Back to Journals » International Journal of Nanomedicine » Volume 21
Multivalent Atezolizumab-Liposome Conjugates as Active Immunotherapeutic Platforms for Enhanced PD-L1 Blockade in Melanoma
Authors Cheng Z ![]() , Amin M, van Brakel M, Sacchetti A
, Amin M, van Brakel M, Sacchetti A ![]() , Kim RQ
, Kim RQ ![]() , Seynhaeve ALB, Debets R
, Seynhaeve ALB, Debets R ![]() , ten Hagen TLM
, ten Hagen TLM ![]()
Received 16 August 2025
Accepted for publication 8 December 2025
Published 12 January 2026 Volume 2026:21 561111
DOI https://doi.org/10.2147/IJN.S561111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Dong Wang
Ziyun Cheng,1,2 Mohamadreza Amin,1 Mandy van Brakel,2 Andrea Sacchetti,3 Robbert Q Kim,4 Ann LB Seynhaeve,1 Reno Debets,2 Timo LM ten Hagen1
1Precision Medicine in Oncology (PrMiO) and Nanomedicine Innovation Center Erasmus (NICE), Department of Pathology, Erasmus MC Cancer Institute, Erasmus MC, Rotterdam, the Netherlands; 2Laboratory of Tumor Immunology, Department of Medical Oncology, Erasmus MC Cancer Institute, Erasmus MC, Rotterdam, the Netherlands; 3Department of Pathology, Erasmus MC, Rotterdam, the Netherlands; 4Department of Cell and Chemical Biology, Leiden University Medical Center, Leiden, the Netherlands
Correspondence: Timo LM ten Hagen, Precision Medicine in Oncology (PrMiO) and Nanomedicine Innovation Center Erasmus (NICE), Department of Pathology, Erasmus MC Cancer Institute, Erasmus MC, Molewaterplein 50, 3015, GE, Rotterdam, the Netherlands, Tel +31 0 614375863, Email [email protected]
Purpose: Anti-programmed death ligand-1 monoclonal antibody (anti-PD-L1 mAb) therapy has demonstrated notable clinical success. However, efficacy is often limited by transient interactions at the tumor-immune interface, limiting sustained immune activation. To address this, we hypothesized that liposomal delivery of anti-PD-L1 mAbs could enhance therapeutic efficacy. Nanosystems may increase binding avidity and prolong cell-surface retention, and enable multivalent antibody presentation through conjugation of multiple antibodies onto a single liposome.
Methods: In this study, we engineered multivalent anti-PD-L1 liposomes (anti-PD-L1 LPs) by conjugating FDA-approved anti-PD-L1 mAbs (atezolizumab) at varying densities onto a clinically relevant liposomal formulation. The biophysical properties, cellular interactions, and therapeutic potential of anti-PD-L1 LPs were evaluated using two melanoma cell line models (BLM and MZ2Mel43), employing multiple analytical techniques.
Results: Our results demonstrated that high-density anti-PD-L1 LPs exhibited superior binding avidity, prolonged membrane retention, and reduced non-specific cellular interaction with PD-L1-expressing cells compared to both low-density and non-targeted LPs. Additionally, in 3D tumor spheroid models, high-density anti-PD-L1 LPs showed deeper penetration, suggesting improved tissue accessibility compared with low-density formulations. Moreover, compared to free antibodies, anti-PD-L1 LPs displayed a higher association rate (k_on) and a significantly lower dissociation rate (k_off), resulting in an overall improved (lower) dissociation constant (K_D). Functional assays confirmed that anti-PD-L1 LPs achieved superior PD-L1 blockade compared to free antibodies. Importantly, in co-cultures of human peripheral blood mononuclear cells and tumor cells, anti-PD-L1 LPs maintained immunomodulatory activity comparable to free anti-PD-L1 antibodies.
Conclusion: This study highlights the critical role of ligand density in enhancing binding strength, tumor retention, and tissue penetration of the anti-PD-L1 LP system. Our nanosystem offers a promising improvement over conventional anti-PD-L1 mAbs, supporting the broader application of this modular liposomal platform to other therapeutic antibodies in melanoma and other solid tumors.
Plain Language Summary: Current limitations and our approach
Cancer immunotherapy drugs like anti-PD-L1 antibodies help the immune system fight cancer, but they often do not stay in tumors long enough to work effectively. To improve this, we developed nanoscale lipid-based carriers, like tiny fat bubbles (called liposomes) densely coated with anti-PD-L1 antibodies. These engineered liposomes bind to tumors more effectively, stay longer, and deliver treatment more precisely than regular antibodies.
Key discoveries
We discovered that packing more drugs onto each tiny carrier makes them stick to tumors better and last longer. Also, how we deliver these drugs matters too—the carrier-delivered versions bind to tumors more effectively than regular antibody drugs. Importantly, in controlled lab tests with cells, these engineered antibodies work just as powerfully as regular immunotherapy—proving we have created a delivery system without sacrificing cancer-fighting strength.
Conclusion and potential benefits
This research introduces an innovative approach to cancer immunotherapy delivery using antibody-coated liposomes. By enhancing tumor targeting and retention while minimizing accumulation in healthy tissues, this method could improve treatment effectiveness while reducing side effects. The platform offers three key advantages: patients may require fewer treatment sessions with more durable results, clinicians gain enhanced options for treating melanoma and solid tumors, and researchers can extend this adaptable platform to other cancer therapies.
Future directions
Our immediate focus will be on validating these findings in animal models and expanding the technology to other immunotherapies. The use of clinically approved components positions this approach for potential rapid translation to human trials, pending successful preclinical results.
Keywords: cancer immunotherapy, melanoma, immune checkpoint inhibitors, liposomes, anti-PD-L1 mAb, targeted drug delivery
Introduction
Immune checkpoint inhibitors (ICIs) have emerged as a powerful class of cancer immunotherapeutics that reverse immune suppression and restore T cell-mediated tumor elimination. By blocking interactions between checkpoint proteins on T cells and their ligands on tumor or antigen-presenting cells, ICIs inhibit suppressive signaling pathways that dampen anti-tumor responses.1,2 A major subclass of ICIs is monoclonal antibodies (mAbs), which have been widely studied in clinical oncology.3 The most clinically validated targets include cytotoxic T lymphocyte-associated antigen-4 and programmed death-1 (PD-1) on T cells, as well as programmed death ligand-1 (PD-L1) on tumor cells. These agents have demonstrated significant success across multiple cancer types.4,5
Despite their clinical success, ICIs face several limitations, including variable patient responses and immune-related toxicities.6,7 Beyond the search for new checkpoint targets and combination therapies, optimizing the clinical application of already-approved ICIs deserves greater attention, given their well-characterized biological activity and safety profiles. Strategies to enhance efficacy should address three critical challenges: precise delivery to the tumor region, effective local immune activation, and minimization of systemic toxicity. Importantly, clinical observations suggest that a robust early immune response and the establishment of durable immune memory may play a greater role in clinical outcomes than prolonged antibody (Ab) exposure. For instance, in the CheckMate-067 trial, patients who discontinued nivolumab plus ipilimumab during the induction phase due to treatment-related adverse events exhibited comparable five-year survival to those who completed the full course of therapy.8
These insights support a shift toward ICI strategies that achieve potent, localized checkpoint blockade with reduced systemic toxicity. Nanoparticle platforms, especially liposomes (LPs), have emerged as promising tools for this purpose.9–11 By exploiting the enhanced permeability and retention effect, LPs can passively accumulate in tumor tissues through the leaky vasculature and impaired lymphatic drainage of the tumor microenvironment.12 Once localized at the tumor site, the therapeutic efficacy of ICIs depends on their interaction with surface-expressed immune checkpoints, a process influenced by epitope accessibility, receptor internalization, and recycling dynamics.13 LP-ICI platforms enable multivalent presentation of Abs on LP surface, effectively mimicking synthetic multivalent mAbs that enhance checkpoint recognition.14,15 Additionally, the LP format can alter cellular uptake pathways, promoting lysosomal localization and reduced recycling, which may prolong the efficacy of checkpoint blockade.16 The relatively large size of LPs (typically 50–1000 nm compared to 10–15 nm for mAb) also contributes to faster clearance from systemic circulation and reduced accumulation in healthy tissues, thereby further limiting on-target, off-tumor immune activation.17–19
While many studies focus on enhancing LP uptake through tissue-specific Ab targeting,20 few have investigated the fate and functional role of the Ab itself, especially in the context of immune checkpoint blockade. Unlike conventional targeting Ab-conjugated LPs, which are designed to promote cellular internalization of their cargo, these therapeutic ICIs function at the tumor-immune interface, where prolonged surface retention and strong avidity are critical. Current research has primarily focused on LP-mediated drug internalization rather than on the binding dynamics and functional behavior of checkpoint inhibitors themselves. These leave a gap in knowledge regarding how ligand density affects LP-ICI binding stability, retention, and functional blockade, and what the potential advantages of LP-ICI against their parental mAbs. Addressing these factors is essential for the rational design of next-generation nanoparticle-ICIs with enhanced performance.
Here, we address this gap by developing PD-L1-targeted LPs through conjugation of atezolizumab, an FDA-approved anti-PD-L1 mAb,4 onto a clinically relevant LP formulation. Using a post-insertion method with DSPE-PEG (2000)-Maleimide linkers, we achieve precise control over Ab surface density. We then systematically investigate how antibody density affects liposome binding avidity, cellular retention, internalization kinetics, and 3D tumor penetration. Importantly, we provide the first comprehensive comparison of binding parameters, functional PD-L1 blockade efficacy, and immune suppression reversal between anti-PD-L1 liposomal conjugates and the parental atezolizumab mAb. Our work offers crucial mechanistic insights into ligand density optimization for liposomal immune checkpoint inhibitor design, while demonstrating the potential to enhance clinical mAb therapy through this engineered liposomal platform.
Methods
Materials and Reagents
Atezolizumab (anti-PD-L1). SPDP (3-(2-Pyridyldithio) propionic acid N-hydroxysuccinimide ester) (Sigma). TCEP (Tris(2-carboxyethyl) phosphine) (Sigma). L-cysteine (Sigma). DSPE-PEG(2000)-Maleimide (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N [maleimide(polyethylene glycol)-2000]), DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), DSPE-PEG(2000) (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000]), Cholesterol, TF-PE (fluorescently labeled lipid, 18:1 TopFluorTM PE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[(dipyrrometheneboron difluoride)butanoyl]) were purchased from Avanti Polar Lipids. Track-etched polycarbonate membranes (Whatman; pore sizes: 200 nm, 100nm, and 50 nm). Amicon filtration system (Millipore). Hepes (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (Sigma). NuPAGE MES SDS running buffer (ThermoFisher). 4 to 12%, Tris-Glycine, 1.0 mm, Mini Protein Gels (novex, ThermoFisher). Tris-glycine SDS sample buffer (novex, ThermoFisher). Coomassie Protein Stain (InstantBlue, Abcam). Recombinant Human B7-H1 (PD-L1, CD274)-Fc Chimera (BioLegend). Goat anti-human IgG (Fab)2 (HRP) (Abcam). TMB (Tetramethylbenzidine) substrate solution (ThermoFisher). Stop Solution for TMB Substrates (ThermoFisher). ELISA Plates, Uncoated (Nunc MaxiSorp, BioLegend). PE anti-human CD274 (B7-H1, PD-L1) Antibody (BioLegend). DMEM (Sigma). FBS (fetal bovine serum) (Sigma). 0.05% and 0.25% Trypsin-EDTA (Gibco). Human serum (Sanquin). Recombinant human IL-2 (Proleukin; Chiron). Sensor Chip CM5 (Cytiva). EDC (1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) (Sigma). NHS (N-Hydroxysuccinimide) (Sigma). Lysotracker DND-99 (ThermoFisher). Hoechst (Invitrogen). Cell culture dishes for confocal (Greiner Bio-One, 627870). Agarose (UltraPure, Thermo Fisher). CellTrackerTM Deep Red Dye (Invitrogen). CD3 Monoclonal Antibody (OKT3) (Life Technologies). Human recombinant IFN-γ (PreproTech).
Production of Antibody-Conjugated Liposomes
Production of Free Sulfhydryl (-SH) Groups on a Parental Antibody
Atezolizumab (anti-PD-L1) was obtained from a hospital pharmacy, and stored at 4 °C. Before conjugation, the anti-PD-L1 mAb was incubated at room temperature with a solution of SPDP in DMSO (20mM stock), at a molar ratio of 12:1 (SPDP: mAb) for 1 h in constant shaking (300 rpm) in HEPES saline buffer (10 mM HEPES, 150 mM NaCl, pH 7.4). The reaction mixture was then washed with HEPES saline buffer using the Amicon filtration system (MWCO 10 kDa). The collected mAb was subsequently incubated with TCEP at room temperature for 1 h at a molar ratio of 1:625 (mAb: TCEP). Afterwards, the mixture was washed with HEPES saline EDTA buffer (10 mM HEPES, 150 mM NaCl, 5 mM EDTA, pH 6.7) using the Amicon filtration system (MWCO 10 kDa). The concentration of the collected mAb was measured using a Nanodrop spectrophotometer and used immediately for the subsequent conjugation step.21
Maleimide Reaction Between the Lipid and the Antibody
DSPE-PEG (2000)-Maleimide was stored in chloroform at −20 °C. Before conjugation, the solvent was evaporated, and the DSPE-PEG (2000)-Maleimide was hydrated with HEPES saline EDTA buffer (10 mM HEPES, 150 mM NaCl, 5 mM EDTA, pH 6.7) at a concentration of 0.5 mg/mL. The DSPE-PEG (2000)-Maleimide and Ab were then mixed at molar ratios of 76:1, 19:1, 9.5:1, and 4.7:1, and incubated for at least 16 h at 4 °C with constant shaking (300 rpm). After 16 h, 1 mM L-cysteine was added to quench any remaining free sulfhydryl groups for 10 min at room temperature.
Liposome Production
LPs were composed of the DSPC, DSPE-PEG (2000), cholesterol, and TF-PE in a molar ratio of 55:4:40:0.1 and were prepared using the thin lipid film hydration method, followed by heated extrusion, as described previously.22 Briefly, the lipids were dissolved in a methanol/chloroform mixture (1:9 v/v) and thereafter evaporated under vacuum to form a thin lipid film for 4h at 42 °C. The lipid film was hydrated in HEPES saline buffer (10 mM HEPES, 150 mM NaCl, pH 7.4) for 20 min at 65°C. Small unilamellar LPs were formed by extrusion under pressure through track-etched polycarbonate membrane filters with pore sizes of 200, 100, and 50 nm, performing five extrusion cycles for each pore size at 65°C. The LP quality was assessed by measuring the size, polydispersity, and zeta potential using dynamic light scattering immediately after synthesis. The phosphorus concentration was determined based on the Bartlett assay.
Post-Insertion of Lipid-Antibody Conjugates and Purification
Following maleimide reaction, lipid-Ab conjugates were directly mixed with preformed LPs at a molar ratio of 1:99 (DSPE-PEG (2000)-Maleimide: LPs). The mixture was incubated at 55 °C for 30 min to facilitate the insertion of lipid-Ab conjugates into the LP membrane. Unincorporated Ab conjugates were removed by ultracentrifugation at 94,000 g for 3 h at 4 °C using a Beckman Optima XE-90 Ultracentrifuge equipped with a 70.1Ti rotor. The pellet was resuspended in HEPES saline buffer (10 mM HEPES, 150 mM NaCl, pH 7.4) overnight with constant shaking at 4 °C.
Sodium Dodecyl-Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Ab-conjugated LPs were mixed with Tris-Glycine SDS sample buffer and heated at 85 °C for 10 min. The samples were then loaded onto 4–12% Tris-Glycine Mini Protein Gels (1.0 mm) and electrophoresed in MES running buffer for 30 min. Following electrophoresis, the gels were stained with Instant Coomassie Blue stain to visualize the proteins for imaging.
Quantitation of Anti-PD-L1 Coupling to Liposomes
The bioactivity of anti-PD-L1 conjugated to LPs was quantified by a functional ELISA assay as previously described.23 Recombinant Human B7-H1 (PD-L1, CD274)-Fc Chimera was pre-coated onto ELISA Plates overnight at a concentration of 1 μg/mL. The following day, the plates were blocked with 2% bovine serum albumin (BSA) in PBS containing 0.05% (v/v) Tween-20 for 1 h to prevent non-specific binding. LP formulations were prepared by mixing anti-PD-L1 LPs with PBS containing 0.5% (v/v) Tween-20 to solubilize the lipids. The solubilized LP samples were then added to the ELISA plate and incubated for 1 h with constant shaking. A detection Ab, HRP-conjugated goat anti-human IgG (Fab)2 (diluted 1:10,000), was added, followed by the HRP-sensitive colorimetric substrate TMB. After the reaction, a stop solution was applied, and absorbance was measured at 450 nm using a Victor NivoTM plate reader. Parental Abs (atezolizumab) were used to generate a standard curve, which was then employed to calculate the Ab concentration in each LP formulation.
Cell Culture (Standard Condition)
Two patient-derived human melanoma cell lines, MZ2Mel43 and BLM, were used in this study. Tumor cells were cultured in DMEM supplemented with 10% FBS, 1% nonessential amino acids, 1% L-glutamine, and 1% penicillin–streptomycin. Cultures were maintained at 37 °C in a humidified incubator with 5% CO2. Cells were passaged by 0.05% Trypsin-EDTA when they reached 80%–90% confluency. Cells were regularly tested for Mycoplasma by PCR. PBMCs were obtained from healthy human donors (Sanquin) using density gradient centrifugation with Ficoll-Isopaque. Cells were cultured in RPMI 1640 medium supplemented with 25 mmol/L HEPES, 6% human serum, 2 mmol/L L-glutamine, 1% penicillin-streptomycin, and 360 IU/mL recombinant human IL-2. T cells were stimulated biweekly using irradiated allogeneic feeder cells, following established protocols.24
Flow Cytometry for Liposome Binding
BLM and MZ2Mel43 cells were seeded under standard conditions. For the IFN-γ treatment groups, cells were stimulated with 50 ng/mL IFN-γ for 48 h before the experiments. Before use, cells were harvested, and 5×105 cells were centrifuged at 450 × g for 5 min. The cell pellet was resuspended in 100 µL of LPs at a concentration of 10 µM and incubated for 1 h at 4 °C. After incubation, the cells were washed and resuspended for data acquisition using a FACSymphonyTM flow cytometer (BB515 channel), with the fluorescent lipid signal from TF-PE being detected.25 Flow cytometry analysis was conducted using FlowJo software. The correlation between protein/lipid ratios and binding capacity was assessed using the Pearson Correlation Coefficient (r) in GraphPad Prism.
For PD-L1 expression analysis, 5×105 cells were centrifuged at 450 × g for 5 min. The cell pellet was resuspended in PE anti-human CD274 (B7-H1, PD-L1) Ab and incubated for 30 min at 4 °C. Data were acquired using a flow cytometer (PE channel).
Binding Avidity by ELISA
Recombinant Human B7-H1 (PD-L1, CD274)-Fc Chimera was pre-coated onto ELISA plates overnight at a concentration of 1 µg/mL. The following day, the plates were blocked with 2% BSA in PBS containing 0.05% (v/v) Tween-20 for 1 h to minimize non-specific binding. LP formulations at 100 µM were prepared in PBS containing 0.05% (v/v) Tween-20. The LP samples were added to the ELISA plates and incubated for 1 h with constant shaking. After incubation, the plates were washed with PBS containing 0.05% (v/v) Tween-20 to remove unbound LPs. Finally, 100 µL of PBS was added to each well for measurement. The fluorescence of LPs containing TF-PE was measured using a plate reader at an excitation/emission wavelength of 480/530 nm.
Cytotoxicity Assay
BLM and MZ2Mel43 cells were cultured under standard conditions. Cells were seeded in 96-well plates at a density of 5×103 cells per well and incubated for 24 h to allow adherence and entry into the exponential growth phase. After 24 h, the medium was replaced with fresh medium containing LPs at concentrations of 1 µM, 10 µM, or 100 µM. Medium without any drugs was used as a negative control. The plates were placed in the Incucyte system for continuous monitoring over 72 h. Cell growth was measured every 12 h using a 10× objective lens. Total cell confluence in bright-field images was analyzed to calculate the growth rate.
Surface Plasmon Resonance Analysis
Surface plasmon resonance (SPR) experiments were performed using the Biacore T200 Control System, and data were analyzed with Biacore T200 Evaluation Software. CM5 sensor chips were employed for all experiments. The running buffer consisted of PBS containing 0.05% (v/v) Tween-20, while a 20 mM sodium acetate buffer at pH 4.0 was used for antigen immobilization.
Antigen immobilization was performed using a CM5 chip, coated with carboxymethylated dextran. The chip was first activated using a mixture of EDC (75 mg/mL) and NHS (11.5 mg/mL) at a flow rate of 10 µL/min for 7 min. Subsequently, the antigen solution (Recombinant Human B7-H1 (PD-L1, CD274)-Fc Chimera at 0.05 mg/mL in sodium acetate buffer) was injected at the same flow rate and duration. The Biacore T200 system divides the chip into two flow channels; both were activated with EDC/NHS, but one was closed during antigen injection to serve as the reference channel. Finally, both channels were blocked with 1M ethanolamine at pH 8.5 (10 µL/min for 7 min) to quench any remaining active esters.
Following chip priming and normalization, samples containing either LPs or free Ab were injected into both reference and detection channels at a flow rate of 30 or 50 µL/min in running buffer. Samples were prepared at various concentrations and analyzed in three different batches. The Biacore T200 recorded sensorgrams representing association (during injection) and dissociation (post-injection) phases for both flow channels. Chip surface regeneration was performed after each injection using 10 mM glycine buffer at pH 1.4. Real-time changes in refractive index were automatically monitored and recorded in response units using Biacore Control Software. Final analysis, including calculation of association and dissociation rate constants and equilibrium binding affinity, was conducted by fitting reference-subtracted curves (Fc2–Fc1) to appropriate binding models using Biacore T200 Evaluation Software (Kinetic Analysis: Langmuir 1:1 binding model; Steady-State Affinity Analysis: Steady-state affinity model). GraphPad Prism was used for additional data visualization and figure preparation.
Flow Cytometry Analysis of Liposome Uptake Under Physiological Conditions
BLM cells were seeded in 12-well plates at a density of 1×105 cells per well and incubated overnight for adherence. After 24 h, the medium was replaced with fresh medium containing LPs at concentrations of 1 µM or 10 µM, followed by incubation for an additional 2 or 24 h at 37 °C in a humidified incubator with 5% CO2. After incubation, the cells were washed and harvested for flow cytometry to detect the fluorescent lipid signal from TF-PE (BB515 channel). Flow cytometry analysis was performed using FlowJo software.
Confocal Microscopy Live Cell Imaging
BLM cells were seeded in cell culture dishes at a density of 2.5×104 cells per chamber and treated with 10 nmol/mL of Ctrl-LP, anti-PD-L1 low-LP, or anti-PD-L1 high-LP for 24 h. After treatment, cells were gently washed three times with PBS to remove unbound LPs. Lysotracker DND-99 (1:100,000) was added for 5 min, followed by three PBS washes. Nuclei were stained with Hoechst (1:10,000) for 10 min before imaging. Images were acquired using a Leica SP8 confocal microscope with a 63× oil immersion objective lens. Data were analyzed using ImageJ software. Co-localization between LPs and lysosomes was quantified using Manders’ coefficient. The co-localized LP signal was calculated as: Total LP signal × Manders’ coefficient.
Flow Cytometry for Liposome Retention and PD-L1 Blocking
MZ2Mel43 cells were cultured under standard conditions. Before use, the cells were harvested, and 5×105 cells were centrifuged at 450 × g for 5 min. The cell pellet was resuspended in 100 µL of LPs at 100 µM and incubated for 30 min at 4 °C. Following incubation, unbound LPs were removed by washing, and the cells were resuspended in DMEM and incubated at 37 °C for an additional 1 h. Parallel groups were kept at 4 °C for the same period. Afterwards, the cells were centrifuged, and the cell pellet was stained with PE anti-human CD274 (B7-H1, PD-L1) antibody and 7AAD for 30 min at 4 °C. The cells were then rewashed and resuspended in PBS for further analysis. Data acquisition was performed using a flow cytometer. Fluorescent signals were recorded for the lipid probe (TF-PE) to detect LP absorption and for PE to evaluate PD-L1 expression. The retention ratio of LPs was calculated as follows: Retention ratio = LP signal after 37 C incubation/LP signal after 4 C incubation.
Liposome in vitro 3D Penetration Assay
To prepare the experiment, 65 µL of 1% agarose was added to a 96-well plate and allowed to solidify for 10 min. Then, 2×104 BLM cells were seeded into each well. Spheroid formation was observed after three days of incubation at 37 °C. Different treatments (Ctrl-LP, anti-PD-L1 low-LP, or anti-PD-L1 high-LP, with lipid concentrations of 10 µM) were then applied and incubated with the spheroids for 24 h to prepare for flow cytometry analysis. CellTracker Deep Red Dye was added to each well 4 h before flow cytometry to define the different layers of LPs. Finally, the spheroids were collected from each well and dissociated into a single-cell suspension using 0.25% Trypsin-EDTA and gentle pipetting. The samples were then directly analyzed by flow cytometry.
PD-L1 Ligand Blocking Comparison of Free Atezolizumab to Atezolizumab-LP
MZ2Mel43 cells were cultured under standard conditions. Before use, the cells were harvested, and 5×105 cells were centrifuged at 450 × g for 5 min. The cell pellet was resuspended in 100 µL of either atezolizumab, atezolizumab low-LP, or atezolizumab with equivalent concentrations of anti-PD-L1 Ab (concentrations: 0.000781 µg/mL, 0.003125 µg/mL, 0.0125 µg/mL, 0.05 µg/mL, 0.2 µg/mL) and incubated for 30 min at 4 °C. After incubation, unbound Ab or LPs were removed by PBS washing. And the cells were centrifuged at 450 × g for 5 min, and the cell pellet was stained with PE anti-human CD274 (B7-H1, PD-L1) antibody and 7AAD for 30 min at 4 °C. The cells were then rewashed and resuspended in PBS for further analysis. Data acquisition was performed using a flow cytometer. Fluorescent signals were recorded for the lipid probe (TF-PE) to detect LP absorption and for PE to evaluate PD-L1 expression.
Immune Suppression Reversal in a Human Peripheral Blood Mononuclear Cell–Tumor Cell Co-Culture System
For activation and PD-1 ligand induction, T cells were preincubated overnight with 0.08 µg/mL aCD3 mAb (OKT3). Target tumor cells (MZ2Mel43) were seeded at 2×104 cells per well in 96-well plates. After 3–4 h, tumor cells adhered firmly to the bottom of the wells. Then, anti-PD-L1, either in free form or encapsulated in LPs, was added to the tumor cells at a final concentration of 0.005 µg/mL (corresponding to the EC50 as determined in a PD-L1 ligand blocking assay). After 1 h of Ab incubation, T cells were added to the wells at an effector-to-target ratio of 1:1 or 3:1 (2 or 6×104 T cells per well) in a final volume of 200 µL. The co-culture was maintained in RPMI 1640 supplemented with 25 mmol/L HEPES, 10% FBS, 2 mmol/L L-glutamine, and 1% antibiotics for 72 h. Real-time monitoring of tumor cell killing was performed using the Incucyte live-cell imaging system. Tumor cells were stably labeled with a PiggyBac-mScarlet plasmid, and mScarlet fluorescence was used to quantify their survival.
Statistics
All results were analyzed using GraphPad Prism and are presented as mean ± SD. Statistical analyses employed Student’s t-test for two-group comparisons and ANOVA for multiple-group comparisons. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS = not significant).
Results
Preparation of Atezolizumab-Conjugated Liposomes with Varying Antibody Concentrations
Ab-conjugated LPs were prepared following a previously published method,21 as summarized in the schematic diagram shown in Figure 1A. Anti-PD-L1 mAbs were conjugated to DSPE-PEG (2000)-Maleimide at four different Maleimide/Ab molar ratios (Supplementary Figure 1A). The conjugation efficiency exhibits a dose-dependent trend, ranging from 40% to 100% (Supplementary Figure 1A–D). Based on these results, Maleimide/Ab molar ratios of 76 and 19 were selected for insertion into PEGylated LPs to produce anti-PD-L1 LPs with low and high Ab concentrations, respectively (Figure 1B; hereafter referred to as anti-PD-L1 low- and high-LPs). Following purification, the final lipid and Ab concentrations were quantified using a phospholipid assay and a functional ELISA (Figure 1C and Supplementary Table 1). By using free atezolizumab as a standard curve, the concentration of atezolizumab on LPs was calculated (Supplementary Figure 2A and B). Across three production batches, the post-insertion method yielded consistent Ab/lipid ratios for both low- and high-Ab-concentration formulations (1.8-fold anti-PD-L1 high-LP vs anti-PD-L1 low-LP; Figure 1C).
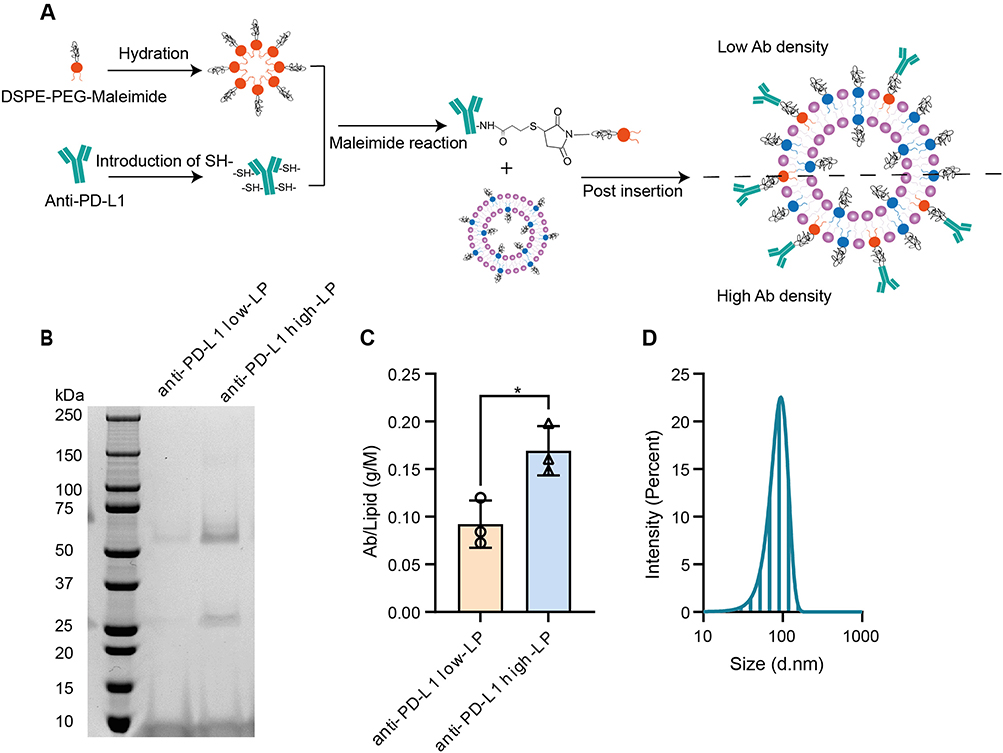 |
Figure 1 Production and basic biophysical properties of atezolizumab-conjugated liposomes. Abbreviations: SPDP, 3-(2-Pyridyldithio) propionic acid N-hydroxysuccinimide ester; TCEP, Tris(2-carboxyethyl) phosphine. Notes: (A) Schematic diagram for atezolizumab (anti-PD-L1)-conjugated liposomes (LPs). Step 1: Production of free sulfhydryl (-SH) groups on a parental antibody (Ab). Step 2: Ab-lipid conjugates formulation. Step 3: LP production. Step 4: Post-insertion of lipid-Ab conjugates to LPs. (B) anti-PD-L1 LPs with low and high Ab concentration were shown on non-reducing SDS-PAGE; Upper (around 50 kDa) and lower (around 25 kDa) bands represent the heavy and light chains, respectively. (C) The Ab-to-lipid ratio for each batch was calculated. Ab concentrations were determined using a functional ELISA (data shown in Supplementary Figure 2), and lipid concentrations were quantified through phosphorus concentrations using the Bartlett assay. (D) Representative size distribution of anti-PD-L1 LP was measured using dynamic light scattering. p-value < 0.05 was considered statistically significant (*p < 0.05). |
Next, we used dynamic light scattering to analyze the physical characteristics of our products (Figure 1D and Supplementary Figure 3A–C). After protein conjugation, we observed a slight increase in particle size (from 83 ± 2 nm to 86 ± 2 nm; Supplementary Figure 3A), which is optimal for exploiting the enhanced permeability and retention effect, given that the endothelial cell gaps in tumor tissues range from 100 to 800 nm.26 Additionally, a polydispersity index (PDI) below 0.1 indicates a highly homogeneous particle population (Supplementary Figure 3B). The slight negative charge of the LPs suggests favorable safety characteristics (Supplementary Figure 3C). These physical properties are critical for LPs, and our formulation demonstrates consistent quality across different batches over at least six months.
In summary, atezolizumab-LP formulations with varying Ab expression can be reliably produced by post-insertion of atezolizumab into stealth LPs.
Liposomes with High Antibody Concentration Exhibit Enhanced Binding Avidity
One of the primary advantages of multivalent Ab-LPs is their potential enhanced apparent binding avidity. This property may lead to a more rapid onset of blocking activity. In our study, two melanoma cell lines, MZ2Mel43 and BLM, were used to assess anti-PD-L1 LPs binding by flow cytometry. Significant differences in binding were observed between control (Ctrl)-LP, anti-PD-L1 low-LP, and anti-PD-L1 high-LP (Figure 2A–D), with the absolute binding capacity strongly correlating with the protein/lipid ratio of each product (Supplementary Figure 4A–D). The data also indicates that in vitro cell binding ability is closely related to the baseline PD-L1 expression on the cell lines. Ab-LP exhibits significantly higher binding to the BLM cell line, which has higher PD-L1 expression, compared to the MZ2Mel43 cell line (Supplementary Table 2). When PD-L1 expression levels on cells were increased via IFN-γ stimulation in the MZ2Mel43 cell line (Supplementary Table 2), the binding capacity of Ab-LP correspondingly increased (Figure 2B). No additional toxicities were observed in any of the formulations (Supplementary Figure 4E–J).
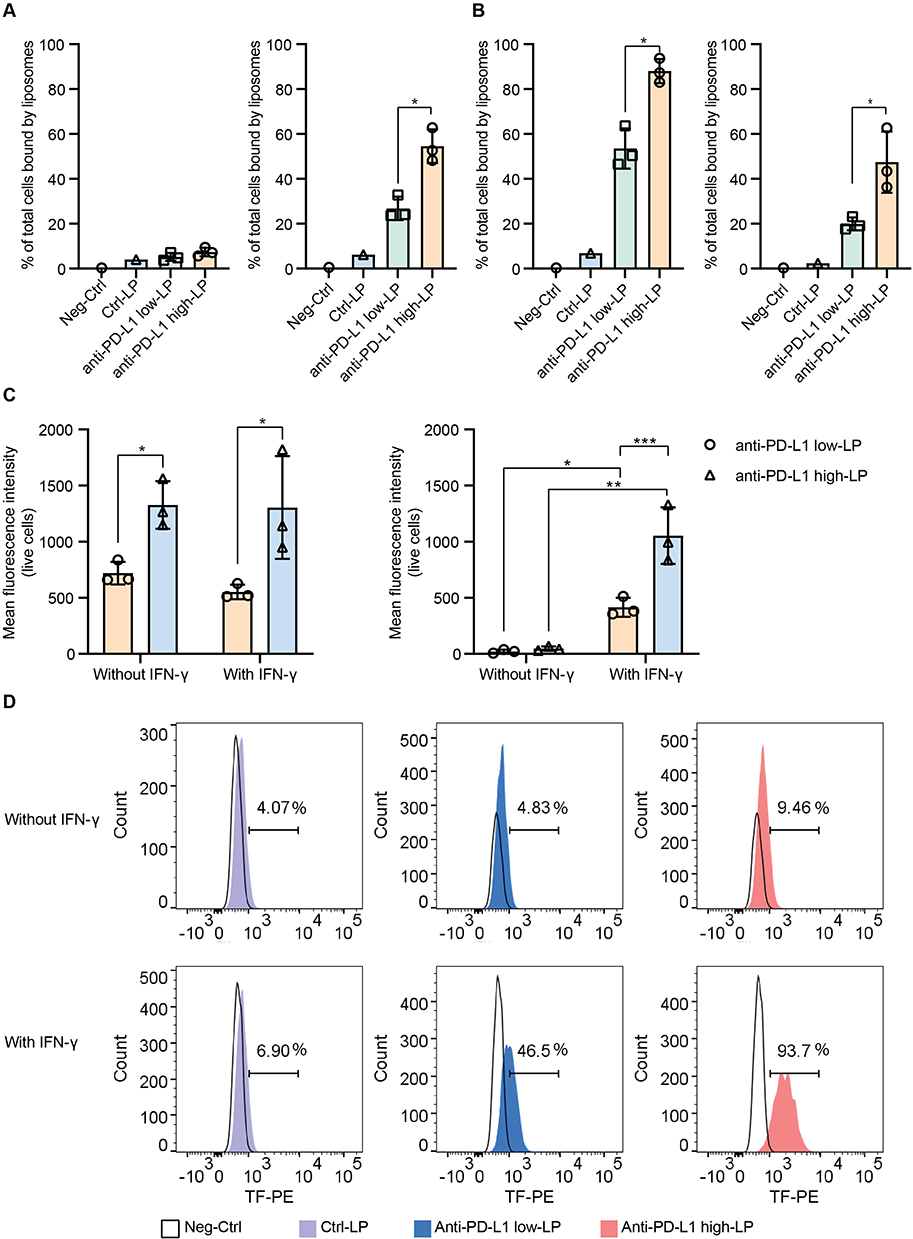 |
Figure 2 In vitro binding avidity of anti-PD-L1 liposomes with varying concentrations. Notes: Negative control (PBS) or 10 µM of Ctrl-liposomes (LP), anti-PD-L1 low-LP, and anti-PD-L1 high-LP were incubated with BLM or MZ2Mel43 cells at 4 °C for 1 h. LP binding was assessed by detecting the fluorescence signal from the fluorescent lipid TF-PE in the formulations, using flow cytometry. (A) Percentage of liposome-positive cells; tumor cells were incubated without IFN-γ stimulation (Left: MZ2Mel43, Right: BLM). (B) Percentage of liposome-positive cells; tumor cells were stimulated with 50 ng/mL IFN-γ for 48 h before the experiments (Left: MZ2Mel43, Right: BLM). (C) Mean Fluorescence Intensity of liposome signal in all live cells, both with or without IFN-γ stimulation (Left: MZ2Mel43, Right: BLM). (D) Representative histogram of liposome binding to MZ2Mel43 cells under different conditions. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). |
To further validate the binding properties of LPs functionalized with different anti-PD-L1 Abs, we next conducted experiments in a non-cellular context. We first used an ELISA-based assay by coating 96-well plates with recombinant human PD-L1-Fc chimera to evaluate the binding avidity of anti-PD-L1 low- and high-LPs (Figure 3A). Anti-PD-L1 high-LPs demonstrated a significantly greater binding signal (1.28-fold increase) compared to anti-PD-L1 low-LPs.
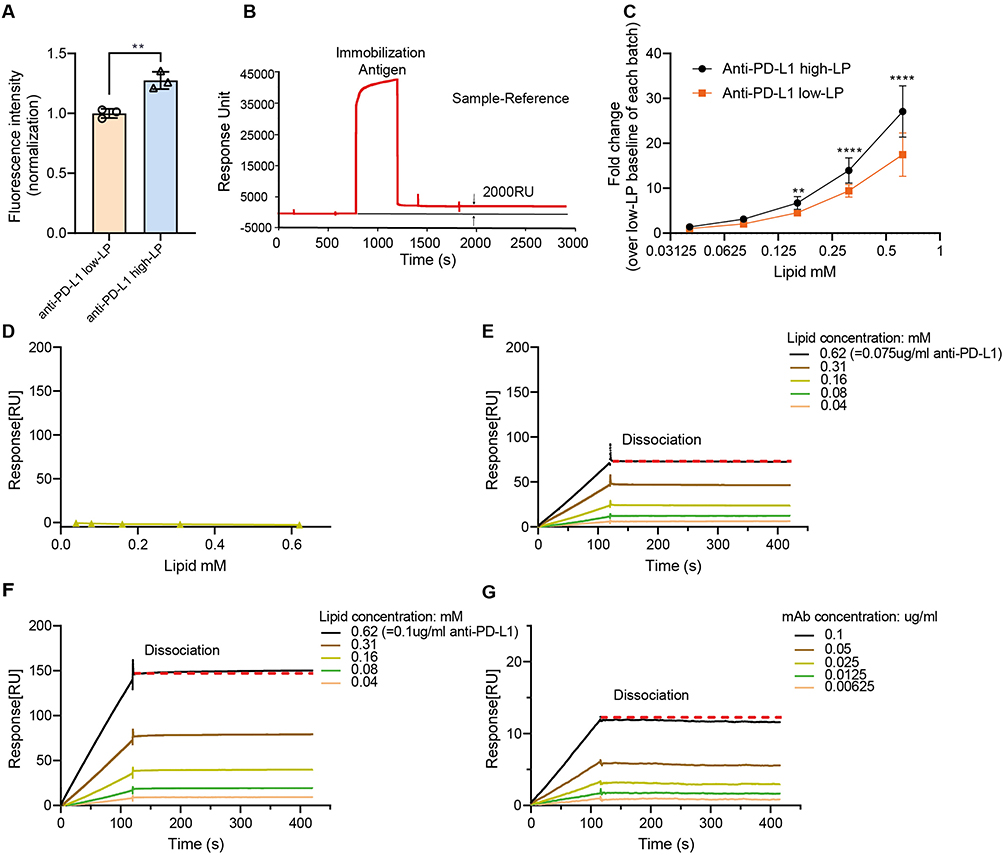 |
Figure 3 In vitro binding avidity of anti-PD-L1 liposomes at the non-cellular level. Notes: (A) Binding avidity was assessed using an ELISA-based assay. Recombinant human PD-L1 (B7-H1/CD274)-Fc chimera protein was immobilized onto 96-well plates. Anti-PD-L1 high liposomes (high-LP) and anti-PD-L1 low-LP (low-LP) containing fluorescent lipid probe TF-PE were added at a concentration of 100 µM. (B–D) Surface plasmon resonance analysis of control (Ctrl)-LP, high-LP, low-LP, and free antibody (Ab) across a range of lipid or Ab concentrations. Samples were injected into both reference and detection channels of a CM5 sensor chip at a flow rate of 30 µL/min using standard running buffer. (B) The CM5 sensor surface was immobilized with recombinant human PD-L1 (B7-H1/CD274)-Fc chimera protein, achieving a final immobilization level of ~2000 response units. (C) Comparison of binding signals from high-LP and low-LP. The y-axis represents the maximum RU obtained from steady-state analysis at each lipid concentration, normalized to the baseline response of low-LP at 0.04 mM within each experimental batch. (D–G) Representative sensorgrams showing response curves of Ctrl-LP (D), low-LP (E), high-LP (F), and free Ab (G) (matched for molar Ab concentration to anti-PD-L1 high-LP) at various lipid or protein concentrations. The red dotted line is horizontal (Indicating no detectable dissociation of the analyte from the ligand). p-value < 0.05 was considered statistically significant (**p < 0.01, ****p < 0.0001). |
Subsequently, we used SPR to analyze binding kinetics. Recombinant PD-L1-Fc was immobilized on a CM5 sensor chip at a surface density of 2000 response units (Figure 3B). Ab-LPs (high and low density) and free Ab (matched for molar Ab concentration to anti-PD-L1 high-LP) were injected across the chip at varying lipid or protein concentrations. Anti-PD-L1 high-LPs consistently showed higher overall avidity compared to anti-PD-L1 low-LPs (Figure 3C). Figure 3D–G displays representative dose-dependent binding curves.
In summary, these data demonstrate that the in vitro binding avidity of anti-PD-L1 LPs can be significantly increased by increasing the PD-L1 numbers on the LP surface. Also, the avidity is influenced by the intrinsic PD-L1 expression levels of the target cells.
High Antibody Concentrations Liposomes Exhibit Enhanced Membrane Binding While Maintaining Similar Lysosomal Interactions
Following the initial binding, the next key factor influencing the function of anti-PD-L1 Ab is cellular dynamics. Specifically, longer presence of the blocking Ab on the cell surface, may potentially lead to a stronger therapeutic effect. We subsequently investigated cellular uptake of different Ab concentration-LPs by flow cytometry. When anti-PD-L1 LPs were added to the BLM cell line (PD-L1 high expression cell line, Supplementary Table 2), the cellular uptake ratio correlated positively with both lipid concentration and anti-PD-L1 Ab concentrations on the liposomal surface (Figure 4A and B). Anti-PD-L1 high-LP is taken up more efficiently by BLM cells compared to anti-PD-L1 low-LP at a lipid concentration of 1 µM (Figure 4A). More pronounced differences in cellular uptake are observed at higher lipid concentrations. At 10 µM of lipid, anti-PD-L1 low- and high-LP showed 39% vs 59% drug uptake after 2 h, and 34% vs 50% after 24 h (Figure 4B). These results demonstrate the importance of a higher ligand number on LPs for these receptor-mediated processes. Interestingly, cellular uptake of LPs was higher at 2 h compared to 24 h—for example, in the case of anti-PD-L1 high-LP at 10 µM, 59% of cells were positive at 2 h versus 50% at 24 h. This decline at the later time point may be attributed to intracellular trafficking and processing of internalized LPs, including endocytosis, degradation, or signal quenching within acidic compartments such as lysosomes. These findings suggest that liposome internalization occurs rapidly (within 24 h), with subsequent intracellular redistribution or clearance contributing to the reduced measurable signal over time.
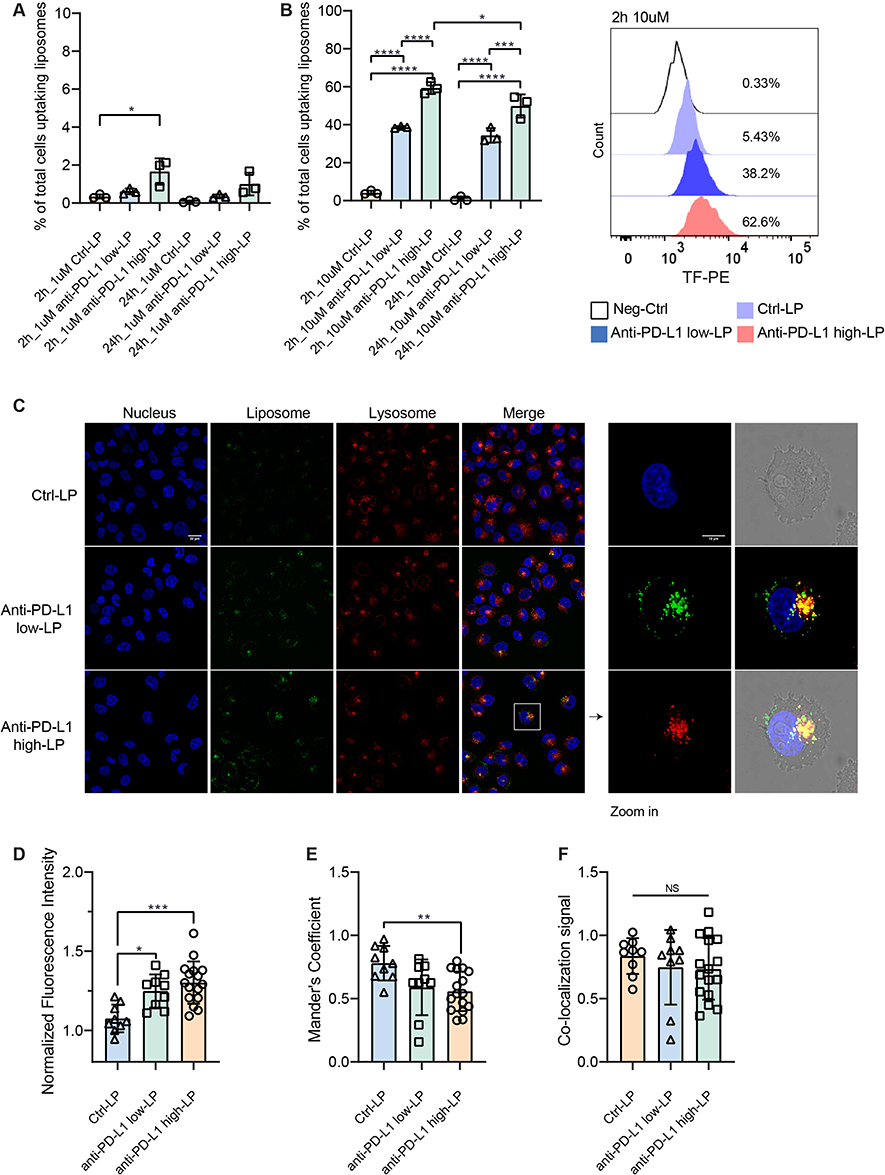 |
Figure 4 Cellular trafficking of anti-PD-L1 liposomes. Notes: (A and B) 1 µM or 10 µM of Control-liposome (Ctrl-LP), anti-PD-L1 low-LP, or anti-PD-L1 high-LP containing fluorescent lipid probe TF-PE were incubated with BLM at 37 °C for 2 h or 24 h and analyzed by flow cytometry. (C–F) BLM cells were incubated with 10 µM of Ctrl-LP, anti-PD-L1 low-LP, or anti-PD-L1 high-LP at 37 °C for 24 h. Signals were then detected using a Leica SP8 confocal microscope. Nucleus (Blue): Signal detected from Hoechst. LP (Green): Signal detected from TF-PE in the formulations. Lysosome (Red): Signal detected from Lysotracker DND-99. (C) All images were captured using a 63× oil immersion objective lens. Zoom in = 3.45 times. (D) Fluorescence signal quantification from confocal images was performed by normalizing the fluorescence intensity values to the no-treatment group (cells stained only with Hoechst and Lysotracker). (E) Co-localization between LPs and lysosomes was quantified using the Manders’ coefficient in ImageJ. (F) LP co-localization signal = Total LP signal × Mander’s coefficient. Confocal images used for quantification were obtained from five independent experiments. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS = not significant). |
The internalization process was further examined using confocal microscopy. Fluorescent LPs, along with lysosome tracker, were added to track their localization. Consistent with the flow cytometry data, anti-PD-L1 LPs demonstrated higher cellular uptake compared to Ctrl-LPs (Figure 4C and D). Images showed clear membrane localization of anti-PD-L1 Abs, with partial co-localization observed in lysosomes (Figure 4C). Co-localization analysis between LPs and lysosomes was performed using ImageJ software. Both Ctrl-LP and Ab-LP exhibited lysosomal co-localization with Mander’s coefficients greater than 0.5 (Figure 4E). Ctrl-LP showed a higher correlation (78%) than anti-PD-L1 LPs (56–59%), reflecting greater non-specific uptake through endocytosis. In contrast, Ab-LPs displayed stronger surface binding and partial internalization, consistent with receptor-mediated interactions. Despite these mechanistic differences, the overall proportion of LPs reaching lysosomes was similar across all groups (Figure 4F). This suggests that antibody density primarily enhances surface avidity without fundamentally altering the intracellular trafficking pathway.
To further investigate cellular uptake, we examined liposome retention by indirectly measuring the amount of PD-L1 ligand remaining on the cell surface after initial binding (Figure 5A). In parallel, cellular uptake of the LPs was assessed by measuring the fluorescence signal of the lipid probe TF-PE incorporated into the formulations. Consistent with the data shown above, anti-PD-L1 high-LP exhibited significantly higher binding capacity (9.3%) compared to the Ctrl- (1.8%) and anti-PD-L1 low-LP (5.3%) at 4 °C (Figure 5B) at 4 °C. For all formulations, a clear decrease in total LP signal was observed after incubation at 37 °C for 1 h (Figure 5C and D). These results indicate that the anti-PD-L1 LP formulation undergoes rapid dissociation or internalization within this timeframe. Notably, the retention ratio of anti-PD-L1 high-LP was higher than that of anti-PD-L1 low-LP (Figure 5E), suggesting that high ligand density not only contributes to the initial high binding avidity but also enhances retention. We further examined PD-L1 expression following treatment with anti-PD-L1 LPs (Figure 5F and G). The lowest PD-L1 expression levels were observed in the anti-PD-L1 high-LP group. However, despite the significant drop in LP signals after 1 h of incubation at 37 °C, no changes in PD-L1 expression were detected across the three groups. This indicates that PD-L1 ligands had not recovered during this time frame.
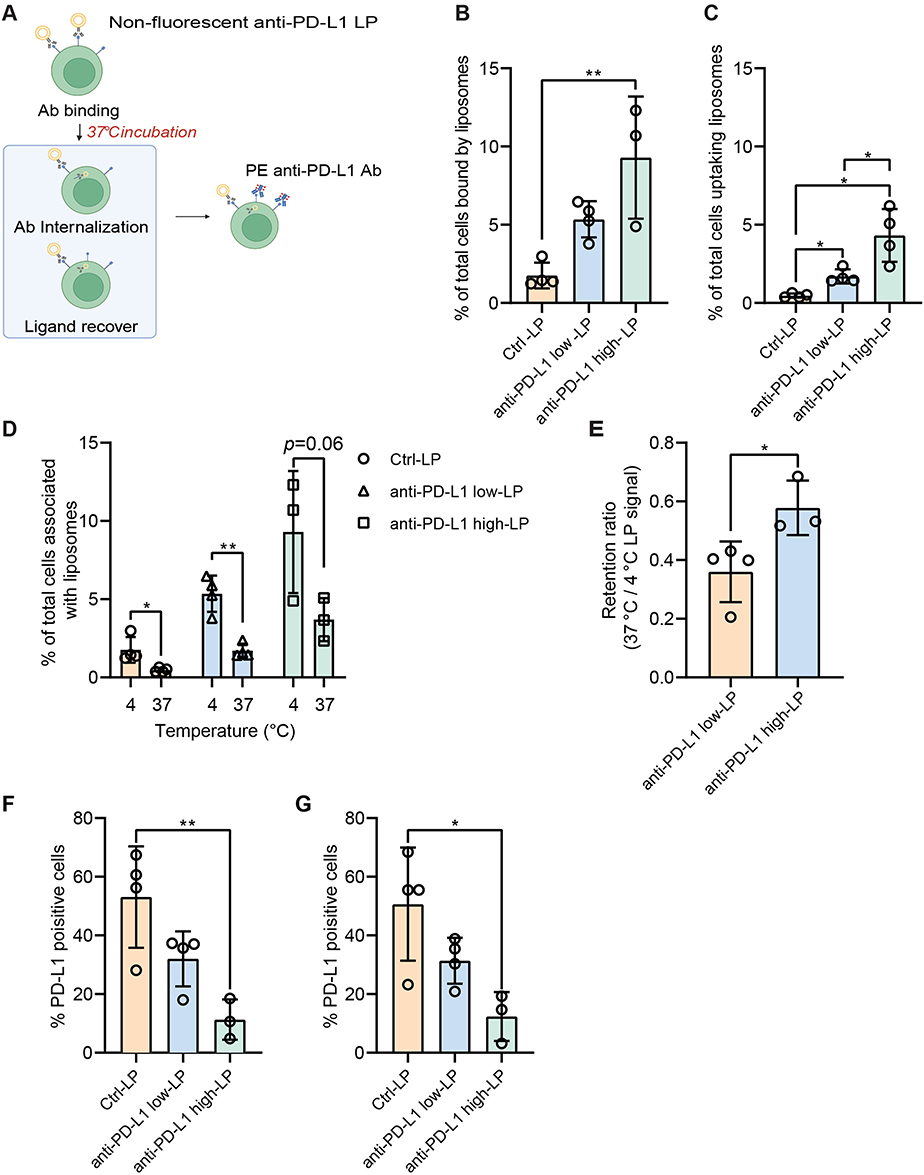 |
Figure 5 Cellular retention and PD-L1 blockade by anti-PD-L1 liposomes. Notes: (A) MZ2Mel43 cells were treated with different liposome (LP) formulations—Control (Ctrl)-LP, anti-PD-L1 low-LP, or anti-PD-L1 high-LP (100 µM lipid)—for 30 min at 4 °C to block PD-L1 receptors, followed by 1 h at 37 °C. Residual PD-L1 was detected using a fluorescent anti-PD-L1 antibody. TF-PE was used to assess LP retention, and PE fluorescence indicated PD-L1 expression, both measured by flow cytometry. (B) LP signal by cells with a 30 min incubation at 4 °C, followed by an additional 1 h incubation at 4 °C. (C) LP signal by cells with a 30 min incubation at 4 °C, followed by an additional 1 h incubation at 37 °C. (D) Data analysis of LP absorption: comparing 4 °C and 37 °C. (E) The retention ratio of LPs was calculated as follows: Retention ratio = LP signal after 37 °C incubation/LP signal after 4 °C incubation. (F) PD-L1 expression of cells with a 30 min incubation at 4 °C, followed by an additional 1h incubation at 4 °C. (G) PD-L1 expression of cells with a 30 min incubation at 4 °C, followed by an additional 1 h incubation at 37 °C. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01). |
In summary, high concentrations of anti-PD-L1 Ab on the liposomal surface enhance initial ligand binding and drug retention under physiological conditions, while showing no significant differences in lysosomal localization (reflecting total intracellular uptake) among the groups.
Liposomes with a High Antibody Concentration Show Improved Penetration in 3D Models
Additionally, we examined the penetration of LPs in 3D spheroids made from the BLM cell line. To differentiate the distinct layers of the spheroids, we used CellTracker Deep Red. This free cell-labeling small molecule dye enables the visualization of three distinct layers: the interior layer (characterized by low CellTracker signal), the middle layer (with moderate signal), and the outer layer (with high signal). Effective penetration requires LPs to reach the interior layers of the spheroids, whereas poorly penetrating LPs remain confined to the outer regions (Figure 6A). To quantify penetration, we analyzed LP signals in each layer using FlowJo software (see analysis strategy in Figure 6B). Regardless of the treatment condition, CellTracker dye consistently reached a similar depth in the spheroids (Figure 6C), confirming that its presence does not interfere with LP delivery. Notably, anti-PD-L1 high-LP demonstrated superior penetration compared to Ctrl-LPs and anti-PD-L1 low-LPs (Figure 6D). Reaching the spheroid core remained challenging, as indicated by a decrease in the penetration ratio from the outer to the interior layers. In the interior layer, anti-PD-L1 high-LP showed the highest penetration; however, fewer than 5% of interior cells exhibited detectable levels.
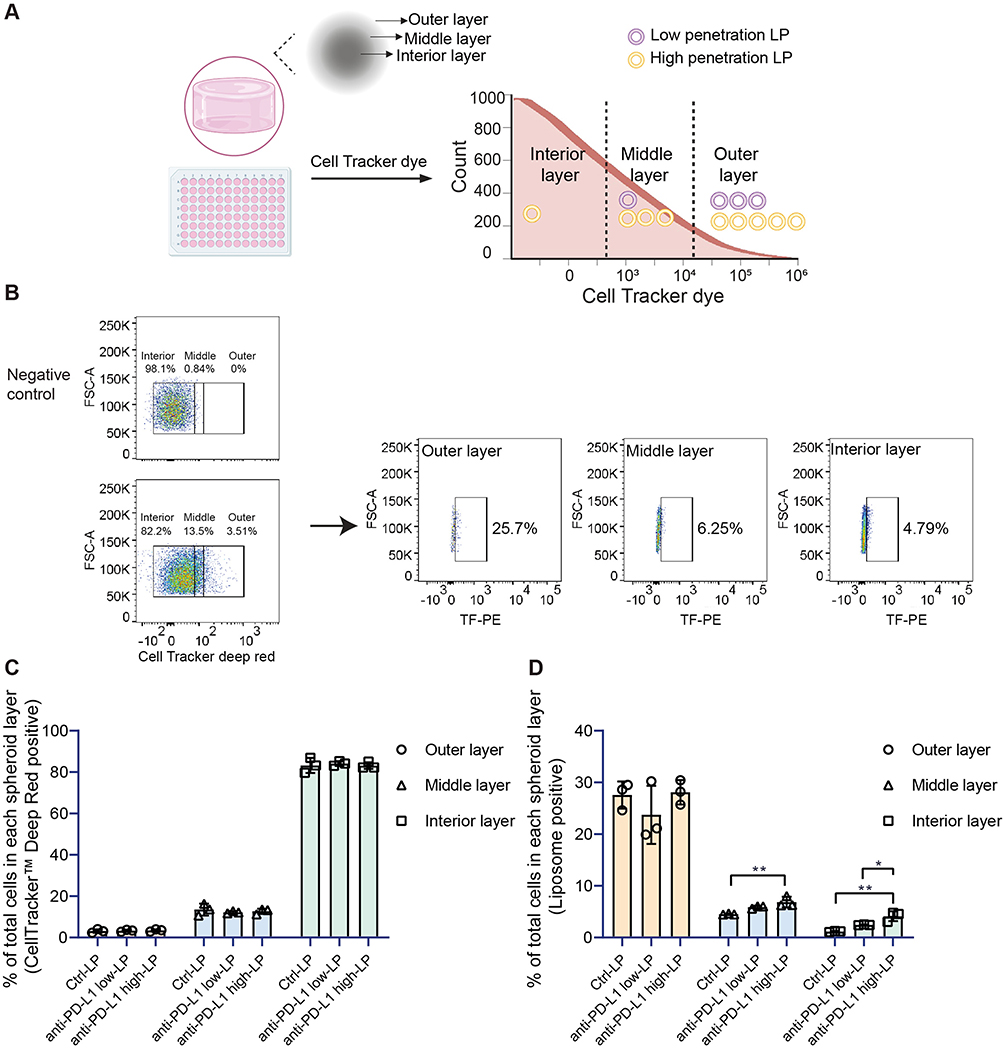 |
Figure 6 Penetration of anti-PD-L1 liposomes in a 3D BLM cell model. Notes: (A) Schematic diagram of anti-PD-L1 liposomes (LPs) in a 3D penetration assay. Different treatments (Control-LP (Ctrl-LP), anti-PD-L1 low-LP, or anti-PD-L1 high-LP, with lipid concentrations of 10 µM) were applied and incubated with the spheroids for 24 h to prepare for flow cytometry analysis. (B) Flow cytometry gating strategy for LP penetration analysis using FlowJo software. (C) Percentage of CellTracker Deep Red Dye signal detected in all cells in combined with the three different LPs within the various layers. (D) Percentage of LP signal detected in all cells from different formulations across the various layers. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01). |
Comparison of the Cellular Dynamics Between Parental Atezolizumab and Atezolizumab-Liposomes
Having characterized the anti-PD-L1 LPs with varying Ab densities, we next aimed to explore potential differences in binding avidity between the free Ab and its liposomal formulations under equivalent protein conditions. In this experiment, the same quantity of atezolizumab was presented in different formats, either as the free Ab or conjugated to the liposomal surface (Figure 7A). We assessed the PD-L1 blocking efficiency across various anti-PD-L1 concentrations. The results demonstrated that presenting the Ab on the liposomal surface significantly enhanced PD-L1 blocking ability compared to the free Ab (Figure 7B and C). Notably, at a concentration of 0.000781 µg/mL, 93% of PD-L1 remained detectable with the free Ab, whereas only 75% was detected with the anti-PD-L1 high-LP formulation (p <0.05), indicating more effective ligand masking by the liposomal format. These findings confirm that the manufacturing process of anti-PD-L1 LP formulations does not compromise their PD-L1 blocking capacity; instead, they exhibit superior binding avidity compared to the parental free Ab. In parallel, we examined LP binding and observed an interesting result: while anti-PD-L1 high-LP exhibited greater PD-L1 blocking capacity, binding to the cell surface was less effective than that of anti-PD-L1 low-LP (Figure 7D). This phenomenon can be explained by the fact that, when the same amount of Ab was used, anti-PD-L1 high-LP had a lower lipid content compared to anti-PD-L1 low-LP (Figure 7E). These results also indicate that increasing the anti-PD-L1 density effectively reduces non-specific binding.
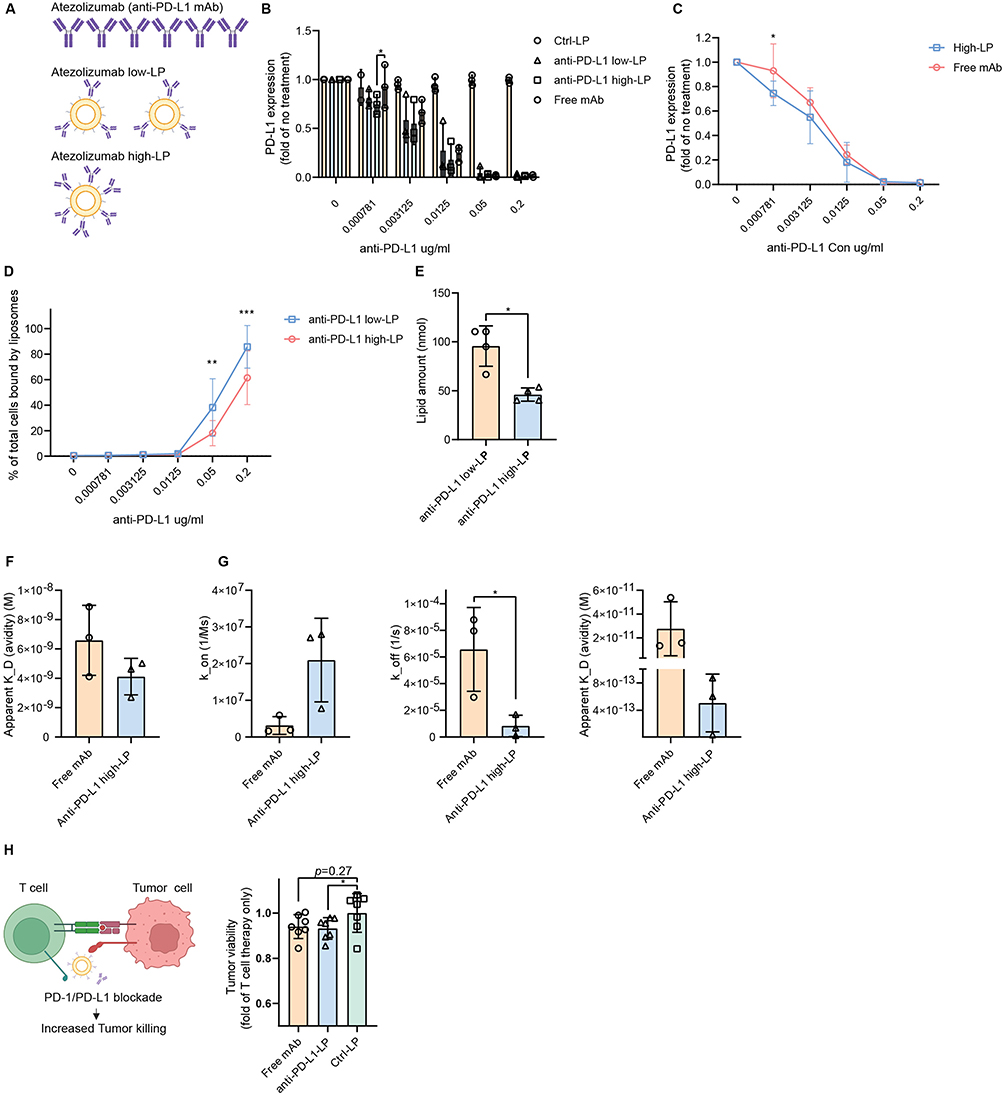 |
Figure 7 Comparative functional activity and binding dynamics of free vs liposomal atezolizumab. Notes: (A–E) PD-L1 blocking capacity and liposome (LP) binding percentages to total MZ2Mel43 cells at equivalent concentrations of anti-PD-L1 antibody (Ab) in atezolizumab (free anti-PD-L1 Ab), anti-PD-L1 low-LP, and anti-PD-L1 high-LP. (A) The same quantity of atezolizumab was presented in different formats. (B) PD-L1 expression after treatment with free anti-PD-L1 Ab, anti-PD-L1 low-LP, and anti-PD-L1 high-LP. (C) PD-L1 expression after treatment with free anti-PD-L1 Ab and anti-PD-L1 high-LP. (D) Binding capacity of anti-PD-L1 low- and high-LPs to MZ2Mel43 cells at different Ab concentrations. (E) Lipid amount of anti-PD-L1 low- and high-LP when the protein concentration is 0.2 µg/mL. (F and G) Surface plasmon resonance analysis of anti-PD-L1 high-LP and free Ab across a range of protein concentrations (0.1 μg/mL, 0.05 μg/mL, 0.025 μg/mL, 0.0125 μg/mL, and 0.00625 μg/mL). Analysis was conducted by fitting reference-subtracted curves to appropriate binding models using Biacore T200 Evaluation Software ((F) Steady-State Affinity Analysis: Steady-state affinity model (G) Kinetic Analysis: Langmuir 1:1 binding model). (H) Tumor cells were treated with free or anti-PD-L1 high-LP (0.005 µg/mL; EC50 calculated based on Figure 7B) for 1 h before adding T cells at an effector-to-target ratio of 1:1 or 3:1. Co-cultures were maintained for 72 h in supplemented RPMI. Tumor cell killing was monitored in real-time using Incucyte imaging by tracking mScarlet fluorescence, which was stably expressed in the tumor cells. p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). |
We further examined binding kinetics using SPR, as previously described. Anti-PD-L1 high-LP and free Ab samples were tested across a range of concentrations (0.1, 0.05, 0.025, 0.0125, and 0.00625 µg/mL). Using a steady-state fitting model, free mAb exhibited a comparable K_D compared to Ab-LP (6.59 × 10−9 M vs 4.12×10 −9 M) (Table 1 and Figure 7F). When applying detailed kinetic parameters, Ab-LP showed a higher association rate (k_on) and a significantly lower dissociation rate (k_off) (p < 0.05), resulting in an overall improved dissociation constant (K_D) (Table 1 and Figure 7G). Notably, the steady-state analysis showed higher apparent K_D values than the kinetic fit. This discrepancy likely results from incomplete equilibrium at the tested liposome concentrations, as the response curves did not reach a clear plateau. Moreover, the classical Langmuir 1:1 model does not fully capture the multivalent binding behavior of Ab-LPs. Therefore, the kinetic parameters are reported as apparent values that reflect overall multivalent avidity rather than true monovalent affinities. Finally, in a human peripheral blood mononuclear cells (PBMCs)–tumor cell co-culture assay, both free mAb and Ab-LP promoted comparable enhancement of tumor cell killing (Figure 7H).
 |
Table 1 Summary of Surface Plasmon Resonance Binding Parameters for Free mAb and Anti-PD-L1 High-LP |
In conclusion, presenting atezolizumab at higher concentrations on LPs enhances PD-L1 blocking efficiency while reducing non-specific binding. Moreover, the Ab-LP formulation exhibits stronger cellular interactions than the free Ab, likely due to its improved K_D.
Discussion
ICIs represent one of the most promising strategies in cancer immunotherapy. For instance, anti-PD-L1 mAbs prevent interaction of tumor cell PD-L1 with PD-1 on T cells, thereby restoring T cell activity and promoting an anti-tumor immune response. Building on this established mechanism, multivalency strategies—long recognized for enhancing binding avidity and therapeutic efficacy27 —offer new opportunities to improve checkpoint blockade. In this study, we developed multivalent anti-PD-L1 formulations by conjugating a clinically approved anti-PD-L1 mAb to a liposome-based drug delivery platform. Our findings demonstrate that liposomal presentation enhances antibody function not only through multivalent binding but also by enhancing drug retention and tumor penetration. Together, these results support liposome-based delivery as a promising strategy for developing next-generation nanoparticle-based ICIs.
Apparent Binding Avidity
Apparent (functional) binding avidity refers to the total strength of multiple binding interactions between molecules. A multivalent Ab system can be achieved by presenting multiple antigen-binding sites on a liposome.28 This design allows multivalent Abs to engage multiple epitopes simultaneously, leading to binding strengths that are orders of magnitude higher than their monovalent counterparts.
When considering ICIs, such as anti-PD-L1 mAbs, classical antagonist Abs typically follow a sigmoidal dose-response curve, where functional activity peaks near 100% receptor occupancy and plateaus at higher doses.29,30 This supports our rationale for developing multivalent Ab-lipid nanoparticle systems: by increasing ligand density, the probability of reaching saturation and therapeutic efficacy can be improved. In our study, we found a linear correlation between protein-to-lipid ratios and binding capacity under both low and high PD-L1 expression conditions in two different melanoma cell lines. Additionally, anti-PD-L1 LPs had superior receptor blockade capacity compared to conventional bivalent anti-PD-L1 mAb, as indicated by both flow cytometry and SPR experiments. Similar findings were reported by Bu et al using dendrimer-based anti-PD-L1 systems,16 and by Mamani et al, who developed a multivalent anti-PD-L1 peptide with ~40-fold higher blocking efficiency and improved stability. Comparable results were also observed for folate-functionalized nanodevices with tunable ligand density.31 However, optimizing ligand density remains challenging due to inter- and intra-patient heterogeneity in PD-L1 expression. Recent research suggests that the degeneracy prefactor, which refers to the number of ways multiple binding interactions can be arranged, is critical in determining multivalent binding behavior.32 This concept is particularly relevant for PD-L1 blockade, as elevated PD-L1 expression during immune activation effectively increases the degeneracy prefactor by providing more available binding configurations. Together, these effects underscore the promise of multivalent strategies in overcoming the limitations of conventional bivalent mAbs for immune checkpoint blockade.
Beyond ligand quantity, apparent binding avidity in Ab-based systems also depends on receptor clustering. For example, in the B7-CD28 co-stimulatory axis, dimeric B7 ligands (eg, B7-1 and ICOSLG) enhance signaling by bridging CD28 or ICOS dimers at the immune synapse, promoting receptor supercluster formation via avidity effects.33 Such systems often follow bell-shaped dose-response profiles,34 and receptor clustering is influenced by factors such as ligand spacing, linker flexibility, and geometric compatibility between ligands and receptors.
Selectivity
In vivo delivery introduces further challenges, where specificity becomes critical. Avidity and selectivity often behave nonlinearly.35 Research has reported that weak but multivalent binding interactions can enhance selectivity. Our findings support this concept: while high-concentration anti-PD-L1 LPs demonstrated stronger PD-L1-specific binding, lower-concentration LPs showed more non-specific cell surface binding. This underscores the importance of optimizing ligand density in liposomal systems to maximize selective delivery.
Intracellular Trafficking
After effectively targeting PD-L1, two key questions arise: (1) how long does the PD-L1 blockade persist, and (2) how long does it take for internalized PD-L1 to recycle back to the cell surface? In our study, we observed a significant increase in total cellular uptake of anti-PD-L1 high-LP compared to both anti-PD-L1 low-LP and Ctrl-LP. This elevated uptake signal primarily resulted from increased membrane binding rather than intracellular delivery, as we found non-targeted LPs demonstrated a similar level of lysosome localization as targeted LPs. Nonetheless, the overall intracellular accumulation of Ab-LPs may still increase over time, as membrane-bound LPs are eventually internalized into the cytoplasm. Additionally, anti-PD-L1 high-LPs exhibited longer surface retention compared to anti-PD-L1 low-LPs.
Although we focus primarily on the anti-PD-L1 LP system itself, combining it with other drugs encapsulated inside the LPs remains a valuable strategy. In such cases, achieving a balance between surface retention and internalization becomes critical: sufficient membrane retention ensures effective PD-L1 blockade, while timely internalization facilitates intracellular drug delivery. Previous studies have shown that atezolizumab undergoes 40–60% internalization after 2 h in multiple cell lines.36 Consistently, we observed 56–59% lysosomal co-localization after 2 h by confocal microscopy, and 42–64% internalization by flow cytometry after 1 h. Free atezolizumab and atezolizumab-conjugated LPs exhibited comparable internalization rates, suggesting that multivalency did not significantly alter the early internalization profile, supporting the feasibility of co-delivery strategies over short periods.18,37 In summary, high-density anti-PD-L1 LPs show enhanced surface binding and prolonged retention, leading to more sustained PD-L1 blockade without significantly altering the overall internalization pathway. These findings highlight the importance of optimizing ligand density to balance membrane retention and intracellular uptake for effective therapeutic and co-delivery applications.
Tissue Penetration and Accessibility
Another challenge in nanomedicine is the limited penetration of nanoparticles into solid tumors, primarily due to their larger size compared to mAbs or small-molecule drugs.18,38 Interestingly, our findings contrast with the commonly discussed “binding site barrier” concept,39 which suggests that high-affinity Abs may hinder deep tissue penetration by saturating receptors near the tumor periphery. In our 3D in vitro tumor model, however, the relatively high nanoparticle concentrations likely saturated peripheral PD-L1 binding sites, allowing additional high-avidity liposomes to diffuse deeper into the tissue. This suggests that the binding site barrier is context-dependent and may be alleviated under conditions of receptor saturation or high local particle concentration. Nonetheless, in vivo drug concentrations are typically much lower than those used in vitro, and under such physiological conditions, high-affinity multivalent nanoparticles may still experience penetration limitations, consistent with the binding site barrier hypothesis.
However, these limitations are less critical for checkpoint blockade therapies, where surface receptor engagement is the primary mechanism of action. In such cases, affinity may be more critical than deep tissue penetration, since functional blockade requires specific binding to and engagement with cell-surface receptors.
Manufacturing Optimization
Successful clinical translation of anti-PD-L1 LPs requires addressing key manufacturing challenges: scalability, reproducibility, accurate ligand quantification, and preservation of Ab functionality. In our study, the post-insertion method was employed to conjugate Abs to preformed LPs, achieving near 100% conjugation efficiency while maintaining Ab activity, consistent with literature reports.40–42 However, precise measurement of Ab density remains challenging, as both the total number of conjugated Abs and the fraction retaining functional activity must be considered, given that conjugation steps may compromise Ab functionality. Factors influencing Ab stability include linker chemistry, ultracentrifugation conditions, and general handling.43–48 Addressing these factors is crucial to developing reproducible, scalable, and effective therapeutic nanoparticles. In addition, lipid composition and structural design represent critical formulation variables that remarkably influence the biological behavior and immune responses elicited by LPs.49–51 Rational selection of lipid components, incorporation of biomembrane hybrids may further enhance circulation stability, tumor penetration, and immune activation. Together, optimization of both antibody conjugation and lipid design will be key to advancing anti-PD-L1 LPs toward clinical translation.
Limitations and Future Perspectives
While this study provides detailed insights into the cellular interactions between anti-PD-L1 LPs and tumor cells, the evaluation of therapeutic efficacy remains limited. We performed a human PBMC–tumor cell co-culture assay where both free mAb and Ab-LP enhanced tumor cell killing to a comparable extent. This outcome likely reflects similar drug availability in solution across the groups. However, the in vitro conditions may not fully capture the kinetic advantages of the liposomal formulation. Importantly, the primary aim of this work was to explore the mechanisms of LP–cell interactions in the context of immune checkpoint blockade, rather than to comprehensively assess biological function or treatment efficacy.
Additionally, the co-culture model lacks the complexity of the tumor microenvironment and immune system interactions found in vivo, which limits the translation of in vitro findings to clinical contexts. The heterogeneity of PD-L1 expression across patient tumors and the involvement of diverse immune cell populations were also not addressed, which may affect therapeutic outcomes.
Although this study focused on the active immunomodulatory function of anti-PD-L1 LPs alone, combination with other therapeutic agents within a single liposomal system represents a promising future direction. In melanoma, approximately 40% of patients have BRAF mutations.52 Recent studies suggest that BRAF/MEK inhibition can enhance tumor immunogenicity and modulate responsiveness to ICIs. Several ongoing clinical trials are evaluating sequential or combination regimens integrating both therapeutic classes.1 Moreover, chemotherapeutic agents capable of inducing immunogenic cell death may further amplify immune activation when co-administered with ICIs.53 Therefore, the development of liposomal delivery systems capable of co-encapsulating agents such as BRAF/MEK inhibitors, ICIs, or immunogenic cell death-inducing drugs offers a powerful platform to achieve synergistic antitumor effects and improve clinical outcomes in advanced melanoma.
Conclusion
In conclusion, our study highlights the potential of atezolizumab-conjugated LPs to integrate immune checkpoint inhibition with nanomedicine. By synthesizing LPs with varying Ab densities, we compared different formulations in terms of apparent binding avidity, tumor retention, and tissue penetration using multiple techniques. The results indicate that increasing ligand density benefits initial cellular binding, immune checkpoint inhibitor retention, and tissue penetration. Compared to free atezolizumab, Ab-LPs demonstrated higher binding avidity and blocking capacity.
These findings provide a strong foundation for further in vivo evaluation of anti-PD-L1 LP formulations, both as monotherapies and in combination with other therapeutic agents. Beyond PD-L1 targeting, this Ab-LP platform is highly adaptable and holds broad potential for other immune checkpoint inhibitors, targeted drug delivery systems, and precision medicine applications. Future work should focus on preclinical studies to validate the therapeutic potential of Ab-LP formulations within complex tumor microenvironments.
Abbreviations
Anti-PD-L1, anti-programmed death ligand-1; LP, Liposome; ICI, immune checkpoint inhibitor; mAb, monoclonal antibody; Ab, antibody; Ctrl, control; PD-1, programmed death-1; PDI, polydispersity index; SPDP, 3-(2-Pyridyldithio)propionic acid N-hydroxysuccinimide ester); TCEP, Tris(2-carboxyethyl)phosphine; PBMC, peripheral blood mononuclear cell; DSPE-PEG(2000)-Maleimide, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000]; DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine; DSPE-PEG(2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000]; TF-PE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[(dipyrrometheneboron difluoride)butanoyl]; -SH, sulfhydryl; SDS-PAGE, sodium dodecyl-sulfate polyacrylamide gel electrophoresis; BSA, bovine serum albumin.
Data Sharing Statement
All data supporting the findings of this study are included within the manuscript and its supplementary information files. Additional data is available from the corresponding author upon reasonable request.
Acknowledgments
The authors would like to thank Dr. Mengqi Huang and Dr. Maayke Kuijten (Erasmus Medical Center) for generously providing the PiggyBac-mScarlet plasmid. Illustrations in the graphical abstract and Figures 5A, 6 A, and 7A were created with BioRender.com.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is based on the research supported in part by the China Scholarship Council (File Number 202008310151).
Disclosure
Prof. Dr. Reno Debets reports a patent WO 2022/158977 issued to Pan Cancer T BV, a patent EP 25163944.9 pending to Pan Cancer T B. The authors declare that they have no other competing interests.
References
1. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet. 2021;398(10304):1002–22. doi:10.1016/S0140-6736(21)01206-X
2. Rui R, Zhou L, He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. 2023;14:1212476. doi:10.3389/fimmu.2023.1212476
3. Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
4. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
5. Upadhaya S, Neftelinov ST, Hodge J, Campbell J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat Rev Drug Discov. 2022;21(7):482–483. doi:10.1038/d41573-022-00030-4
6. Holder AM, Dedeilia A, Sierra-Davidson K, et al. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat Rev Cancer. 2024;24(7):498–512. doi:10.1038/s41568-024-00705-7
7. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38(3):326–333. doi:10.1016/j.ccell.2020.07.004
8. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–1546. doi:10.1056/NEJMoa1910836
9. Lu Q, Kou D, Lou S, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J Hematol Oncol. 2024;17(1):16. doi:10.1186/s13045-024-01535-8
10. Guo Z, Zhu AT, Fang RH, Zhang L. Recent developments in nanoparticle-based photo-immunotherapy for cancer treatment. Small Methods. 2023;7(5):e2300252–e2300252. doi:10.1002/smtd.202300252
11. Cheng Z, Fobian S-F, Gurrieri E, et al. Lipid-based nanosystems: the next generation of cancer immune therapy. J Hematol Oncol. 2024;17(1):53. doi:10.1186/s13045-024-01574-1
12. Cheng Y, Wang C, Wang H, et al. Combination of an autophagy inhibitor with immunoadjuvants and an anti-PD-L1 antibody in multifunctional nanoparticles for enhanced breast cancer immunotherapy. BMC Med. 2022;20(1):411. doi:10.1186/s12916-022-02614-8
13. Li L, Li Y, Yang CH, et al. Inhibition of immunosuppressive tumors by polymer‐assisted inductions of immunogenic cell death and multivalent PD‐L1 crosslinking. Adv Funct Mater. 2020;30(12):1908961. doi:10.1002/adfm.201908961
14. Radford DC, Yang J, Doan MC, et al. Multivalent HER2-binding polymer conjugates facilitate rapid endocytosis and enhance intracellular drug delivery. J Control Release. 2020;319:285–299. doi:10.1016/j.jconrel.2019.12.049
15. Miller A, Carr S, Rabbitts T, Ali H. Multimeric antibodies with increased valency surpassing functional affinity and potency thresholds using novel formats. mAbs. 2020;12(1):1752529. doi:10.1080/19420862.2020.1752529
16. Bu J, Nair A, Iida M, et al. An avidity-based PD-L1 antagonist using nanoparticle-antibody conjugates for enhanced immunotherapy. Nano Lett. 2020;20(7):4901–4909. doi:10.1021/acs.nanolett.0c00953
17. Zhang Y, Li N, Suh H, Irvine DJ. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat Commun. 2018;9(1):6. doi:10.1038/s41467-017-02251-3
18. Nobbmann U, Connah M, Fish B, et al. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol Genet Eng Rev. 2007;24(1):117–128. doi:10.1080/02648725.2007.10648095
19. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6. doi:10.3389/fphar.2015.00286
20. Lin YL, Tsai NM, Chen CH, et al. Specific drug delivery efficiently induced human breast tumor regression using a lipoplex by non-covalent association with anti-tumor antibodies. J Nanobiotechnology. 2019;17(1):25. doi:10.1186/s12951-019-0457-3
21. Merino M, Contreras A, Casares N, Troconiz IF. A new immune-nanoplatform for promoting adaptive antitumor immune response. Nanomedicine. 2019;17:13–25. doi:10.1016/j.nano.2018.12.016
22. Saeed M, van Brakel M, Zalba S, et al. Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody. Int J Nanomed. 2016;11:955–975. doi:10.2147/ijn.S96123
23. Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32(22):5134–5147. doi:10.1016/j.biomaterials.2011.03.067
24. Van de Griend RJ, Van Krimpen BA, Bol SJ, Thompson A, Bolhuis RL. Rapid expansion of human cytotoxic T cell clones: growth promotion by a heat-labile serum component and by various types of feeder cells. J Immunol Methods. 1984;66(2):285–298. doi:10.1016/0022-1759(84)90340-5
25. Huang W-Q, Burgers PC, Amin M, Luider TM, ten Hagen TLM. Precision localization of lipid‐based nanoparticles by dual‐fluorescent labeling for accurate and high‐resolution imaging in living cells. Small Sci. 2023;3(8). doi:10.1002/smsc.202300084
26. Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42(5):439–462. doi:10.1016/S0163-7827(03)00032-8
27. Ehrlich PH. The effect of multivalency on the specificity of protein and cell interactions. J Theor Biol. 1979;81(1):123–127. doi:10.1016/0022-5193(79)90085-7
28. Tjandra KC, Thordarson P. Multivalency in drug delivery–when is it too much of a good thing? Bioconjugate Chem. 2019;30(3):503–514. doi:10.1021/acs.bioconjchem.8b00804
29. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi:10.1200/JCO.2009.26.7609
30. Xiang Y, Kiseleva R, Reukov V, et al. Relationship between targeting efficacy of liposomes and the dosage of targeting antibody using surface plasmon resonance. Langmuir. 2015;31(44):12177–12186. doi:10.1021/acs.langmuir.5b01386
31. Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14(1):107–115. doi:10.1016/j.chembiol.2006.11.015
32. Kitov PI, Bundle DR. On the nature of the multivalency effect: a thermodynamic model. J Am Chem Soc. 2003;125(52):16271–16284. doi:10.1021/ja038223n
33. van der Merwe PA, Davis SJ. The immunological synapse--a multitasking system. Science. 2002;295(5559):1479–1480. doi:10.1126/science.1069896
34. Lühder F, Huang Y, Dennehy KM, et al. Topological requirements and signaling properties of T cell–activating, anti-CD28 antibody superagonists. J Exp Med. 2003;197(8):955–966. doi:10.1084/jem.20021024
35. Martinez-Veracoechea FJ, Frenkel D. Designing super selectivity in multivalent nano-particle binding. Proc Natl Acad Sci. 2011;108(27):10963–10968. doi:10.1073/pnas.1105351108
36. Xiao D, Luo L, Li J, et al. Development of bifunctional anti-PD-L1 antibody MMAE conjugate with cytotoxicity and immunostimulation. Bioorg Chem. 2021;116:105366. doi:10.1016/j.bioorg.2021.105366
37. Zanello A, Bortolotti M, Maiello S, Bolognesi A, Polito L. Anti-PD-L1 immunoconjugates for cancer therapy: are available antibodies good carriers for toxic payload delivering? Front Pharmacol. 2022;13:972046. doi:10.3389/fphar.2022.972046
38. Sindhwani S, Syed AM, Ngai J, et al. The entry of nanoparticles into solid tumours. Nat Mater. 2020;19(5):566–575. doi:10.1038/s41563-019-0566-2
39. Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Delivery Rev. 2008;60(12):1421–1434. doi:10.1016/j.addr.2008.04.012
40. Mehrabian A, Dadpour S, Mashreghi M, et al. The comparison of biodistribution of glutathione PEGylated nanoliposomal doxorubicin formulations prepared by pre-insertion and post-insertion methods for brain delivery in normal mice. IET Nanobiotechnol. 2023;17(2):112–124. doi:10.1049/nbt2.12111
41. Nielsen UB, Kirpotin DB, Pickering EM, et al. Therapeutic efficacy of anti-ErbB2 immunoliposomes targeted by a phage antibody selected for cellular endocytosis. BBA. 2002;1591(1–3):109–118. doi:10.1016/s0167-4889(02)00256-2
42. Mao Y, Triantafillou G, Hertlein E, et al. Milatuzumab-conjugated liposomes as targeted dexamethasone carriers for therapeutic delivery in CD74+ B-cell malignancies. Clin Cancer Res. 2013;19(2):347–356. doi:10.1158/1078-0432.CCR-12-2046
43. Woythe L, Tito NB, Albertazzi L. A quantitative view on multivalent nanomedicine targeting. Adv Drug Delivery Rev. 2021;169:1–21. doi:10.1016/j.addr.2020.11.010
44. Woythe L, Madhikar P, Feiner-Gracia N, Storm C, Albertazzi L. A single-molecule view at nanoparticle targeting selectivity: correlating ligand functionality and cell receptor density. ACS nano. 2022;16(3):3785–3796. doi:10.1021/acsnano.1c08277
45. Ansell SM, Harasym TO, Tardi PG, Buchkowsky SS, Bally MB, Cullis PR. Antibody conjugation methods for active targeting of liposomes. Methods Mol Med. 2000;25:51–68. doi:10.1385/1-59259-075-6:51
46. Maslanka Figueroa S, Fleischmann D, Beck S, Goepferich A. The effect of ligand mobility on the cellular interaction of multivalent nanoparticles. Macromol Biosci. 2020;20(4):e1900427–e1900427. doi:10.1002/mabi.201900427
47. Vu TQ, Sant’Anna LE, Kamat NP. Tuning targeted liposome avidity to cells via lipid phase separation. Biomacromolecules. 2023;24(4):1574–1584. doi:10.1021/acs.biomac.2c01338
48. Münter R, Simonsen JB. Comment on “Optimal centrifugal isolating of liposome-protein complexes from human plasma” by L. Digiacomo, F. Giulimondi, A. L. Capriotti, S. Piovesana, C. M. Montone, R. Z. Chiozzi, A. Laganá, M. Mahmoudi, D. Pozzi and G. Caracciolo, Nanoscale Adv. 2021, 3, 3824. Nanoscale Adv. 2022;5(1):290–299. doi:10.1039/d2na00343k
49. Zhu Y, Ma J, Shen R, et al. Screening for lipid nanoparticles that modulate the immune activity of helper T cells towards enhanced antitumour activity. Nat Biomed Eng. 2024;8(5):544–560. doi:10.1038/s41551-023-01131-0
50. Chang JJ, Wang YC, Yang SH, Wu JY, Chang MW, Wang HD. Pioneering astaxanthin-tumor cell membrane nanoparticles for innovative targeted drug delivery on melanoma. Int J Nanomed. 2024;19:2395–2407. doi:10.2147/IJN.S439476
51. Zhao Y, Hou X, Wang Z, et al. A mechanical immune checkpoint inhibitor stiffens tumor cells to potentiate antitumor immunity. Angew Chem Int Ed Engl. 2025;64(5):e202417518. doi:10.1002/anie.202417518
52. Carlino MS, Long GV, Kefford RF, Rizos H. Targeting oncogenic BRAF and aberrant MAPK activation in the treatment of cutaneous melanoma. Crit Rev Oncol Hematol. 2015;96(3):385–398. doi:10.1016/j.critrevonc.2015.08.021
53. Ma GL, Lin WF. Immune checkpoint inhibition mediated with liposomal nanomedicine for cancer therapy. Mil Med Res. 2023;10(1):20. doi:10.1186/s40779-023-00455-x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.