")
Back to Journals » Cancer Management and Research » Volume 11
Mutation spectrum of germline cancer susceptibility genes among unselected Chinese colorectal cancer patients
Authors Gong R, He Y, Liu XY, Wang HY, Sun LY , Yang XH, Li B, Cao XK, Ye ZL, Kong LH, Zhang DD, Li YH, Xu RH, Shao JY
Received 9 November 2018
Accepted for publication 15 February 2019
Published 29 April 2019 Volume 2019:11 Pages 3721—3739
DOI https://doi.org/10.2147/CMAR.S193985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Rui Gong,1,2 Yuan He,1,2 Xiao-Yun Liu,1,2 Hai-Yun Wang,1,2 Li-Yue Sun,1,2 Xin-Hua Yang,1,2 Bin Li,3 Xin-Kai Cao,3 Zu-Lu Ye,1,2 Ling-Heng Kong,1,4 Da-Dong Zhang,3 Yu-Hong Li,1,5 Rui-Hua Xu,1,5 Jian-Yong Shao1,2
1State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 2Department of Molecular Diagnostics, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 3Research and Development Institute of Precision Medicine, 3D Medicine Inc., Shanghai, 201114, People’s Republic of China; 4Department of Colorectal Surgery, Sun Yat-sen University Cancer Center, Guangzhou, People’s Republic of China; 5Department of Medical Oncology, Sun Yat-sen University Cancer Center, Guangzhou, People’s Republic of China
Background: Genetic factors play an important role in colorectal cancer (CRC) risk, yet the prevalence and spectrum of germline cancer susceptibility gene mutations among unselected Chinese CRC patients is largely undetermined.
Methods: We performed next-generation sequencing with a 73-genes panel and analyzed the prevalence and spectrum of germline mutations in 618 unselected Chinese CRC patients. We classified all identified germline alterations for pathogenicity and calculated the frequencies of pathogenic mutations. Clinical characteristics were assessed by age and mutation status. Protein expressions and interactions of MLH1 missense variants were evaluated by western blot and co- immunoprecipitation.
Results: Overall, 112 (18.1%) of 618 unselected Chinese CRC patients were found to carry at least one pathogenic or likely pathogenic variant (totaling 97 variants), including 70 (11.3%) Lynch syndrome (LS) mutation carriers and 42 (6.8%) non-LS mutation carriers. LS mutation carriers were significantly younger at CRC diagnosis and were more likely to have right-sided, poorly differentiated, early stage, high-frequency microsatellite instability (MSI-H) or dMMR CRC and a family history of cancer compared with noncarriers. Non-LS mutation carriers were more likely to be proficient mismatch repair (pMMR) than noncarriers (p=0.039). We found four clinical variables (gender, tumor histological stage, cancer stage and mutation status) that showed significant differences between patients younger and older than 50 years old. Higher mutation rates were found in patients under 50 years old (p=0.017). Thirty-three novel variants were discovered and evaluated as pathogenic mutations by our study.
Conclusion: Given the high frequency and wide spectrum of mutations, genetic testing with a multigene panel should be considered for all Chinese CRC patients under 50 years old and is also needed to determine whether a gene is associated with CRC susceptibility and to promote clinical translation.
Keywords: genetic factor, germline mutations, Lynch syndrome, next-generation sequencing
Introduction
Colorectal cancer (CRC) is the third most common malignancy and the fourth leading cause of cancer-related mortality worldwide.1 The majority of CRC cases are sporadic, but inherited factors contribute to approximately 30–35% of CRC cases.2 Between 5–10% of CRCs are associated with high-risk mutations in known CRC susceptibility genes, predominantly the mismatch repair (MMR) genes (Lynch Syndrome, LS), APC (Familial Adenomatous Polyposis, FAP) and MUTYH (MUTYH - associated Polyposis, MAP).3–5 Overall, mutation carriers have an increased risk of CRC (lifetime risk 30–70%).6,7 Genetic factors play an important role in CRC risk and predisposition.8,9 The identification of individuals at high risk for CRC, especially those who carry mutations in a CRC susceptibility gene, is important to provide various options for risk management and targeted screening for cancer prevention.10
While hereditary CRC has been a common indicator for germline genetic evaluation since MMR genes were identified, many additional genes have subsequently been implicated in CRC.11 Patients who performed germline genetic testing for CRC were typically tested for a limited number of genes that were strongly associated with CRC, such as MMR genes, APC and MUTYH. With the advent of next-generation sequencing (NGS), germline genetic testing for CRC has shifted from a limited number of phenotype-specific gene assessments to broad panels with multiple genes indicating various hereditary cancer syndromes. Since NGS reduces costs and increases the capacity to analyze multiple genes in parallel, there is an opportunity to provide more information on a large number of genes, allowing for more accurate cancer surveillance and tailored prevention options.12,13 The genes included in different panels vary, ranging from well-established cancer susceptible genes with quantifiable risk levels to less well-defined genes that are not traditionally associated with CRC.12,13
Herein we used NGS with a 73-genes panel associated with various hereditary cancer syndromes to determine the prevalence and spectrum of germline mutations in a consecutive series of 618 CRC patients. This may help to shape a more comprehensive understanding of genetic structure of CRC and generate accurate individualized risk management strategies for mutation carriers.14
Materials and methods
Patients and methods
We reviewed the genetic test results and clinical data from a consecutive series of 618 CRC patients evaluated by an NGS hereditary cancer panel between September 2014 and September 2017 at the Sun Yat-sen University Cancer Center (SYSUCC) (Guangzhou, China). All patients submitted a peripheral blood sample, from which germline DNA was isolated for clinical sequencing. Germline DNA was tested for mutations in 73 cancer susceptibility genes using the NGS method (gene list is detailed in Table 1). Patient demographics, medical history, family history (three generation), colonoscopy or resection findings, tumor location, tumor histology and phenotype, MMR status and CRC stage were obtained through review of medical records and through detailed patient inquiries. The SYSUCC ethical review board approved this study, and written informed consents were obtained.
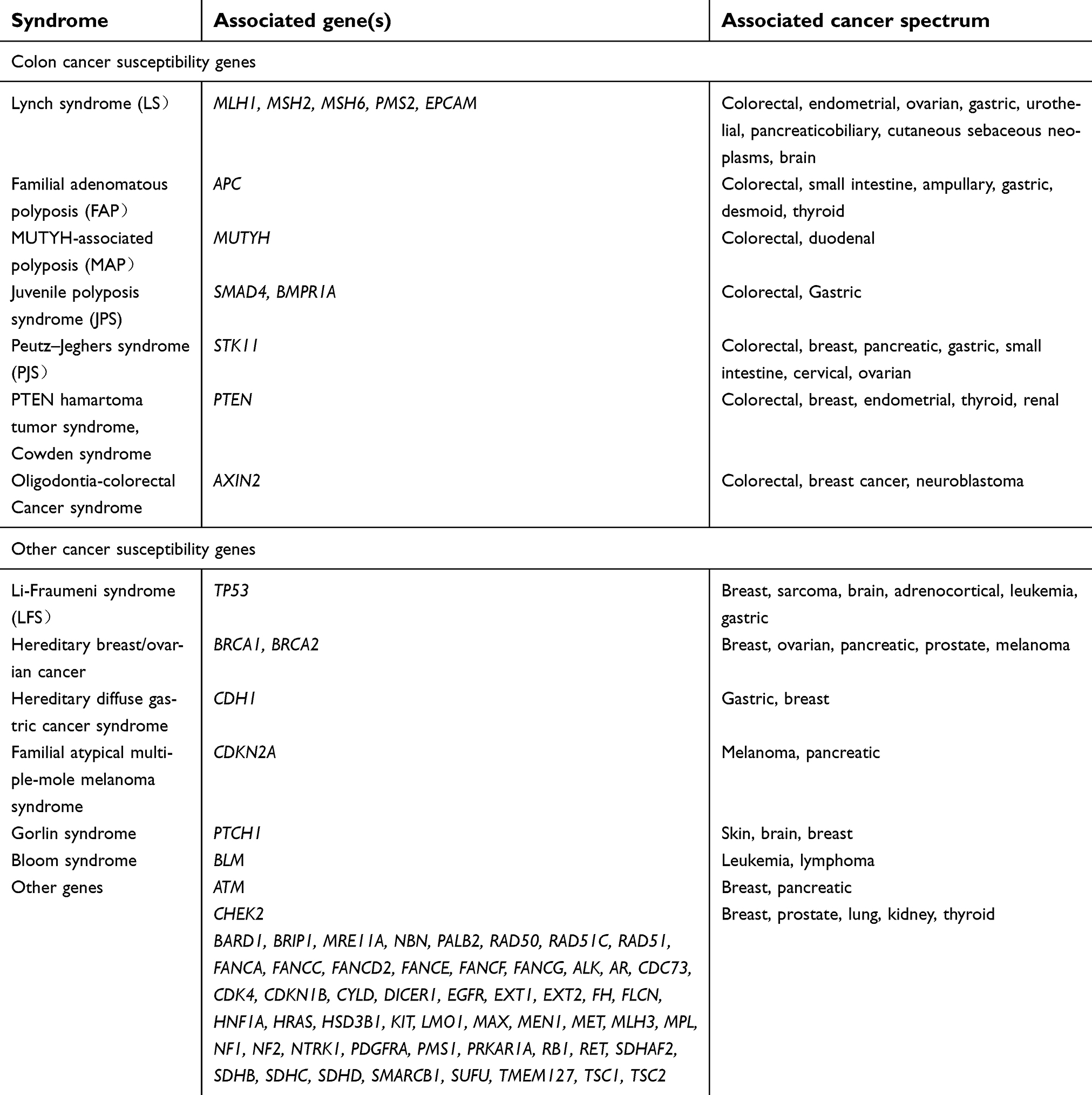 | Table 1 Genes analyzed by a multigene hereditary cancer panel |
Clinical genetic testing
NGS-based clinical sequencing of germline DNA for mutations was performed at Clinical Laboratory Improvement Amendments (CLIA)-certified commercial genetic testing laboratory (The Beijing Genomics Institute, BGI, Shenzhen, China) using sequencing by synthesis (SBS) in accordance with current practice standards. NGS was performed to detect single nucleotide variations (SNVs), copy number variants (CNV) and insert/deletion mutations (indels) within 20 bp for all exon regions as well as a portion of intronic regions (±10 bp) for all of the genes in the panel. The average depth was more than 300× in all samples and the coverage of the target regions was approximately 99.5%.
IHC and MSI analysis
MMR status were determined by immunohistochemistry (IHC) testing for MLH1, MSH2, MSH6 and PMS2 protein (antibody: Roche, 07862237001, 078622530, 07862245001, 07862261001) expression in tumor tissue. MSI status were tested using the Bethesda consensus panel (mononucleotide repeats BAT25 and BAT26, and dinucleotide repeats D2S123, D5S346, and D17S250) by multiplex fluorescent PCR and capillary electrophoresis. Tumors showing MSI at 0 marker were classified as microsatellite stable (MSS). Tumors showing MSI at 1 marker were classified as low-frequency microsatellite instability (MSI-L). Tumors showing MSI at 2 or more markers were classified as high-frequency microsatellite instability (MSI-H).
Data analysis
The outcomes of clinical genetic testing were analyzed by us with the assistance of 3D Medicines (Shanghai, China) according to the 2015 American College of Medical Genetics and Genomics (ACMG) standards and guidelines15 for variant classification. Genetic variants were classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign or benign. Every variant was analyzed by at least a master- or PhD-level analyst and a PhD-level Fellow clinical molecular geneticist. We also referred to specific tools and resources include the Exome Sequencing Project (ESP),16 The 1,000 Genomes Projects, The Exome Aggregation Consortium (Ex AC), The Single Nucleotide Polymorphism Database (dbSNP), ClinVar, SIFT17 and Polyphen.18
PCR amplification and sanger validation
To validate the results of NGS, sequence-specific PCR was used to amplify the target fragment with specific primers designed with PelPrimer. Total PCR volume was 25 μl, including12.5 μl 2×Taq PCR MasterMix (KT201, Tiangen Biotech, Beijing), 1 μl template DNA, 9.5 μl ddH2O, 1 μl PCR Primer F and 1 μl PCR Primer R. The first step in the PCR was performed at 94 °C for 3 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 1 min and a final extension at 72 °C for 5 min. Agarose gel electrophoresis and a DNA purification kit were used to isolate and purify the PCR products. Sanger sequencing with an ABI 3500 (Applied Biosystems 3500) genetic analyzer was used to validate all of the pathogenic and likely pathogenic variants. Finally, the sequences were aligned to the Nucleotide Database with NCBI Blast online software.
Statistical analysis
The primary outcome of this study was the detection of cancer susceptibility gene mutations. Information related to patient numbers and demographics were presented using descriptive statistics. Data for qualitative variables were reported as percentages. The association of mutation status with clinical characteristics was analyzed using the chi-squared test or Fisher’s exact test (when cells have an expected count of less than 5) to determine p-values (qualitative variables) and level of significance was set at 0.05. Patient age at CRC diagnosis was analyzed as a continuous variable and assessed by the two-sample t-test, reported as median and range. Wilson score intervals with continuity correction were used to compute confidence intervals. Statistical analysis was performed using SPSS 20.0.
Site-directed mutagenesis and expression plasmid construction
pcDNA3.1B was kindly provided by Dr Xiao-Feng Zhu (State Key Laboratory of Oncology in Southern China, Cancer Center, Sun Yat-sen University). To analyze protein expression in human cells, human hMLH1/MSH2/MSH6 cDNA was synthesized by PCR and then cloned into the plasmid pcDNA3.1B by double enzyme digestion (EcoR-I/BamH-I restriction enzyme from NEB) and homologous recombination (CloneExpress II One Step Cloning Kit, C112, Vazyme Biotech, Nanjing). Selected MLH1 variants were constructed by site-directed mutagenesis using a PCR-based protocol. In addition, eight plasmids were constructed and tagged with Flag protein (N-DYKDDDDK-C): MLH1-WT, MLH1 c.1153C>T (p.R385C), MLH1 c.1230_1232delinsTG (p.I411Vfs*80), MLH1 c.1573T>G (p.L525V), MLH1 c.1713delT (p.F571Lfs*2), MLH1 c.1866delT (p.A623Qfs*14), MLH1 c.278delG (p.S93Ifs*15) and MLH1 c.522delG (p.I176Ffs*26). pcDNA3.1B (empty vector) used as a negative control and MLH1-WT was used as a positive control.
HEK293T cell culture and transfection
HEK293T cell line was purchased from the Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). HEK293T cells were cultured in DMEM with 10% fetal bovine serum (FBS) at 37 °C in a humidified 5% CO2 atmosphere. HEK293T cells were transfected at 60–70% confluence with expression plasmids using Lipofectamine 3,000 reagent (L3000015, Invitrogen, Shanghai) according to the manufacturer’s instructions. After 48 h, the cells from each group were harvested for western blot.
Western blot analysis and co-immunoprecipitation
Cells were harvested and washed with phosphate-buffered saline (PBS). The lysates were obtained with RIPA lysis buffer (containing 1 mM PMSF) followed by centrifugation (4 °C, 12,000 g, 15 min). Total protein concentrations in the supernatant were examined using an Enhanced BCA Protein Assay Kit (Beyotime Biotechnology, P0010, China). For co-immunoprecipitation, 1 mg of lysate was incubated with anti-Flag Ab or control mouse IgG overnight at 4 °C with rotation and then for 2 h at 4 °C with 20 μl Protein A/G PLUS-Agarose (Santa Cruz Biotechnology). Immunoprecipitates were collected by centrifugation and washed five times with ice-cold PBS. After the final wash, the supernatant was discarded and the pellet was resuspended in SDS lysis buffer, then boiled in 5× SDS loading dye for 5 min. Western blot was performed according to the standard procedure. Briefly, proteins were normalized to 25 μg/lane and loaded on a 10% SDS-polyacrylamide gel for electrophoresis and then transferred to PVDF membranes. The membranes were blocked with 5% BSA in TBST (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20) for 1 h. The membranes were incubated with primary antibodies (anti-MLH1, CST3515T, mouse, 1:1,000; anti-PMS2, Abcam, ab110638, rabbit, 1:1,000 and anti-β-actin, 60008-1-Ig, Proteintech, mouse, 1:1,000) overnight at 4 °C, then washed three times with TBST for 10 min each time and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotech) at room temperature for 1 h. After washing the secondary antibody, the bands in the membrane were detected using chemiluminescence ECL kit. Finally, the membranes were processed using an enhanced chemiluminescence detection system.
Results
Clinical characteristics
A total of 77.8% (481/618) CRC patients were from south China in the Pearl River Valley and 12.5% (n=77) of them were from Yangtze River Valley and the rest were from other catchment areas. Patient characteristics including gender, age at CRC diagnosis, family history of cancer, primary tumor site, tumor histological stage, cancer stage, MMR and MSI status are summarized in Table 2. Men accounted for 59.2% (n=366) of patients and 44.7% (n=276) had a family history of cancer. The mean age at CRC diagnosis was 49.8 years and 48.7% (n=301) patients were diagnosed before 50 years old.
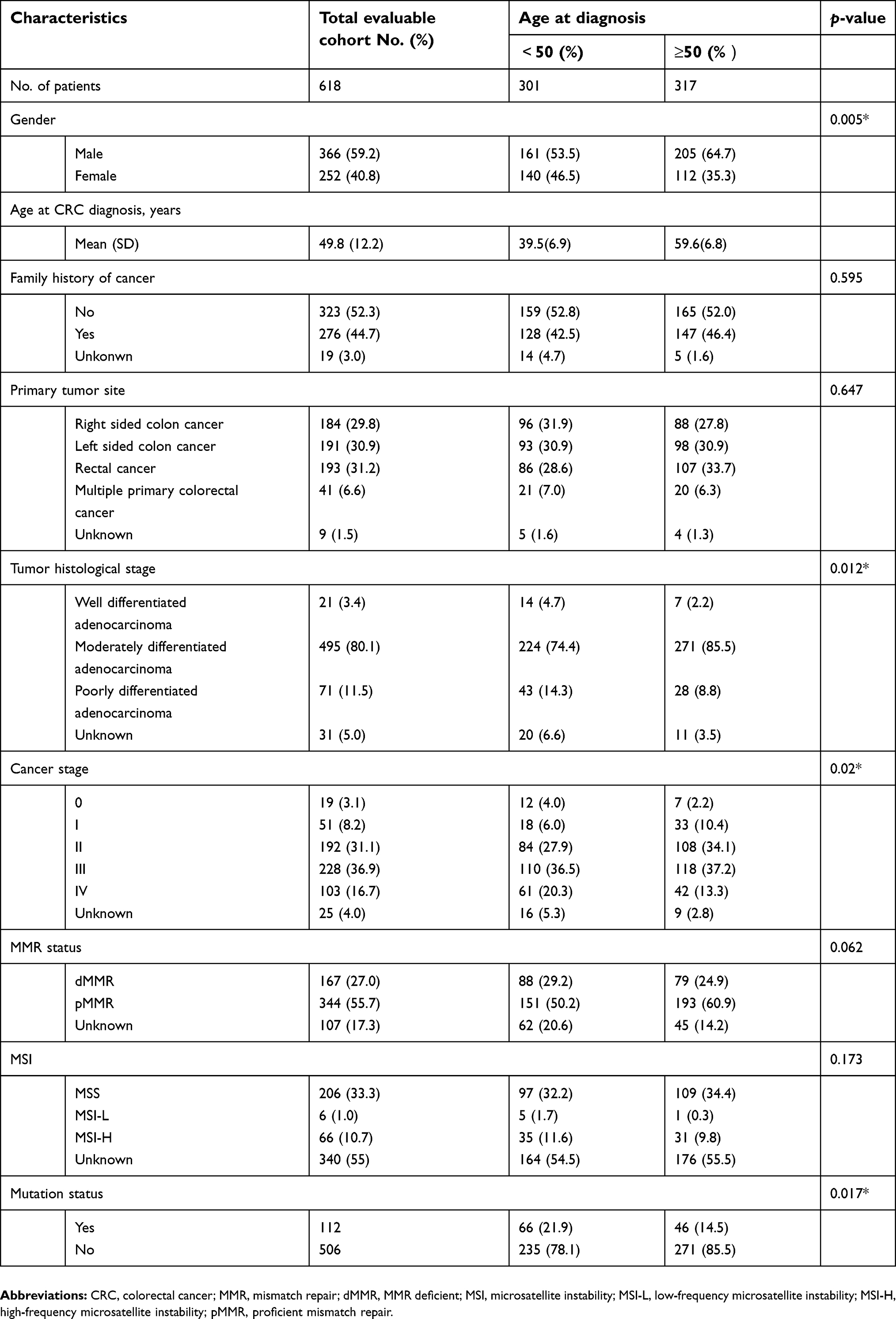 | Table 2 Clinical characteristics of 618 CRC patients |
A total of 82.7% (n=511) CRC patients had tumors with MMR IHC data available, of which 27% (n=167) were MMR deficient (dMMR). The most common pattern of MMR protein loss at all sites was MLH1±PMS2-. 45% (n=278) patients had tumors with MSI data available, of which 10.7% (n=66) were MSI-H. Most tumor screening results (MSI and IHC) were consistent for each patient, except for 12 patients (2 MSI-H, pMMR and 10 MSS/MSI-L, dMMR).
According to the clinical characteristics analysis by age (Table 2), we found 4 clinical variables (gender, tumor histological stage, cancer stage, mutation status) that showed significant differences between patients younger and older than 50 years old. There were more male patients than females in both groups, and the difference was more significant in patients over 50 years old. 20.3% (n=61) of patients under 50 and 13.3% (n=42) of patients over 50 were diagnosed with CRC at stage IV. Patients under 50 tended to have more poorly differentiated tumor than patients over 50 years. Patients under 50 years old were significantly more likely to carry pathogenic or likely pathogenic mutations than patients older than 50 (21.9% vs 14.5%, respectively). LS mutation carriers account for 14.3% (n=43) and non-LS mutation carriers account for 7.6% (n=23) of 301 patients under 50 years old.
Germline findings
Overall, 460 patients carried at least one pathogenic or likely pathogenic or VUS mutation (Figure 1A and C). A total of 596 VUS germline variants were detected among 419 patients (67.8%, 95% CI, 63.9–71.4%). The genes most likely to have a VUS variant discovered included ATM (n=42), FANCA (n=37), and BRCA2 (n=31) (Figure 1B). The highest VUS frequency was observed in the ATM gene. A total of 97 pathogenic and likely pathogenic germline variants were detected among 112 (18.1%; 95% CI, 15.2–21.4%) of the 618 patients, including 80 (12.9%; 95% CI, 10.5–15.9%) with high-penetrance mutations (one with a concurrent moderate-penetrance mutation) and 19 (3.1%; 95% CI, 1.9–4.8%) with only moderate-penetrance mutations. Additionally, 33 pathogenic or likely pathogenic mutations were newly discovered by our study which were unreported in public data bases (Table 3). They were not listed in ClinVar 20150330, dbSNP 138 databases or other literatures and their frequency in the databases of 1,000 Genomes Projects 2015 Aug, ESP 6500 and Ex AC is 0. The rest of the pathogenic and likely pathogenic germline mutations were detailed in Table S1. Evidence of pathogenicity were according to the 2015 American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
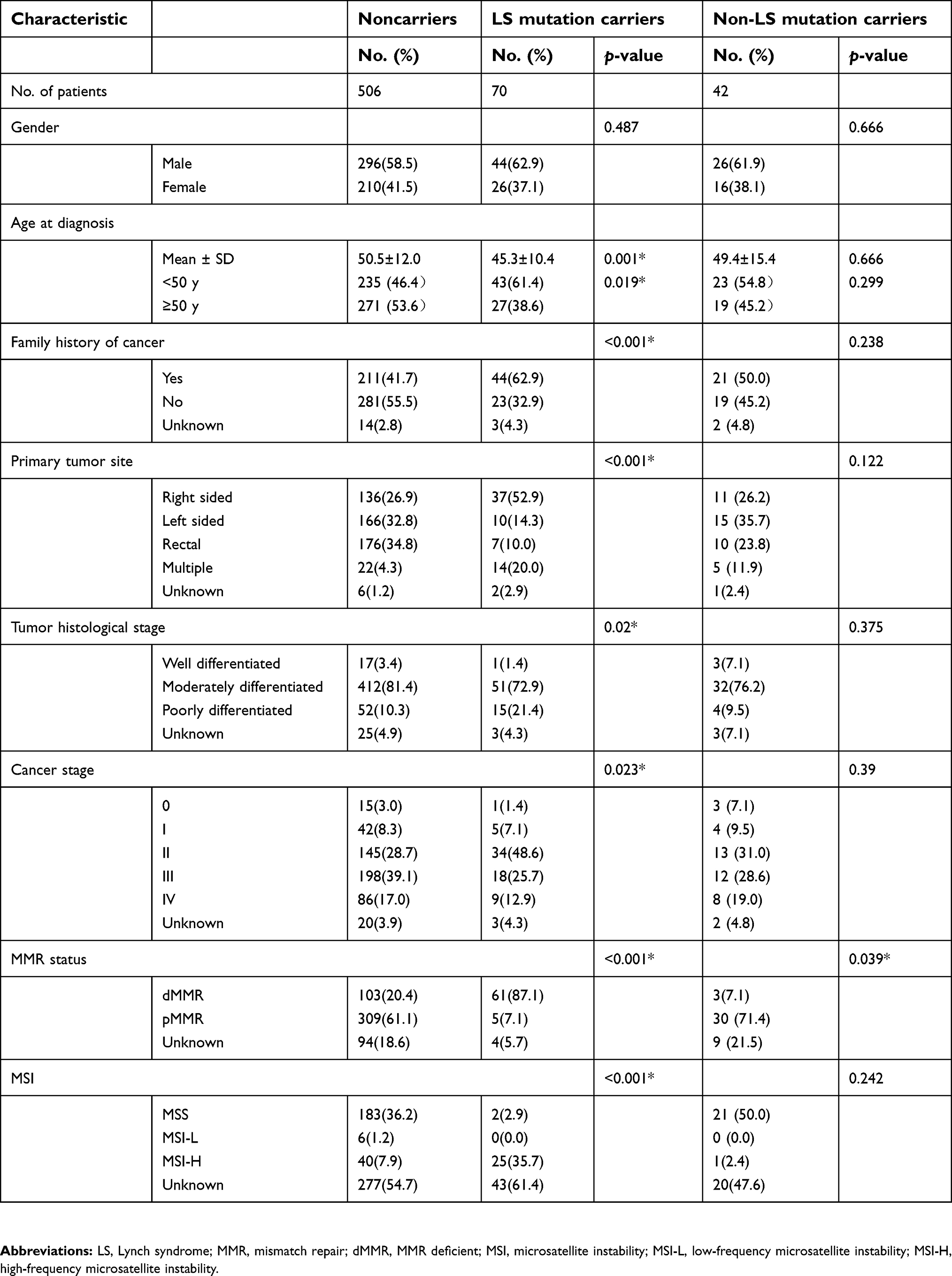 | Table 4 Clinical characteristics analysis of mutation carriers compared with noncarriers |
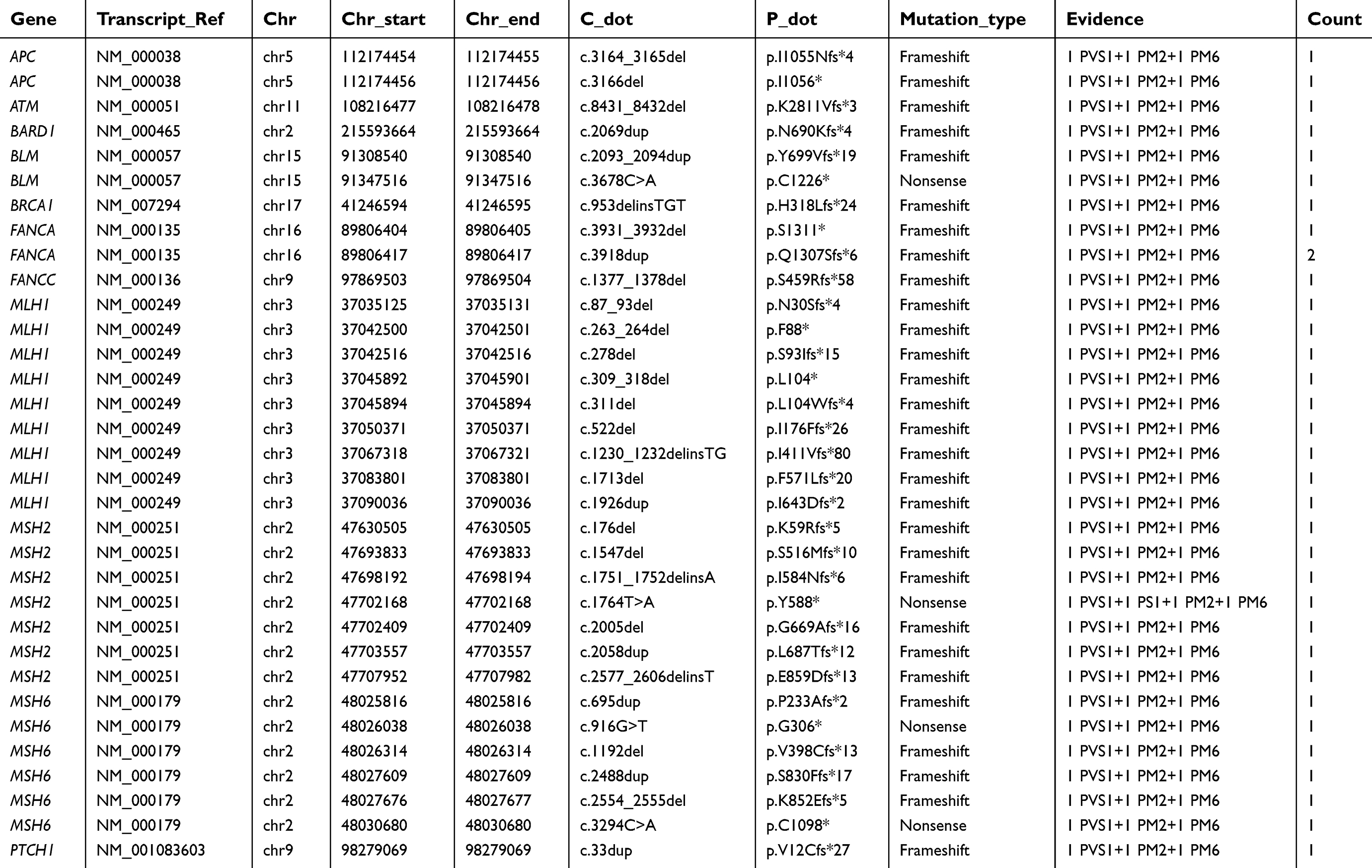 | Table 3 Summary of novel pathogenic germline mutations |
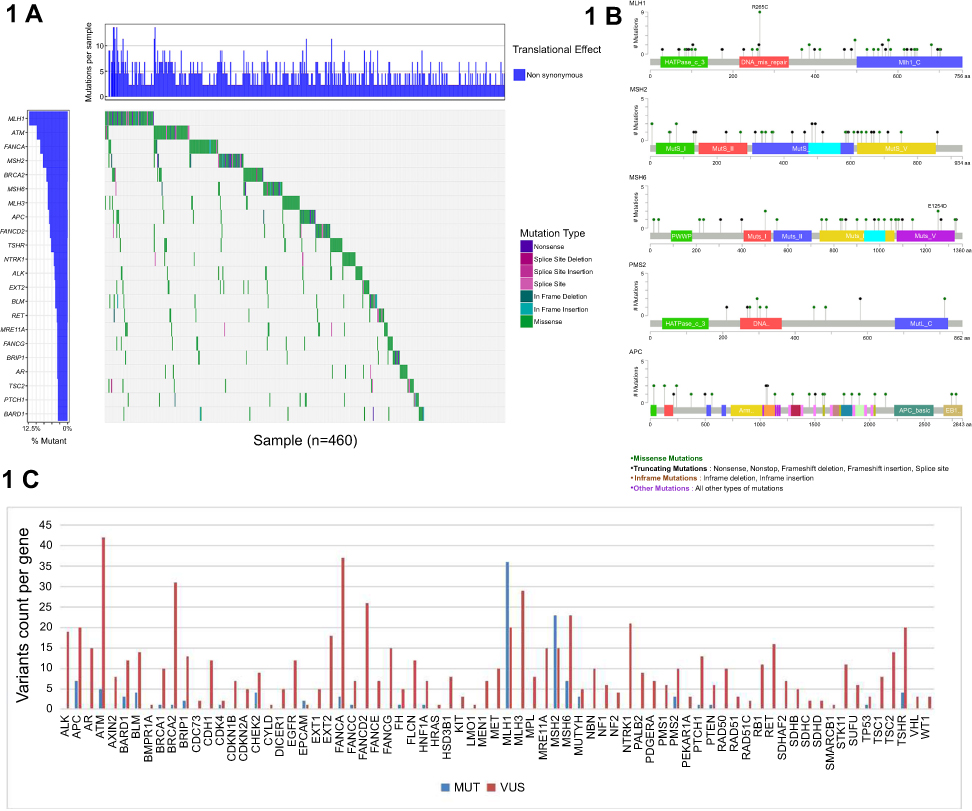 | Figure 1 The mutations found in 618 CRC patients. (A) Heatmap of genes (mutation frequency >3%) with germline non-benign variants identified among 618 unselected colorectal cancer patients. Each column represents a patient, and each row represents a gene with multiple germline variants. (B) A lollipop diagram of germline non-benign variants of MLH1, MSH2, MSH6, PMS2 and APC genes identified among 618 unselected colorectal cancer patients. (C) The number of MUT and VUS variants per gene detected with a multigene panel among 618 unselected colorectal cancer patients. The MUT group includes both pathogenic and likely pathogenic variants. Abbreviations: MUT, mutation; CRC, colorectal cancer ; VUS, variants of uncertain significance. |
Seventy patients (11.3%, 95%CI 9% to 14.2%) carried LS mutations (36 MLH1, 23 MSH2, 7 MSH6, and 3 PMS2, 2 deletions of the 3ʹ-end of EPCAM, including one patient with both MSH2 and EPCAM mutations) and 42 (6.8%, 95% CI, 5–9.15%) carried non-LS mutations (including one patient with both a LS and a non-LS mutations, MLH1/BLM). Thirteen patients (2.1%; 95% CI, 1.2–3.7%) carried high penetrance non-LS mutations (7 APC, 1 PTCH1, 1 PTEN, 1 TP53, 1 BRCA1, 1 BRCA2 and 1 biallelic MUTYH). Twenty patients (3.2%; 95% CI, 2–5.1%) carried mutations in moderate-penetrance genes (5 ATM, 3 BARD1, 4 BLM, 2 BRIP1, 1 CDK4, 4 CHEK2 and 1 Monoallelic MUTYH). The rest carried mutations in less well-defined genes. The spectrum of pathogenic and likely pathogenic germline mutations is shown in (Figure 2). 61 (87.1%; 95% CI, 76.5–93.6%) of 70 LS mutation carriers demonstrated MSI-H and/or dMMR (2 pMMR or MSS; 6 had missing MSI/MMR data). All the pathogenic and likely pathogenic mutations detected by NGS were validated by Sanger sequencing or qPCR.
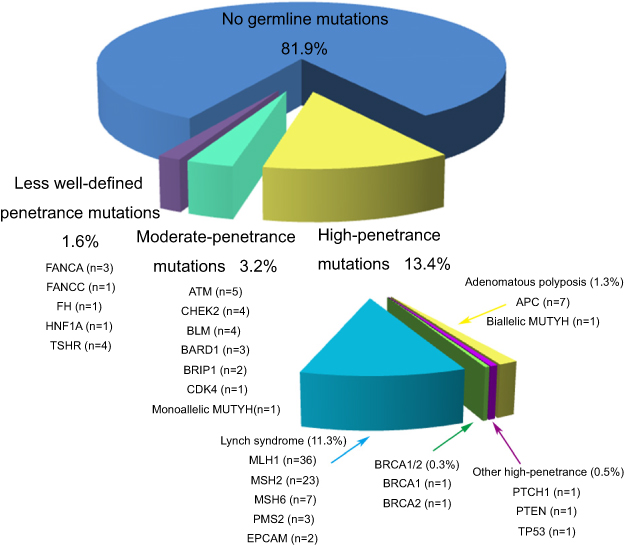 | Figure 2 The identified pathogenic and likely pathogenic germline mutations. There was one patient with both MSH2 and EPCAM mutations and one patient with both MLH1 and BLM mutations. |
In the clinical characteristics analysis of mutation carriers compared with noncarriers, LS mutation carriers were significantly younger at CRC diagnosis and more likely to have right-sided, poorly differentiated, early stage, MSI-H, dMMR CRC and family history of cancer compared with noncarriers (Table 4). Non-LS mutation carriers were more likely to be pMMR than noncarriers. Age at CRC diagnosis, gender, primary tumor site, tumor histological stage, cancer stage and family history of cancer was not significantly associated with the presence of a non-LS mutation. However 23 (54.8%) of 42 non-LS mutation carriers and 40 (59.7%) of LS mutation carriers were given a diagnosis of CRC under 50 years old. Eleven (26.2%) of 42 non-LS mutation carriers were given a diagnosis of CRC at age ≥50 years old and lacked a family history of cancer.
Pedigree analysis
Two families that had a typical family cancer history and novel mutations were taken into the study after informed consent. Analysis of the candidate variant was performed in additional family members by using NGS or Sanger sequencing.
In family W, the proband was diagnosed with sigmoid colon cancer at 48 years old (pMMR/MSI unknown) and ovarian cancer at 61 years old. The patient carried 3 mutations of uncertain significance (MUTYH c.924+7C>T, MLH1 c.T1573G, EGFR c.A1703G). The patient’s father, mother and 4 siblings were diagnosed with different types of cancer (Figure 3A).
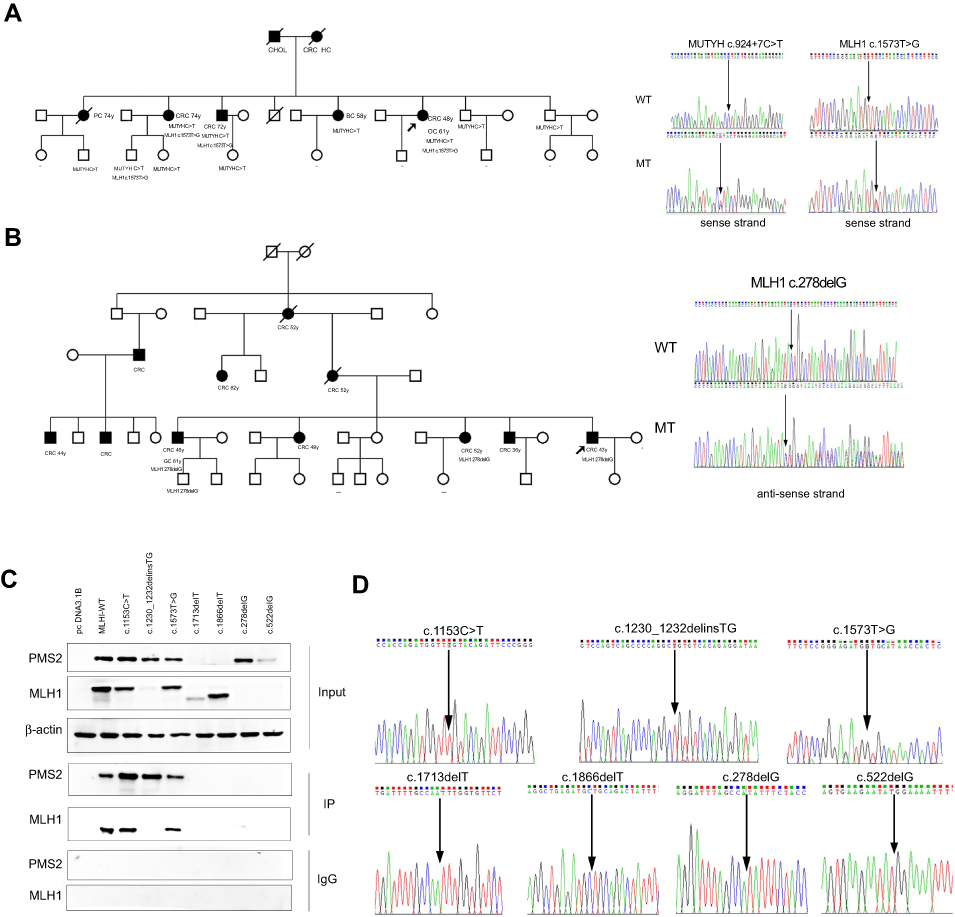 | Figure 3 Evidence to identify germline variants for pathogenicity. (A) Pedigree of family W. (B) Pedigree of family T. (C) Western-blot analysis of MLH1 and PMS2 proteins. (D) Characterization of constructed MLH1 mutated plasmids. Numbers represent age at diagnosis. Minus signs indicate that the individual was confirmed not to carry the specific mutation. Shading indicates that the individual was affected with cancer. The arrow heads indicate the proband for that family.Abbreviations: CRC, colorectal cancer; CHOL, cholangiocarcinoma; HC, hepatic cancer; BC, breast cancer; PC, pancreatic carcinoma; OC, ovarian cancer; GC, gastric cancer. |
In family T, the proband was diagnosed with ascending colon cancer at 43 years old. Immunohistochemistry showed a lack of staining of MLH1 and PMS2 proteins. The MSI status was unknown. The patient carried one novel pathogenic mutation (MLH1 c.278delG). Eleven family members in 3-generations were diagnosed with CRC (Figure 3B).
The mutation status of the pedigrees suggests that these novel mutations may be pathogenic (supporting evidence, PP4).
In vitro protein analysis of MLH1 missense variants
Expression was determined by transient transfection of mutated plasmid (Characterization of constructed MLH1 mutated plasmids were showed in Figure 3D) into HEK293T cells, which do not express the endogenous MLH1 gene due to promoter hyper-methylation.19 The c.1230_1232delinsTG (p.I411Vfs*80), c.1713delT (p.F571Lfs*2), , c.278delG (p.S93Ifs*15) and c.522delG (p.I176Ffs*26) mutations were novel MLH1 variants that were unreported in public databases and were classified as pathogenic mutations by our study. The MLH1 c.1866delT (p.A623Qfs*14) mutation were pathogenic MLH1 variant that has been previously reported. The c.1153C>T (p.R385C) and c.1573T>G (p.L525V) mutations were VUS MLH1 variants that have been previously reported. The c.1230_1232delinsTG, c.278delG and c.522delG variants showed a strong reduction of MLH1 expression, and the c.1713delT and c.1866delT variants showed protein truncation at approximately 65 kd and 70 kd. PMS2 was not properly co-expressed in the truncated c.1713delT, c.1866delT and c.522delG MLH1 variants since PMS2 was destabilized on the protein level in the absence of proper dimerization with the MLH1 C-terminus, which was absent in these variants. Immunoprecipitation experiment showed that the truncating variants MLH1 c.1713delT, c.1866delT, c.278delG and c.522delG inhibited the interaction between MLH1 and PMS2. In contrast, we found that the c.1153C>T and c.1573T>G variants had no effect on protein expression and interactions (Figure 3C).
The function analysis suggests that MLH1 c.1230_1232delinsTG (p.I411Vfs*80), c.1713delT (p.F571Lfs*2), c.1866delT (p.A623Qfs*14), c.278delG (p.S93Ifs*15) and c.522delG (p.I176Ffs*26) may be pathogenic (moderate evidence).
Discussion
Using multigene panel testing, we determined the prevalence and spectrum of germline mutations in 73 genes associated with various hereditary cancer syndromes in 618 unselected Chinese CRC patients. One hundred twelve patients (18.1%) carried at least a pathogenic or likely pathogenic germline mutation, most of which were in high-penetrance cancer susceptibility genes. One of every 5 patients with CRC diagnosed younger than 50 years had at least 1 pathogenic or likely pathogenic germline mutation (21.9%). The mutation rate was between Pearlman’ (16%)9 and Stoffel’s studies (25.1%).20 This differences may due to different testing panels or populations. Therefore, we recommend colon cancer screening in the general population starting at age 50.
The prevalence of LS mutation carriers reported in this study (11.3%) was slightly higher than previous publications.21,22 All the CRC patients in our cancer center were recommended for immunohistochemistry and genetic testing. However practical challenges in implementation and concerns regarding cost-effectiveness may result in certain selection bias, which means the patients who participated in the NGS are not completely random. After all, 44.7% of CRC patients in the cohort had a family cancer history and 46.4% of them were under 50, which was also higher than the previous publications. Therefore the prevalence we found for pathogenic and likely pathogenic mutations probably represents the maximum prevalence. Consistent with prior studies that performed germline LS testing after preselection with MSI/MMR tumor testing,23–25 36.5% of dMMR CRC patients (61/167) in the current study had LS mutations and 87.1% of LS mutation carriers (61/70) demonstrated MSI-H and/or dMMR. These results support the current practice of performing systematic MSI/MMR immunohistochemistry for all CRCs to screen for LS.26,27 For screening, immunohistochemistry is almost equally sensitive as MSI. However limiting tumor analysis to patients who fulfill the Bethesda criteria or the Amsterdam II criteria would fail to identify 20% (14/70) of LS cases.
In addition to LS, our study also determined the prevalence and spectrum of other hereditary cancer syndromes found in 618 unselected CRC patients. Among 618 CRC patients, 6.8% (42/618) carried non-LS mutations (one with both LS and non-LS mutations), accounting for 37.5% (42/112) of all positive variants identified in our overall testing population. The large number of non-LS mutations found in this cohort suggests that MMR/MSI testing alone is insufficient to identify cancer risk in unselected CRC patients. Furthermore, 26.2% (n=11) of non-LS mutation carriers in this study also lacked traditional phenotypic characteristics of hereditary CRC risk (age under 50 years or family history of cancer), which made it more difficult to identify non-LS mutations. ATM mutations were found in 0.8% (n=5) of CRC patients in our study which was higher than the estimated 0.37% (455/123136) of general population prevalence in a previous publication28 and raises questions about whether such mutations predispose to CRC. Other moderate genes that were not traditionally associated with CRC, such as CHEK2 and BLM (0.6%, 4/618), also showed a possible link between CHEK2/BLM mutations and CRC risk. These findings further support the hypothesis that the analysis of genes currently excluded from routine molecular diagnostic screens may be predisposed to a wider range of cancers, potentially including CRC.28 While studies support an association with cancer, the magnitude of the risk and complete cancer spectrum for variants in these genes is unclear.29
In our study, pedigree analysis and western blot analysis also provided some moderate or supporting evidence to identify germline variants for pathogenicity. Families W and T had phenotypes and family histories that were highly specific for CRC with a single genetic etiology. The protein expression and interaction of MLH1 and PMS2 were affected by some inframe variants especially those that led to protein truncation or had an impact on the interacting domains which is consistent with previous studies.30,31
Our study has certain limitations. We could not detect a large deletion (>20 bp), methylation of MLH1 or the BRAF V600E mutation to confirm that there were no such mutations in the patients who were non-carriers especially non-LS mutation carriers with dMMR. We failed to obtain a comprehensive gene mutation status of the pedigrees due to patient compliance.
In conclusion, the advancements in NGS have led to a refined understanding of the genomics of colorectal cancer. The prevalence and spectrum of germline cancer susceptibility gene mutations have been investigated in previous studies among unselected or high-risk Caucasian, American and Asian populations, yet these conditions among unselected Chinese CRC patients are largely undetermined. Our study is the first to our knowledge to determine the prevalence and spectrum of germline cancer susceptibility gene mutations in unselected CRC patients in Chinese population using an NGS panel of 73 genes. Multigene panel testing facilitated the identification of germline mutations in patients who may have otherwise been missed. Only through extensive testing and the accumulation of large international datasets will sufficient information be generated to provide overwhelming evidence to determine whether a gene is associated with CRC susceptibility. Despite these improvements, further studies are needed to determine the function of various mutations in each gene which will enable us to promote clinical translation.
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki. The ethical review board of Sun Yat-sen University Cancer Center approved this study (grant No. C2018-013-01).
Patient consent
Written informed consent were obtained.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors are grateful to the family members who participated in this study and we would like to thank Dr. Xiang-Feng Zhu for the gift of pcDNA3.1B plasmids and the assistance from their laboratory. This work was supported by the Guangzhou Science and Technology Plan Projects (Health Medical Collaborative Innovation Program of Guangzhou) (grant No. 201803040019) and the National Natural Science Foundation of China (No. 81472522).
Author contributions
RG and YH contributed equally to this study. JYS and RHX conceived and designed the experiments; RG and YH performed the experiments; RG, XHY, XYL XKC and BL performed the bioinformatics analysis; LYS, LHK and ZLY collected the samples; RG, YH, YHL and DDZ analyzed the data. HYW and RG wrote the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
2. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi:10.1056/NEJM200007133430201
3. Valle L. Recent discoveries in the genetics of familial colorectal cancer and polyposis. Clin Gastroenterol Hepatol. 2017;15(6):809–819. doi:10.1016/j.cgh.2016.09.148
4. Lorans M, Dow E, Macrae FA, Winship IM, Buchanan DD. Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin Colorectal Cancer. 2018;17(2):e293–e305. doi:10.1016/j.clcc.2018.01.001
5. Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33(2):209–217. doi:10.1200/JCO.2014.58.1322
6. Balmana J, Balaguer F, Cervantes A, Arnold D. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol. 2013;24(Suppl 6):vi73–80. doi:10.1093/annonc/mdt209
7. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–932. doi:10.1056/NEJMra012242
8. Obuch JC, Ahnen DJ. Colorectal cancer: genetics is changing everything. Gastroenterol Clin North Am. 2016;45(3):459–476. doi:10.1016/j.gtc.2016.04.005
9. Pearlman, R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3(4):464–471. doi:10.1001/jamaoncol.2016.5194
10. Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. 2017;35(10):1086–1095. doi:10.1200/JCO.2016.71.0012
11. Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology. 2015;149(3):604–13.e20. doi:10.1053/j.gastro.2015.05.006
12. Gallego CJ, Shirts BH, Bennette CS, et al. Next-generation sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: a cost-effectiveness analysis. J Clin Oncol. 2015;33(18):2084–2091. doi:10.1200/JCO.2014.59.3665
13. Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18(8):823–832. doi:10.1038/gim.2015.166
14. Hahn MM, de Voer RM, Hoogerbrugge N, Ligtenberg MJL, Kuiper RP, van Kessel AG. The genetic heterogeneity of colorectal cancer predisposition - guidelines for gene discovery. Cell Oncol (Dordr). 2016;39(6):491–510. doi:10.1007/s13402-016-0284-6
15. Sue Richards NAPS. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;5(17):405–423. doi:10.1038/gim.2015.30
16. Casper J, Zweig AS, Villarreal C, et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018;46(D1):D762–D769. doi:10.1093/nar/gkx1020
17. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi:10.1038/nprot.2009.86
18. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
19. Trojan J, Zeuzem S, Randolph A, et al. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology. 2002;122(1):211–219.
20. Stoffel EM, Koeppe E, Everett J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology. 2018;154(4):897–905.e1. doi:10.1053/j.gastro.2017.11.004
21. Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354(26):2751–2763. doi:10.1056/NEJMoa053493
22. Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. doi:10.1200/JCO.2008.17.5950
23. Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol. 2013;31(20):2554–2562. doi:10.1200/JCO.2012.46.8454
24. Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308(15):1555–1565. doi:10.1001/jama.2012.13088
25. Kastrinos F, Ojha RP, Leenen C, et al. Comparison of prediction models for Lynch syndrome among individuals with colorectal cancer. J Natl Cancer Inst. 2016;108(2):djv308. doi:10.1093/jnci/djv308
26. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–262. quiz 263. DOI:10.1038/ajg.2014.435.
27. Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014;109(8):1159–1179. doi:10.1038/ajg.2014.186
28. Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319(23):2401–2409. doi:10.1001/jama.2018.6228
29. Ma X, Zhang B, Zheng W. Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut. 2014;63(2):326–336. doi:10.1136/gutjnl-2012-304121
30. Kondo E, Horii A, Fukushige S. The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Res. 2001;29(8):1695–1702.
31. Guerrette S, Acharya S, Fishel R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999;274(10):6336–6341.
Supplementary material
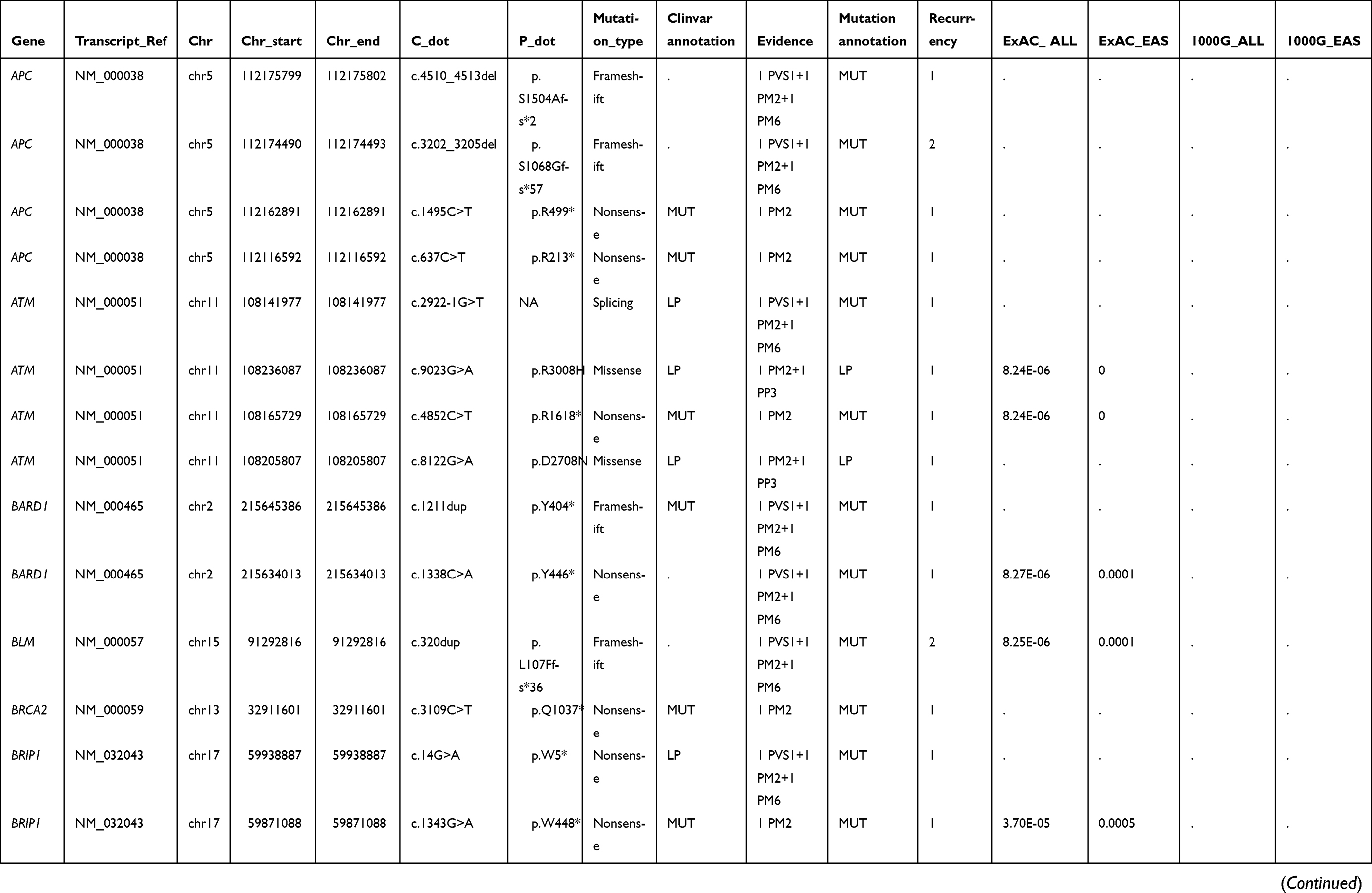 | 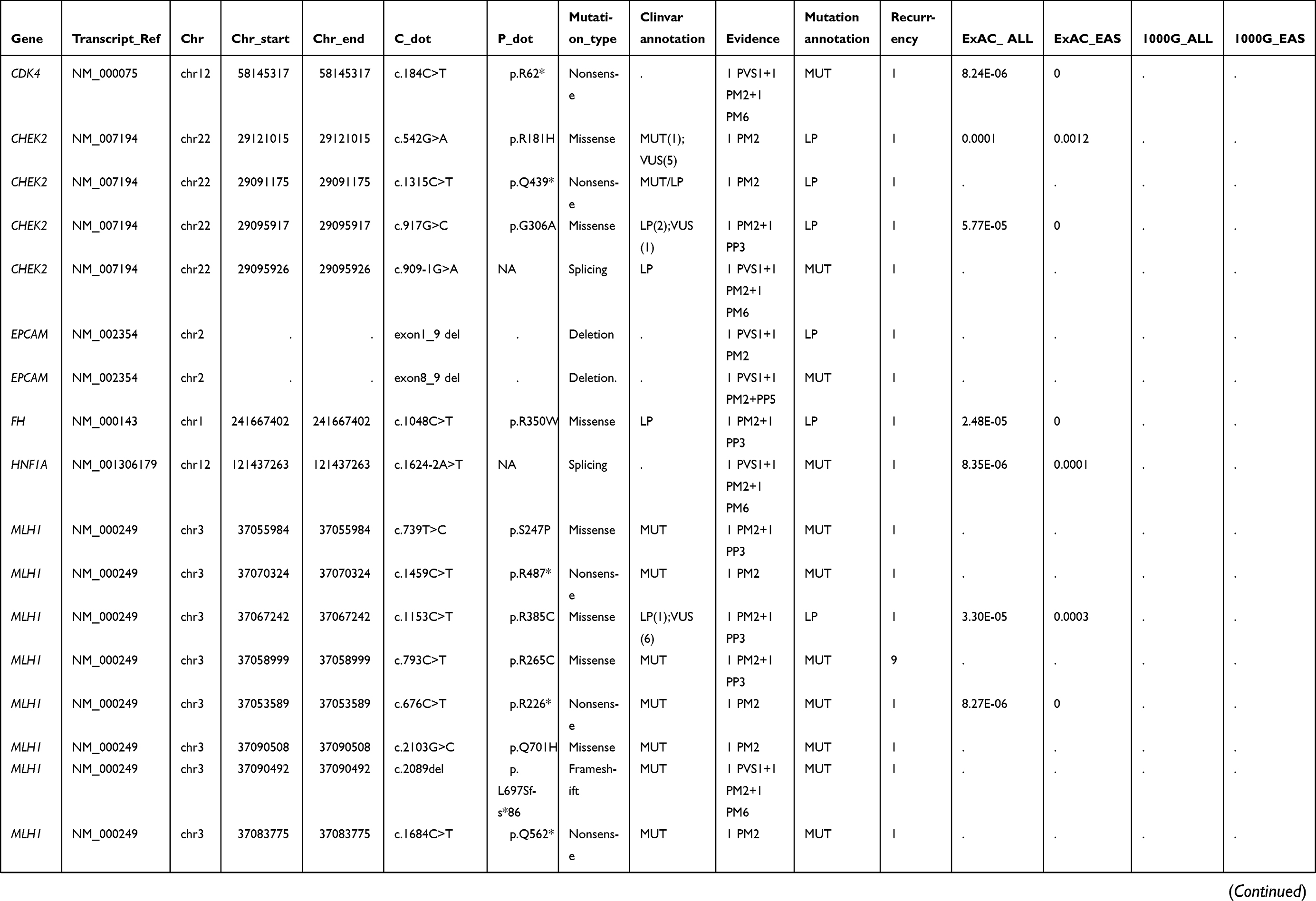 | 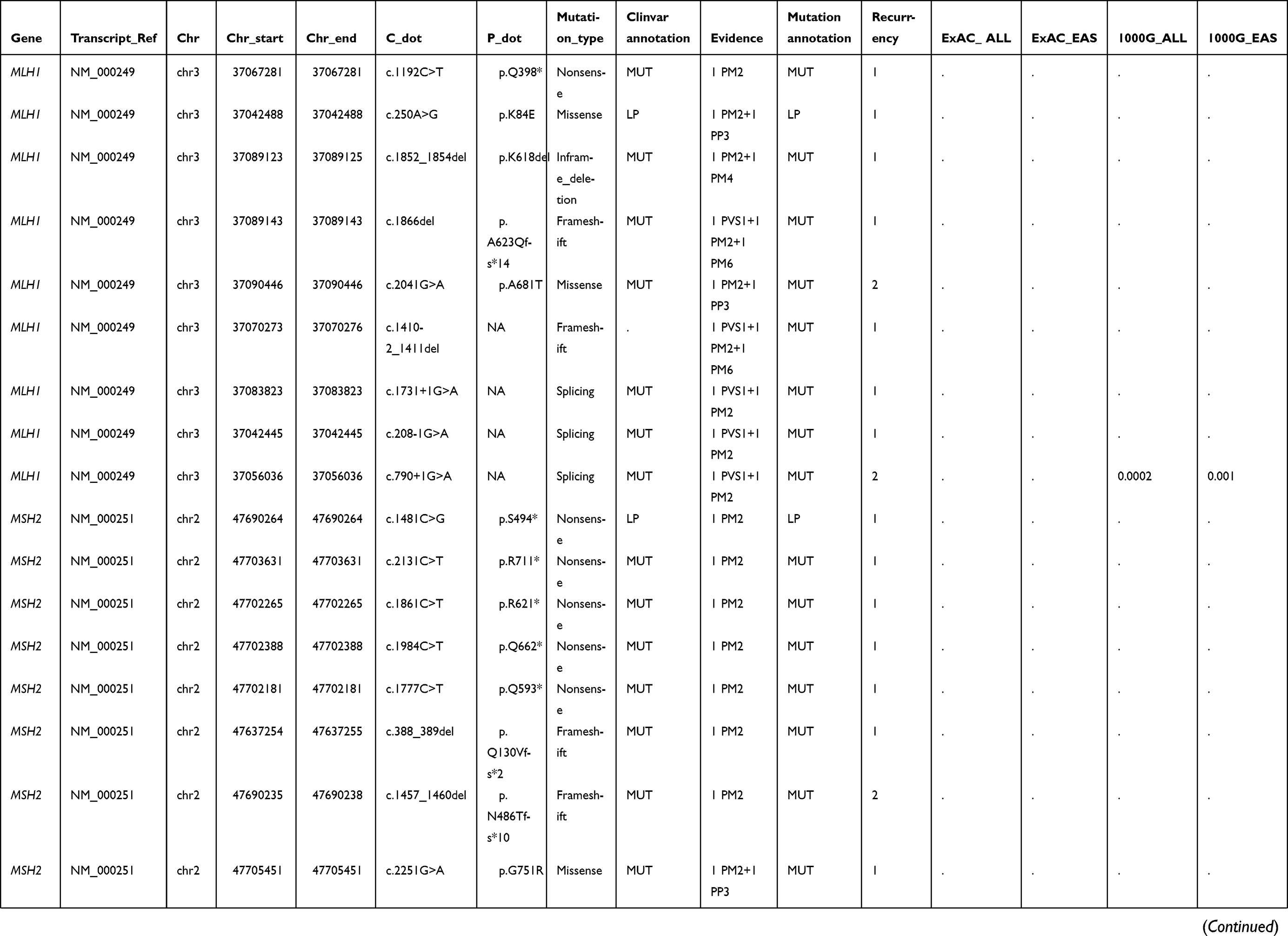 | 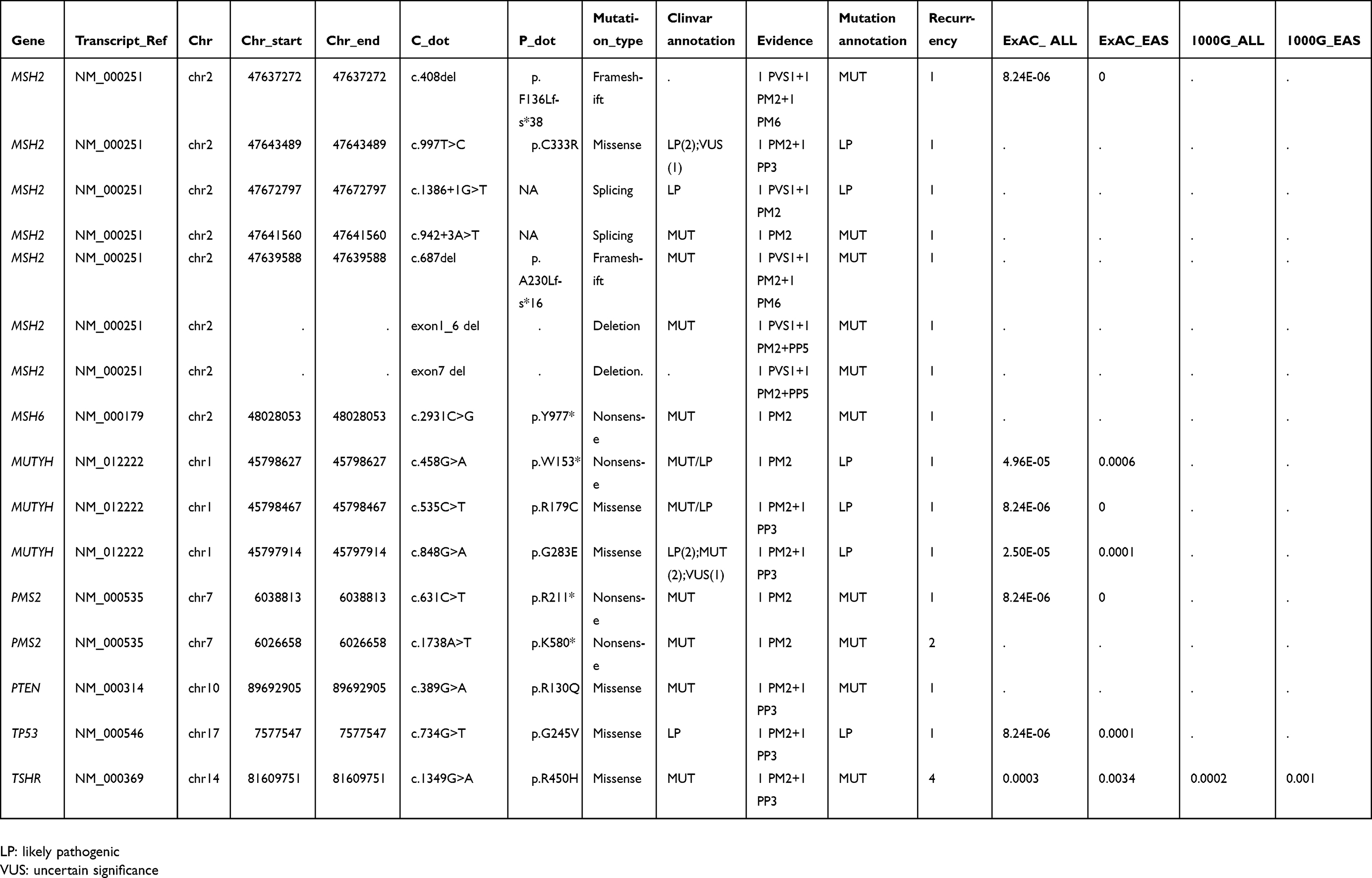 | Table S1 Summary of other detected pathogenic or likely pathogenic germline mutations |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.