoV-2 variants, immune escape, and countermeasures. Front Med. 2022;16(2):196–207.
18. Kandeel M, Mohamed ME, El‐Lateef HM A, Venugopala KN, El‐Beltagi HS. Omicron variant genome evolution and phylogenetics. J Med Virol. 2022;94(4):1627–1632.
19. Gao SJ, Guo H, Luo G. Omicron variant (B. 1.1. 529) of SARS‐CoV‐2, a global urgent public health alert! J Med Virol. 2022;94(4):1255.
20. Kumar A, Parashar R, Kumar S, et al. Emerging SARS‐CoV‐2 variants can potentially break set epidemiological barriers in COVID‐19. J Med Virol. 2022;94(4):1300–1314.
21. Brookman S, Cook J, Zucherman M, Broughton S, Harman K, Gupta A. Effect of the new SARS-CoV-2 variant B. 1.1. 7 on children and young people. Lancet Child Adolescent Health. 2021;5(4):e9.
22. Kustin T, Harel N, Finkel U, et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2-mRNA-vaccinated individuals. Nat Med. 2021;27(8):1379–1384.
23. Gao Q, Hu Y, Dai Z, Xiao F, Wang J, Wu J. The epidemiological characteristics of 2019 novel coronavirus diseases (COVID-19) in Jingmen, Hubei, China. Medicine. 2020;99(23):e20605. doi:10.1097/md.0000000000020605
24. Van Der Made CI, Simons A, Schuurs-Hoeijmakers J, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324(7):663–673.
25. Caputo E, Mandrich L. Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life. 2022;12(8):74.
26. Tian D, Sun Y, Xu H, Ye Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J Med Virol. 2022;94(6):2376–2383. doi:10.1002/jmv.27643
27. Rochman ND, Wolf YI, Faure G, Mutz P, Zhang F, Koonin EV. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. bioRxiv. 2021. 10.1101/2020.10.12.336644.
28. Braeye T, Catteau L, Brondeel R, et al. Vaccine effectiveness against onward transmission of SARS-CoV2-infection by variant of concern and time since vaccination, Belgian contact tracing, 2021. Vaccine. 2022;40(22):3027–3037. doi:10.1016/j.vaccine.2022.04.025
29. Parlikar A, Kalia K, Sinha S, et al. Understanding genomic diversity, pan-genome, and evolution of SARS-CoV-2. PeerJ. 2020;8:e9576. doi:10.7717/peerj.9576
30. McLean G, Kamil J, Lee B, et al. The impact of evolving SARS-CoV-2 mutations and variants on COVID-19 vaccines. MBio. 2022;13(2):e02979–21.
31. Wang P, Nair MS, Liu L, et al. Antibody resistance of SARS-CoV-2 variants B. 1.351 and B. 1.1. 7. Nature. 2021;593(7857):130–135.
32. Shen X, Tang H, McDanal C, et al. SARS-CoV-2 variant B. 1.1. 7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe. 2021;29(4):529–539. e3.
33. Edara VV, Hudson WH, Xie X, Ahmed R, Suthar MS. Neutralizing antibodies against SARS-CoV-2 variants after infection and vaccination. JAMA. 2021;325(18):1896–1898.
34. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveillance. 2020;25:54.
35. Oliveira RRM, Costa Negri T, Nunes G, et al. PipeCoV: a pipeline for SARS-CoV-2 genome assembly, annotation and variant identification. PeerJ. 2022;10:e13300. doi:10.7717/peerj.13300
36. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi:10.1089/cmb.2012.0021
37. Merkel D. Docker: lightweight Linux containers for consistent development and deployment. Linux Journal. 2014;2014:2.
38. O’Toole Á, Pybus OG, Abram ME, Kelly EJ, Rambaut A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genomics. 2022;23(1):121. doi:10.1186/s12864-022-08358-2
39. Robert CE. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797.
40. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659.
41. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
42. Singh H, Dahiya N, Yadav M, Sehrawat N. Emergence of SARS-CoV-2 New Variants and Their Clinical Significance. Canadian j Infect Dis Med Microbiol. 2022;2022:7336309. doi:10.1155/2022/7336309
43. Mannar D, Saville JW, Zhu X, et al. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science. 2022;375(6582):760–764. doi:10.1126/science.abn7760
44. Harris E. Study: no New SARS-CoV-2 Variants in Recent Outbreak in China. JAMA. 2023;329(10):788. doi:10.1001/jama.2023.2089
45. Silva S, Kohl A, Pena L, Pardee K. Recent insights into SARS-CoV-2 omicron variant. Rev Med Virol. 2023;33(1):e2373. doi:10.1002/rmv.2373
46. Vieillard-Baron A, Flicoteaux R, Salmona M, et al. Omicron Variant in the Critical Care Units of the Paris Metropolitan Area: the Reality Research Group. Am J Respir Crit Care Med. 2022;206(3):349–363. doi:10.1164/rccm.202202-0411LE
47. Mohseni Afshar Z, Tavakoli Pirzaman A, Karim B, et al. SARS-CoV-2 Omicron (B.1.1.529) Variant: a Challenge with COVID-19. Diagnostics. 2023;13(3):43.
48. Mourier T, Shuaib M, Hala S, et al. SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Nat Commun. 2022;13(1):601. doi:10.1038/s41467-022-28287-8
49. Darwish DBE. Insight into SARS-CoV-2 Omicron variants in Saudi Arabian genomic isolates. Saudi Med J. 2022;43(11):1276–1279. doi:10.15537/smj.2022.43.11.20220381
50. Fonager J, Bennedbæk M, Bager P, et al. Molecular epidemiology of the SARS-CoV-2 variant Omicron BA.2 sub-lineage in Denmark, 29 November 2021 to 2 January 2022. Euro Surveillance. 2022;27(10). doi:10.2807/1560-7917.es.2022.27.10.2200181
51. Yao L, Zhu KL, Jiang XL, et al. Omicron subvariants escape antibodies elicited by vaccination and BA.2.2 infection. Lancet Infect Dis. 2022;22(8):1116–1117. doi:10.1016/s1473-3099(22)00410-8
52. Tegally H, Moir M, Everatt J, et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat Med. 2022;28(9):1785–1790. doi:10.1038/s41591-022-01911-2
53. Feng Z, Shen Y, Li S, et al. The First Outbreak of Omicron Subvariant BA.5.2 - Beijing Municipality, China, July 4, 2022. China CDC Wkly. 2022;4(30):667–668. doi:10.46234/ccdcw2022.136
54. Ong CP, Ye ZW, Tang K, et al. Comparative analysis of SARS-CoV-2 Omicron BA.2.12.1 and BA.5.2 variants. J Med Virol. 2023;95(1):e28326. doi:10.1002/jmv.28326
55. Cao Y, Yisimayi A, Jian F, et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022;608(7923):593–602. doi:10.1038/s41586-022-04980-y
56. Shrestha LB, Foster C, Rawlinson W, Tedla N, Bull RA. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: implications for immune escape and transmission. Rev Med Virol. 2022;32(5):e2381. doi:10.1002/rmv.2381
57. Zhao F, Ma Q, Yue Q, Chen H. SARS-CoV-2 Infection and Lung Regeneration. Clin Microbiol Rev. 2022;35(2):e0018821. doi:10.1128/cmr.00188-21
58. Pinto D, Park YJ, Beltramello M, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583(7815):290–295. doi:10.1038/s41586-020-2349-y
59. Callaway E. Heavily mutated Omicron variant puts scientists on alert. Nature. 2021;600(7887):21. doi:10.1038/d41586-021-03552-w
60. Cui Z, Liu P, Wang N, et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell. 2022;185(5):860–871.e13. doi:10.1016/j.cell.2022.01.019
61. Wang L, Cheng G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J Med Virol. 2022;94(4):1728–1733. doi:10.1002/jmv.27516
62. Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J Infect. 2020;81(1):e24–e27. doi:10.1016/j.jinf.2020.03.058
63. Ippoliti C, De Maio F, Santarelli G, et al. Rapid Detection of the Omicron (B.1.1.529) SARS-CoV-2 Variant Using a COVID-19 Diagnostic PCR Assay. Microbiology Spectrum. 2022;10(4):e0099022. doi:10.1128/spectrum.00990-22
64. Willett BJ, Grove J, MacLean OA, et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nature Microbiology. 2022;7(8):1161–1179. doi:10.1038/s41564-022-01143-7
65. Zahradník J, Marciano S, Shemesh M, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nature Microbiology. 2021;6(9):1188–1198. doi:10.1038/s41564-021-00954-4
66. Korber B, Fischer WM, Gnanakaran S, et al. Tracking Changes in SARS-CoV-2 Spike: evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4):812–827.e19. doi:10.1016/j.cell.2020.06.043
67. Huang F, Chen L, Guo W, et al. Identifying COVID-19 Severity-Related SARS-CoV-2 Mutation Using a Machine Learning Method. Life. 2022;12:6.
68. Alquraan L, Alzoubi KH, Rababa’h SY. Mutations of SARS-CoV-2 and their impact on disease diagnosis and severity. Inform Med Unlocked. 2023;39:101256. doi:10.1016/j.imu.2023.101256
69. Wise J. Covid-19: the E484K mutation and the risks it poses. BMJ. 2021;372:n359. doi:10.1136/bmj.n359
70. Liu Z, VanBlargan LA, Bloyet LM, et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29(3):477–488.e4. doi:10.1016/j.chom.2021.01.014
71. Lan J, He X, Ren Y, et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022;32(6):593–595. doi:10.1038/s41422-022-00644-8
72. Singh A, Steinkellner G, Köchl K, Gruber K, Gruber CC. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci Rep. 2021;11(1):4320. doi:10.1038/s41598-021-83761-5
73. Barnes CO, West AP Jr, Huey-Tubman KE, et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell. 2020;182(4):828–842.e16. doi:10.1016/j.cell.2020.06.025
74. Vo GV, Bagyinszky E, An SSA. COVID-19 Genetic Variants and Their Potential Impact in Vaccine Development. Microorganisms. 2022;10(3):e46. doi:10.3390/microorganisms10030598
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Comparative Structural Analysis of Human ACE2 Receptor with Spike Protein of SARS-CoV-2 Variants: Implications to Understand Infectivity of the Virus
Koley T, Goswami A, Kumar M, Upadhyay N, Hariprasad G
Advances and Applications in Bioinformatics and Chemistry 2022, 15:21-27
Published Date: 16 June 2022
Old Age is an Independent Risk Factor for Pneumonia Development in Patients with SARS-CoV-2 Omicron Variant Infection and a History of Inactivated Vaccine Injection
Tong X, Huang Z, Zhang X, Si G, Lu H, Zhang W, Xue Y, Xie W
Infection and Drug Resistance 2022, 15:5567-5573
Published Date: 21 September 2022
Clinico-Epidemiological Profile of COVID-19 Patients with Omicron Variant Admitted in a Tertiary Care Center, South India
Ethirajan T, Natarajan G, Velayudham R, Jayakumaran P, Karnan I, Rajendran K, Doraisamy S, Chenakeswarar Sridhar S, Kumaran P, Kamaraj K, Kandasamy A, Natarajan M
International Journal of General Medicine 2023, 16:185-191
Published Date: 17 January 2023
A Nomogram for Predicting Delayed Viral Shedding in Non-Severe SARS-CoV-2 Omicron Infection
Yu T, Dong J, Qi Q, Lv Q, Li J, Huang C, Cai X
Infection and Drug Resistance 2023, 16:2487-2500
Published Date: 27 April 2023