Back to Journals » Infection and Drug Resistance » Volume 14
Multifunctional Non-Coding RNAs Mediate Latent Infection and Recurrence of Herpes Simplex Viruses
Authors Zhang Y, Zeng LS, Wang J, Cai WQ, Cui W ![]() , Song TJ, Peng XC, Ma Z, Xiang Y, Cui SZ, Xin HW
, Song TJ, Peng XC, Ma Z, Xiang Y, Cui SZ, Xin HW
Received 18 August 2021
Accepted for publication 30 November 2021
Published 14 December 2021 Volume 2021:14 Pages 5335—5349
DOI https://doi.org/10.2147/IDR.S334769
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Ying Zhang,1– 3,* Li-Si Zeng,4,* Juan Wang,5,* Wen-Qi Cai,2,3 Weiwen Cui,6 Tong-Jun Song,7 Xiao-Chun Peng,2,8 Zhaowu Ma,2,3 Ying Xiang,2,3 Shu-Zhong Cui,4 Hong-Wu Xin2,3
1Department of Gastroenterology, Chun’an County First People’s Hospital (Zhejiang Provincial People’s Hospital Chun’an Branch), Hangzhou, Zhejiang Province, 311700, People’s Republic of China; 2Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, Jingzhou, Hubei, 434023, People’s Republic of China; 3Department of Molecular Biology and Biochemistry, School of Basic Medicine, Health Science Center, Yangtze University, Jingzhou, Hubei, 434023, People’s Republic of China; 4State Key Laboratory of Respiratory Disease, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Guangzhou, 510095, People’s Republic of China; 5Department of Obstetrics and Gynecology, Lianjiang People’s Hospital, Guangdong, 524400, People’s Republic of China; 6Department of Bioengineering, University of California, Berkeley, CA, 94720, USA; 7Department of Neurosurgery, Shenzhen Hospital of Integrated Traditional Chinese and Western Medicine, Shenzhen, Guangdong Province, 518104, People’s Republic of China; 8Department of Pathophysiology, School of Basic Medicine, Health Science Center, Yangtze University, Jingzhou, Hubei, 434023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong-Wu Xin; Ying Xiang
Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, 1 Nanhuan Road, Jingzhou, Hubei, 434023, People’s Republic of China
Tel +86 13311055391
; +86 15107212530
Fax +0716-8062633
Email [email protected]; [email protected]
Abstract: Herpes simplex viruses (HSVs) often cause latent infection for a lifetime, leading to repeated recurrence. HSVs have been engineered as oncolytic HSVs. The mechanism of the latent infection and recurrence remains largely unknown, which brings great challenges and limitations to eliminate HSVs in clinic and engineer safe oHSVs. Here, we systematically reviewed the latest development of the multi-step complex process of HSV latency and reactivation. Significantly, we first summarized the three HSV latent infection pathways, analyzed the structure and expression of the LAT1 and LAT2 of HSV-1 and HSV-2, proposed the regulation of LAT expression by four pathways, and dissected the function of LAT mediated by five LAT products of miRNAs, sRNAs, lncRNAs, sncRNAs and ORFs. We further analyzed that application of HSV LAT deletion mutants in HSV vaccines and oHSVs. Our review showed that deleting LAT significantly reduced the latency and reactivation of HSV, providing new ideas for the future development of safe and effective HSV therapeutics, vaccines and oHSVs. In addition, we proposed that RNA silencing or RNA interference may play an important role in HSV latency and reactivation, which is worth validating in future.
Keywords: non-coding RNA, small RNA, oncolytic virus, herpes simplex virus, HSV, latent infection, latency-associated transcripts, LATs
Introduction
Herpes viruses are classified into three subfamilies: α, β and γ,1 The α herpes viruses include herpes simplex virus type 1 and 2 (HSV-1/2). HSV typically goes through lytic infection, latent infection, and reactivation. No effective vaccine has been reported.2 HSV infection is very common in human population with an infection rate of approximately 70%. Most HSV infections cause asymptomatic or weak symptomatic illnesses; however, some cause severe illnesses, including viral encephalitis. HSV-1 commonly infects facial areas, such as cold sores, preferentially becomes latent in and reactivates from the trigeminal ganglion (TG) latency; HSV-2 infects genital areas, such as genital herpes, and becomes latent in and reactivated from the sensory dorsal sacral root ganglia.3,4 HSV likes to establish long-term latent infections in the nuclei of sensory neurons, which possess a natural protective barrier from immune responses.5 The reactivation and recurrence of HSV can be caused by a variety of non-specific stimulation after latent infection, leading to related clinical symptoms, such as herpes labialis, herpangina, herpetic encephalitis, gastrointestinal diseases, esophageal diseases, and genital herpes, with increasing morbidity in young populations.6 There are still low levels of lytic gene transcription during the latent stage in mouse models.7 When the factors regulating viral latency change, the viral gene expression level increases, and large-scale reactivation is initiated. Interestingly, HSV is reactivated in response to various factors such as ultraviolet radiation, emotional stress, fever, tissue damage, and immunosuppression. Today, anti-herpes virus drugs can target only lytic infections; therefore, treating latent viral infection remains a challenge.
Since HSV infection can be lytic, inducing rapid and efficient cell killing, HSVs have been used as oncolytic herpes simplex viruses (oHSVs). HSVs can be ideal oHSVs because they have a wide host range, and the lytic virus infection can be controlled by effective anti-herpetic agents.8,9 Engineered oHSVs have been reported in many clinical trials.10 T-VEC is an engineered oHSV with deletion of the genes ICP34.5 and ICP47 and insertion of the gene encoding human GM-CSF.11 In 2015, T-VEC was approved to treat the advanced melanoma in US.12 Although oHSV is an effective treatment for tumors, the challenge is that HSV infection can be latent in the nervous system, prone to reactivate and relapse, which cannot be cured. Therefore, the idea oHSVs should not be able to establish latency. Latency-associated transcript (LAT) is considered to play a very important role in HSV latency and is deleted in some oHSVs to prevent its latency.13 However, the mechanisms of HSV latency are not completely understood, which is a barrier to make safe oHSVs.
Therefore, to better treat HSV latent infection and make better oHSVs or other HSV vectors for gene therapy, we need to further understand the mechanisms of HSV latent infection. In this review, we will introduce latest development of research on the lytic and latent infections of HSV, mechanisms of HSV latency, and constructing safe oHSVs, with emphasis on the role of LAT in latent infection. We highlight a broad repertoire of multifaceted structure, expression regulation, expression products (including non-coding RNAs), function of LAT. Highlighting these quantifiable observations will provide a new prospect for better treatment of HSV latency and recurrence, and cancers by oHSV.
Lytic Infection
When HSV enters the human body, it initially infects epithelial tissues or cells for efficient lytic infection, and then spreads to nerve axons to form latent infection in sensory neurons.14 When HSVs cause lytic infections, HSVs express immediate early genes (IE or α), early genes (E or β), and late genes (L or γ), which play important roles in viral replication.15 IE genes are the first transcribed and expressed, and encode five proteins, ICP0, ICP4, ICP22, ICP27, and ICP47.16 Among them, ICP4 and ICP27 play an indispensable role in virus replication. They are directly related to the synthesis of viral DNA. Once IE proteins have been produced in adequate amounts, infected cells typically transcribe the viral E genes, followed by DNA replication and synthesis of progeny virions. If the IE protein is lacking, the virus does not develop a productive infection.17
The transcription of the IE genes is activated by VP16, which is a major virion protein of HSV. VP16 acts through the target sequence TAATGARAT, which is present in the HSV IE promoter. TAATGARAT is the binding site of cytokine OCT-1, which is able to bind to the octamer sequence ATGCAAAT.18 The Oct family of proteins share a DNA-binding structure called the POU domain, which allows for great flexibility in the sequence recognition of various Oct proteins.19 HCF exists only in the cytoplasm and mainly transports VP16 protein to the nucleus. Therefore, HCF may play an important role in IE gene expression.17 The complex of VP16/OCT-1/HCF is associated with histone acetyltransferase and demethylase, and interacts with IE gene promoters, preventing the formation of heterochromatin and activating IE gene expression,20 such as ICP0 and ICP4, which allows early and late gene transcription and lytic viral infection to occur. In short, HSV will enter the lytic infection successfully if there is enough VP16 entering the nucleus.21
Latent Infection
Latent infection is a typical characteristic of HSV, primarily because the virus limits its own gene expression through its interaction with host cells. During the latent infection, the gene transcription of the virus enters a silent state.21 When HSV enters sensory neurons, the VP16/OCT-1/HCF complex cannot form to promote IE gene expression, since the VP16 protein is degraded in the axons of neurons.17 Instead, the viral genome combines with the nucleosome to form a compact chromatin structure. Histones associated with the promoters of the IE genes are in an inhibitory configuration, preventing IE gene expression. However, the LAT promoter is in the permitted configuration, allowing the transcription of LAT, so that LATs are the only abundant transcription products of HSVs during latent infection of sensory ganglia. LAT plays a crucial role in establishing latency,22 maintaining latency, reactivating the virus from latency, and protecting neurons from apoptosis.23,24 HSV lytic and latent infections are regulated partly by the interaction of different viral gene products.25 Next, we will discuss the pathways and mechanisms of the latent infection of HSV in depth.
The Three Pathways to HSV Latent Infection
After the initial infection of a cell with HSV, the viral genome remains in the nonreplicating state or latent state as in sensory neurons, where the HSV genome is not transcribed owing to insufficient IE proteins.17 It is known that inefficient IE gene expression or function is a prerequisite for the virus to establish latency. The IE gene expression is theoretically regulated by the appropriate viral and cellular signaling pathways and should be hypothetically different in sensory neurons from other cell types. For example, Katz JP et al26 found that the transcription efficiency of the IE gene expression was initially extremely low and E gene expression was not enough to initiate viral DNA synthesis; however, IE and E gene expression rise rapidly once viral DNA synthesis begins. This trigger is attributed to the initiation and extension of DNA synthesis. It was noted that IE genes, especially the IE gene encoding the ICP4 protein is located near the initiation region of DNA replication. Hence, early DNA synthesis may cause structural changes that activate ICP4 transcription. In mice, as with LATs, deletion mutants of HSV-1 ICP4 ORFs, O and P, can establish a significantly more latent infection.27 In addition, the ICP0 expression also regulates the establishment of HSV latent infection. HSV-1 ICP0 defective mutants are approximately 1000 times less effective in establishing an incubation period than wild-type viruses, and this deficiency can be overcome by co-infection with an adenovirus expressing ICP0. This result highlights the pivotal role of ICP0 in determining the outcome of tissue culture cell infections: in the absence of ICP0, the infected cell was transformed into a static state, while in the presence of ICP0, the transformation was prevented or even reversed, leading to lytic infection. Data by Sawtell28 showed that a high level of viral DNA copy number was related to HSV reactivation in the TG. It is hypothesized that transcriptional restoration of a single gene that had not been fully suppressed initiates a chain reaction. Therefore, once ICP0 is synthesized, other genes that have been completely suppressed can be transcribed. Here, we propose three pathways to HSV latent infection below (Figure 1).
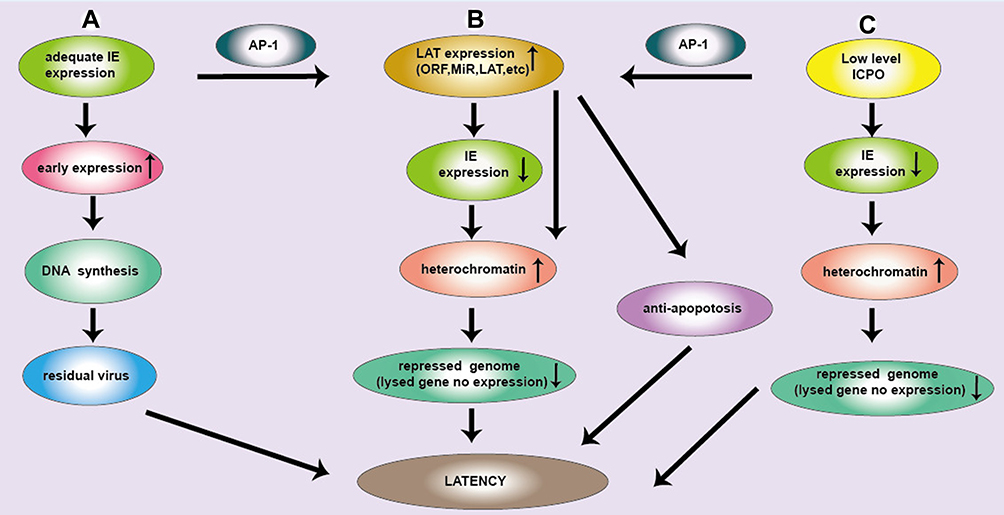 |
Figure 1 Three pathways of latent infection of HSV. (A) IE gene is adequate, the early expression gene is increased and largely neurons are cleaved and subsequently cleared. Due to the low copy number of HSV DNA in neurons, DNA synthesis is reduced and residual viruses are latent infection. (B) IE gene expression can produce ICP0 protein. ICP0 activates the JNK signaling pathway, and AP-1 is phosphorylated, then activate LAT. LAT sequence is complementary to the IE gene encoding ICP0, which continuously consumes ICP0, so decrease IE gene expression. ICP0 and LAT increase histone occupancy and heterochromatin levels of lytic genes and block their transcription, causing HSV latency. (C) Low levels of ICP0 can promote the expression of LAT and lytic genes in latently infected ganglion, where the loading of total histone and heterochromatin on HSV genome was increased, maintaining latency. LAT transcription can downregulate lytic gene transcription during latency. Moreover, ICP0 promoted AP-1 nuclear translocation, which promotes the expression of LAT. Subsequently many products of LAT (LAT, MiR and ORF) are produced, which will make HSV latent through anti-apoptotic pathway. |
(a) When there is adequate IE gene expression, more E genes are expressed, so that a large percentage of neurons may be lysed and subsequently cleared. However, few percentage of neurons may become latently infected likely due to the low copy number of HSV DNA in the neurons. It is difficult to rule out the notion that some of the dormant genomes are retained after viral DNA replication.27
(b) A sufficient amount of IE gene expression can produce a large amount of ICP0 protein. ICP0 activates the JNK signaling pathway, and then AP-1 is phosphorylated by JNK and traffics into the neural nucleus, where AP-1 binds to the transcription regulatory sequence of LAT and activates LAT transcription as we reported recently,10 leading to latent infection of HSV. At the same time, LAT transcription sequence is complementary to the IE gene encoding ICP0, which continuously consumes ICP0, resulting in the corresponding decrease in IE gene expression. However, ICP0 and LAT increase histone occupancy and heterochromatin levels of lytic genes and block their transcription,14 causing HSV latency.
(c) Low levels of ICP0 were found to promote the expression of LAT and lytic genes in latently infected ganglion, where the loading of total histone and heterochromatin on HSV genome was increased, maintaining latency. LAT transcription was shown to downregulate lytic gene transcription during latency through the deposition of repressive histone marks on the lytic IE gene promoters.29 Moreover, low levels of ICP0 promoted AP-1 nuclear translocation, which promotes the expression of LAT. Subsequently, many products of LAT (LAT, MiR and ORF) are produced, which will make HSV latent through many mechanisms, including the anti-apoptotic pathway.
Taken together, ICP0 acts as a “switch” in the balance between the lytic and latent states.17,30 LATs have a variety of potential roles in promoting latent infection by inhibiting apoptosis, inhibiting the expression of lysed genes and promoting viral latency. This is closely related to the biological structure of LAT as we will describe below.
The Structure and Expression of the LAT
It is believed that the LAT presence indicates HSV latency. LAT plays a key role in latent infection, which is attributed to the genetic structure of LAT. During HSV latent infection, LAT is the only transcription products that are highly expressed in the nucleus of host cells. Interestingly, Ying Zhang et al10 showed that HSV LAT has two intervals, LAT fragment 1 (LAT1) and LAT fragment 2 (LAT2), and their respective promoters, LAT promoter 1 (LAP1) and LAT promoter 2 (LAP2) (Figure 2). LAP1 has been shown to be the critical promoter for activating expression of LAT during latency. LAP2 also has been shown to possess promoter activity and augment HSV-1 LAP1 activity during latency. LAT is also divided into non-major LAT (minor LAT or primary LAT) and major LAT, which are produced by different ways of splicing. HSVs have serotypes HSV-1 and HSV-2, which share many similarities in their morphology, molecular structure and drug sensitivity, but are different in their LAT. HSV-1 LAT1/2 includes a family of transcripts comprising an 8.3 kb primary transcript in low levels and three introns (Figure 2A). Among the three introns, the 2.0 kb LAT was highly expressed in both lytic and latent infection; the 2.0 kb LAT was found only at low levels during latency; the predicted 0.5 kb LAT was not detected.31 LAT2 splicing produced a 6.3 kb exon product, which cannot easily be detected, although the 2.0 and 1.5 kb introns can easily be seen in Northern blot analysis.10 Translation of a protein from the spliced LAT exon mRNA remains to be convincingly shown,32 nevertheless it is clear that the exon 1 region of the LAT is required for protection from apoptosis.23,33 The function of the 1.5 kb LAT introns is unclear. However, the 2 kb LAT is upregulated in some cells expressing heat shock proteins.34 Interestingly, the 1.5 kb LAT intron is completely encoded in the 2.0 kb intron and is, thus, called a twintron.35 Both introns are present in the nucleus during latency. The LAT introns can be described as a complex twintron as three overlapping introns (2.0/1.5/0.5kb) can be produced from the LAT transcript. Likewise, HSV-2 LAT2 is transcribed to generate approximately a 9.0 kb of non-primary LAT and a stable 2.2 kb of major LAT. However, transcripts of HSV-2 LAT1 are rarely reported (Figure 2B).
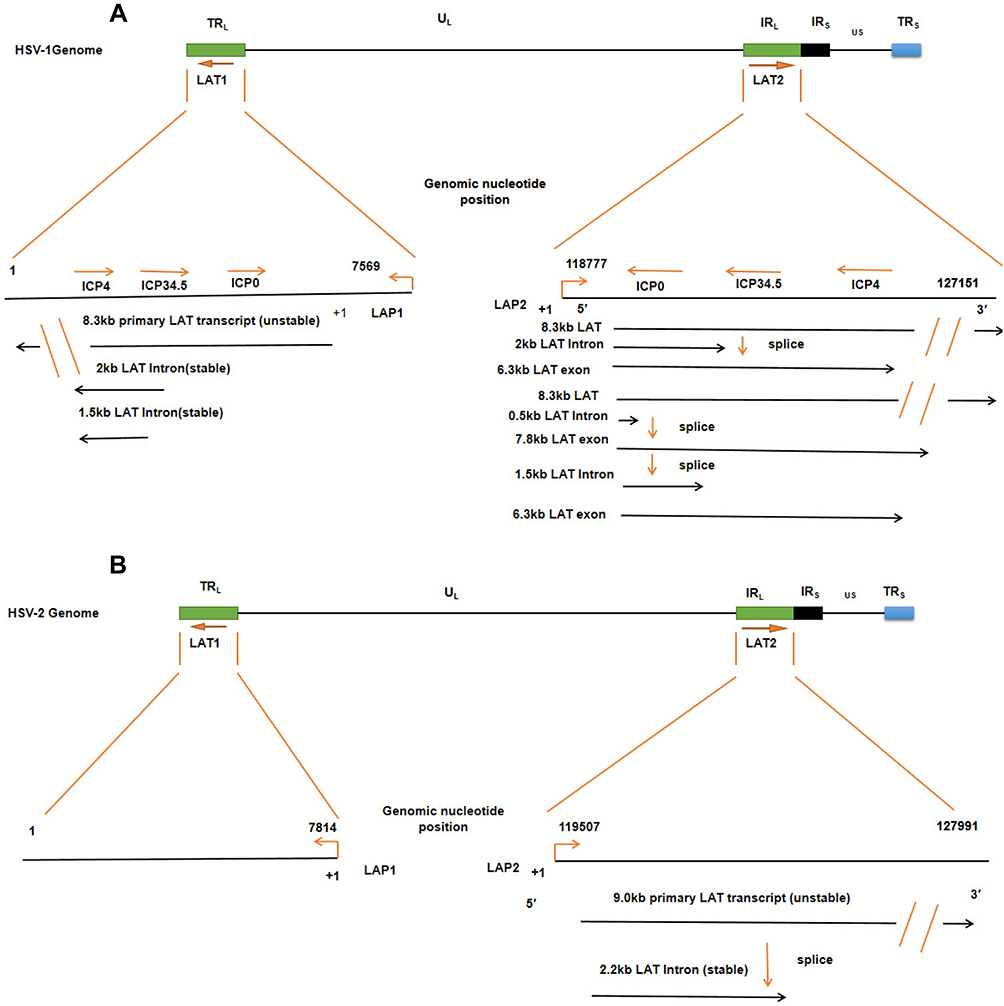 |
Figure 2 The LAT locus and (twintron) splicing of the HSV genome. (A) HSV-1 includes UL and US regions with terminal and internal repeats (TRL, IRL, IRS and TRS). LAT1 is in TRL and UL, LAT2 is in IRL and UL connection region. An TRL fragment, overlapping ICP0, ICP34.5, and ICP4, is expanded to show LAT1, including 8.3 kb original LAT, and 2 kb and 1.5 kb of the LAT intron. The 8.3 kb LAT2 was spliced into a 2.0 kb intron and a 6.3 kb mRNA or spliced as twintron introns into a 0.5 kb unstable intron and a 7.8 kb RNA. (B) HSV-2, like HSV-1, contains the LAT1 and LAT2. Their transcripts include an unstable 9.0 kb primary LAT and a stable 2.2 kb primary LAT. |
Regulation of LAT Expression by Four Pathways
HSV can establish latency in neurons, thus maintaining a latent state for years, or even a lifetime.36 This characteristic is related to the structure, expression and regulation of LATs. Here, we summarize the regulation of LAT expression of HSV (Figure 3) by four pathways.
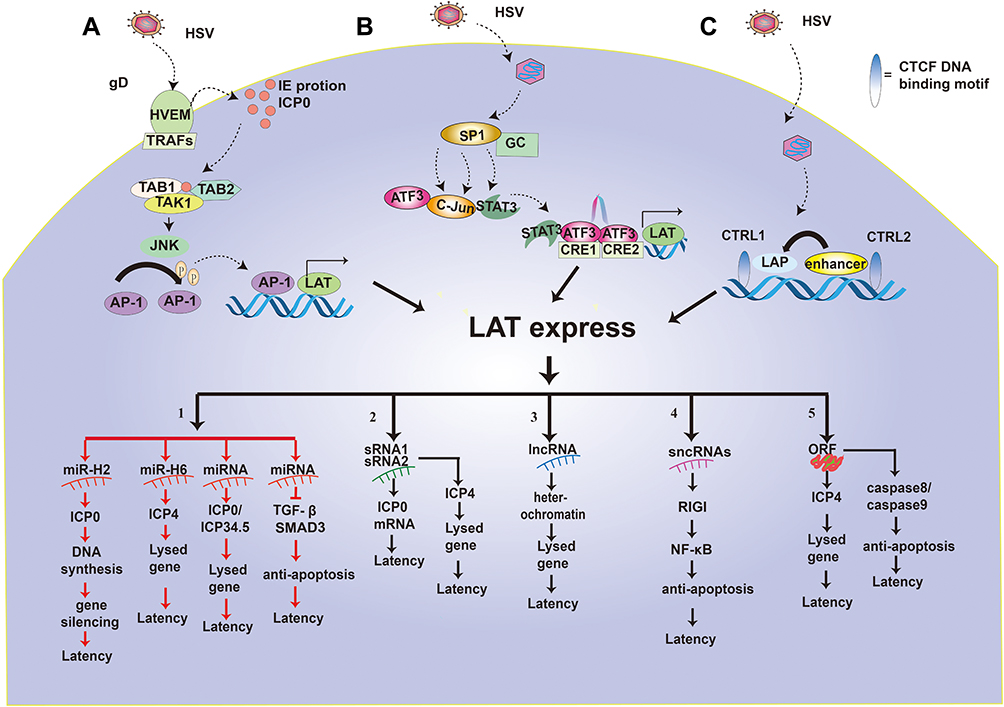 |
Figure 3 Regulation of LAT expression and function of LAT. (A–C) highlight the pathways that promote the transcriptional expression of LATs. 1–5 shows the five functional mechanisms of LAT latency. 1. LAT miR-H2 can inhibit ICP0 protein so that viral DNA cannot be synthesized, causing the silencing of the lysed gene and HSV latency. LAT miRNA-H6 inhibit the expression of ICP4 and lysed genes. LAT miRNA blocks ICP0/ICP34.5 production and reduces viral DNA synthesis, which in turn inhibits the expression of the lysed gene and renders the virus latent. LAT miRNA can also achieve viral latency by targeting transformed growth factor (TGF)-β and SMAD3 in the TGF-β pathway to resist apoptosis. 2. LAT sRNA1 and sRNA2 can inhibit ICP0 mRNA expression by partial base complementation to maintain latency. LAT sRNA2 can also inhibit the expression of ICP4 and lysed genes to help establish and maintain virus latency. 3. LAT lncRNA can induce facultative heterochromatin to promote lytic gene silencing during latency. 4. LAT sncRNA collaborates with RIG-I to stimulate NF-kB dependent transcription for anti-apoptosis and latency. 5. LAT ORFs inhibit ICP4 and then down-regulates lysed genes to make the virus latent. At the same time, LAT ORFs inhibit caspase 8 or caspase 9 for anti-apoptosis and latency. |
(A) The ICP0-JNK-AP1-LAT pathway. The envelope glycoprotein gD of HSV binds to the herpes virus entry mediator (HVEM) on cell membrane, and the transmembrane protein HVEM binds to TRAFs in the cytoplasm, then the HSV enters the cell.37 HSV-1 infection results in the synthesis of IE proteins, such as the ICP0. Subsequently, ICP0 was able to catalyze the K63-linked polyubiquitin chains with the help of the Ubc13ev1A enzyme as it recognizes and interacts with TAK1.38 Then, activated TAK1 phosphorylates MKK6, which subsequently activates the JNK kinase pathway.38 The JNK pathway phosphorylates the AP-1 transcription factor, then the phosphorylated AP-1 translocated into the nucleus to bind to the LAT promoter sequence and promoted LAT expression, leading to HSV latency.14
(B) The Sp1-Stat3-ATF3-CRE-LAT pathway. HSV dissociates the capsid and enters the nerve SK-N-SH cells. Sp1 protein is abundant in SK-N-SH cells.39 However, Sp1 protein interacts with a variety of GC rich promoter sequences, and Sp1 recruits STAT3, ATF3, and c-jun complexes.39 In neurons, STAT3 exists in the cytoplasm and travels to the nucleus under neuronal stress.40 STAT3 binds to ATF3:c-jun heterodimer in the nucleus without DNA binding activity or phosphorylation. ATF3 is a member of the ATF/CREB transcription factor family.41 The current consensus is that ATF3 is an immediate early-response gene, a regulator of stress response, and a factor in determining cell fate.42 ATF3 binds to a cAMP response element (CRE) with the canonical sequence 5′-TGACGTCA-3′ in the form of homodimers or heterodimers with other members of the ATF/CREB family.43 HSV-1 DNA contains eight CRE sites; however, only two of these are in a promoter, specifically in promoter 1 of LAT (LAP1). Results showed that ATF3 bound to both CRE sites in LAP1, formed a homodimer, and fostered increased accumulation of LAT.43 Furthermore, another study found that ATF3 enhanced the expression of LAT,44 and played a role in the maintenance of HSV-1 in a latent state.43
(C) The CTRL-CTCF-LAT pathway. When HSV enters the cytoplasm and the capsid breaks away, the IE gene begins to express ICP0. Interestingly, the ICP0 region is flanked by two CTCF binding motifs, namely CTRL1 and CTRL2.45 CTRL binds to CTCF and activates LAT promoter (LAP), making the virus latent. Recent studies have shown that the CTCF-insulator’s role in lytic gene inhibition of β and γ herpes viruses further supports the possibility that CTCF-insulators also regulate α herpes virus latency via lytic gene repression.45
(D) The VP16-LowICP0-LAT pathway. Multi-protein complex of VP16, Oct-1 and HCF initiate IE gene transcription. However, as a virion protein, VP16 cannot be successfully transported to ganglion nuclei with viral genomes because of certain physical losses. In the absence of VP16, multi-protein complex (VP16, Oct-1 and HCF) cannot be formed, thus reducing IE gene transcription.46 Therefore, the IE gene encoding the ICP0 protein would be low. Low levels of ICP0 can promote both LAT and lytic gene expression as well as total histone and heterochromatin loading on the HSV genome in latently infected ganglia.14 In contrast, in latently infected neuron nuclei, the low levels of ICP0 expression can promote the expression of LAT and the accumulation of H3K27me3/H3K9me3 heterochromatin to silence lytic gene expression.47,48 Together, LAT expression may play a pivotal role in the establishment and maintenance of latency through regulation of lytic gene expression.49
LAT Function Mediated by Its miRNAs, sRNAs, lncRNAs, sncRNAs and ORFs
The expression of LAT is crucial. LAT has several other expression products, such as microRNA (miRNA), small RNA (sRNA), long non-coding RNA (lncRNA), short non-coding RNA (sncRNA), and LAT open reading frames (ORF). Numerous studies have supported that LATs encode multiple RNAs or ORFs. It is noteworthy that these RNAs or ORFs may play an crucial role in the latency-reactivation cycle.50 Here, we summarize the five main types of LAT expression products that are closely related to HSV latency (Figure 3).
MicroRNAs
MicroRNAs (miRNAs, miRs) are a class of 21 to 24 nucleotides (nt) noncoding RNAs that regulate the stability or the translational efficiency of target messenger RNAs (mRNAs).51 In transfection assays, miRNAs originating from the LAT region can repress expression of viral lytic proteins.31 LAT can promote the establishment of latent infection via LAT-encoded miRNAs.52 Firstly, HSV-1-miR-H2, which has been associated with the regulation of latency, binds to viral ICP0 mRNA and inhibits its expression.53 In addition, LAT miR-H2 has been experimentally shown to inhibit the production of ICP0 protein by downregulating ICP0 translation, but not transcription.54,55 Some report that LAT miR-H2 functions to decrease HSV-1 pathogenesis and to help maintain HSV latency in Swiss Webster mice.56 HSV-1-miR-H6 can target ICP4 mRNA, inhibiting the expression of the transcription factor ICP4 crucial to E and L gene transcription, leading to blocking virus lytic infection and decreasing IL-6 expression; therefore, HSV-1-miR-H6 promotes HSV-1 latency.57
Interestingly, miRNA derived from the LAT region helps to inhibit the expression of cleavage function by blocking the synthesis of two viral proteins, ICP0 and ICP34.5, consequently facilitating the maintenance of latency.4 Recently, acutely and latently expressed miRNAs encoded by HSV-2 LAT was reported to inhibit ICP34.5 expression, whereas two other miRNAs encoded by HSV-1 LAT also mapped antisense to the ICP34.5 region.58 In HSV-2 LAT miRNAs, miR-H3 and miR-H4, as well as the corresponding HSV-1 LAT-encoded miRNAs, were processed from the primary LAT and a L/ST that traverses the viral long/short repeat connection targeting ICP34.5.59 However, LAT miRNAs are negatively regulated by ICP4.54 ICP4 inhibits miRNAs targeted at ICP34.5 and activates lytic virus genes, which suggests that ICP4 plays a critical role in the transition between HSV latency and activation.59 Meanwhile, the LAT plays an anti-apoptotic role by encoding miRNA, which prevents neuronal apoptosis and maintains the integrity of the neuron to preserve HSV in a latent state.60
In addition, studies have shown that miRNAs can protect nerve cells from apoptosis mediated by CD8+ T cells.61 During HSV latency, some neurons are surrounded by CD8+ T cells, which are thought to infiltrate neurons to help maintain HSV latency by inhibiting virus reactivation.61 Surprisingly, when large numbers of CD8+ T cells were injected into cytotoxic lytic granules of latently infected neurons, apoptosis and neuronal death did not occur. Therefore, it has been hypothesized that the protective effect of latently infected neurons from cytotoxic CD8+ T cell killing is attributed to the anti-apoptotic activity of LAT.61 The ability of LAT to block granzyme B induced apoptosis was certified,62 given that granzyme B is the major factor released by CD8+ T cells and responsible for cytotoxic and soluble granulocyte-mediated apoptosis. Granzyme B can directly cleave and activate caspase-3 as well as other proteins known to regulate apoptosis.63 In addition, LAT encoded miRNA plays an anti-apoptotic role by down-regulating the expression of functional transforming growth factor (TGF)-β and SMAD3 in the TGF-β pathway.64 Some results indicate that miRNAs encoded by HSV-1 LAT gene regulates apoptosis of infected cells by modulating TGF-β signaling pathway, leading to the persistence of HSV in a latent form in sensory neurons.65
In conclusion, LAT encoded miRNAs make HSV latent through the downregulation of ICP0, ICP34.5, ICP4, TGF-β and SMAD3.
sRNA
HSVs encode small RNAs (sRNA1 and sRNA2), which have extensive secondary structure and a stem-loop structure and are larger than micro-RNAs. LAT sRNA may interact with mitochondria, inhibit the release of pro-apoptotic factors, and enable cells to avoid the fate of apoptosis and may survive, thus giving the HSV an opportunity to be latent in host cells.66 Further, LAT sRNA1 and sRNA2 play essential roles in the latency-reactivation cycle because the sRNAs inhibit productive infection and apoptosis, which is consistent with previous studies showing that the same mutation affects its anti-apoptotic function in a large LAT fragment.67,68 In addition, sRNA1-LAT and/or sRNA2-LAT inhibit the ICP0 gene expression by partial base complementation to maintain latency.50 Surprisingly, when the wild-type LAT sRNA1 was co-transfected with the plasmid expressing LAT sRNA2, the intrinsic pathway of neuro-2A cells apoptosis was blocked, thereby protecting neuro-2A cells from cold-shock induced apoptosis.67 Finally, LAT sRNA1 was also predicted to hybridize with VP16 and UL8 mRNA (proteins essential for viral DNA replication), suggesting that LAT sRNA1 may reduce the expression of related viral genes. Further, LAT sRNA2 and miRNA-H6 inhibit the expression of ICP4, which may contribute to establishing and maintaining viral latency via inhibiting the expression of lysed genes and protecting infected neurons from cell death.50,67 In short, LAT sRNAs can successfully make HSV latent due to its anti-apoptotic and anti-lytic effects.
lncRNA
HSV LAT is a set of long non-coding RNA (lncRNA), which is the only transcript known to accumulate at a large amount during latency.69 LncRNA play a key role in maintaining latent infected neurons.43 During latency, neurons accumulate 1.5/2.0kb of non-coding RNA of LAT. Relevant studies have reported that the transcription of viral noncoding RNAs can also promote the lytic gene silencing during the incubation period by inducing facultative heterochromatin.47 LncRNAs help form heterochromatin in HSV chromosome, inhibit the expression of lytic genes, and make the virus latent.47 Furthermore, lncRNAs are also considered to play a critical role in promoting the activation of HSV.70 Together, lncRNAs of LAT makes HSV latent or reactivated by modification of heterochromatin.
SncRNAs
The herpes virus entry mediator (HVEM) is one of several cell surface proteins that HSV uses for attachment/entry. HVEM regulates cellular immune responses and can also increase cell survival. SncRNAs of LAT were distributed on HVEM promoter. The increase of HVEM would down-regulate the immune spongy body in the latent microenvironment and increase the survival rate of latent infected cells.71 Experiments showed that two sncRNAs encoded by LAT can induce the expression of HVEM in latently infected mice in the trigeminal ganglia.50 In addition, the effect of LAT on latency was significantly reduced in HVEM-deficient mice. The results showed that LAT regulated latent infection and reactivation of HSV to some extent by upregulating the expression of HVEM mRNA levels,72 thereby increasing the survival of latently infected cells and preventing the activation of effector T cells.71 Therefore, the results indicate that the LAT-HVEM relationship is a new mechanism directly involved in the HSV latency steady state pathway.71 Interestingly, one study found that LAT sncRNA1 cooperates with the RNA sensor, retinoic acid-inducible gene I (RIG-I), to stimulate IFN-β promoter activity and nuclear factor-kB (NF-kB) dependent transcription in human or mouse cells.73 LAT sncRNA2 stimulated RIG-I induction of NF-kB dependent transcription in mouse neuroblastoma cells (Neuro-2A) but not human 293 cells.73 In Neuro-2A cell model, both sncRNAs work together with RIG-I to suppress cold-shock induced apoptosis.73 Together, two sncRNAs derived from LAT contribute to the establishment of virus latency by combining RIG-I to IFN-β.
LAT ORF
All the LATs of HSV-1/2 have multiple ORFs, which encode proteins related to activation and function, and are similar to ICP0 and of great significance in the process of latent infection.74 The expression of LAT ORFs can complement the mutation of ICP0, which instead greatly reduces the growth of the virus.75 Furthermore, LAT ORFs can activate wild-type phenotype in the rabbit ocular model.76 In addition, LAT ORF overexpression could down-regulate the expression of virus lytic genes by downregulating the ICP4.77 Interestingly, LAT ORF has a significant role in the HSV life cycle. LAT ORF can interact with HSV-1 proteins, RNA or DNA, to promote viral growth. Studies have shown that LAT-encoded ORF enhances HSV-1 replication in epithelial and neuronal cells, especially in cells that normally do not allow for virus growth.78 Further, LAT-encoded ORF2 can enhance the transactivation of lytic proteins.76 Experiment results showed that ORF knocking-down reduced the protective effect of LAT on caspase 8- or caspase 9-induced apoptosis.68 Knocking out all eight ORFs blocked the antiapoptotic activity of LAT more significantly and effectively than knocking out only ORFs 1–2 or ORFs 3–8.
In summary, we summarized the function of LAT mediated by LAT encoded miRNA, sRNA, lncRNA, sncRNAs and ORFs, and elaborated on how they regulate gene expression at both transcriptional and post-transcriptional levels to influence the latency and reactivation of HSVs (Figure 3).
Application of HSV LAT Deletion Mutants in HSV Vaccines and oHSVs
After HSV infection, it may become latent in the ganglia, and then reactivate and relapse under some non-specific stimulation, causing associated clinical symptoms. Because HSVs have a latency and easy activation mechanism, it cannot achieve a radical cure in clinical practice. Therefore, it is necessary to study the latency and reactivation mechanisms of HSVs. Many researchers have paid great attention to the vital role of LATs in latency and recurrence of HSVs for the effective targeted HSV treatment and making safe oHSVs. Here, we have extensively outlined the different effects of LAT deletions from HSVs and oHSVs as shown in Tables 1 and 2.
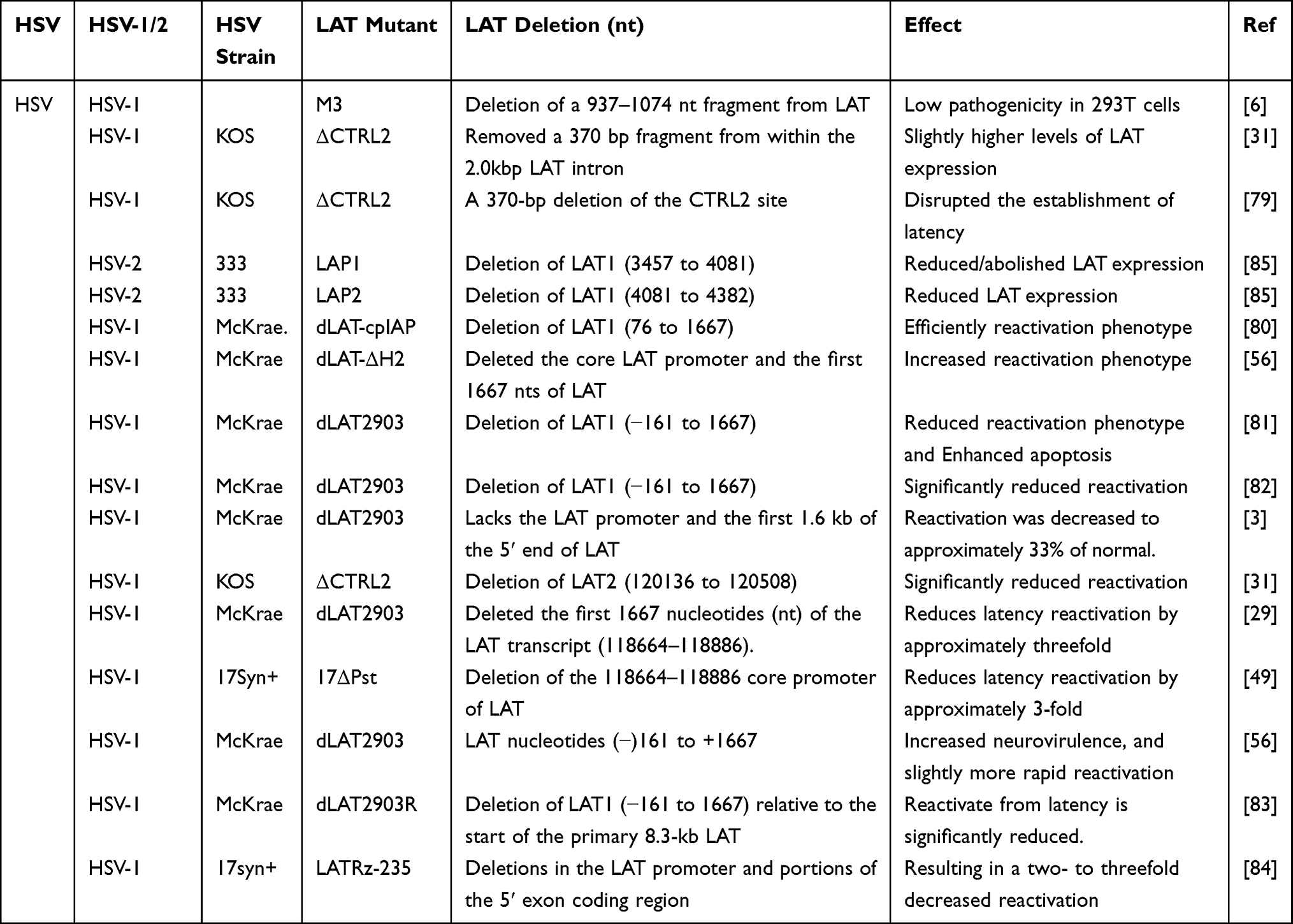 |
Table 1 Application of Deleted LAT in HSV |
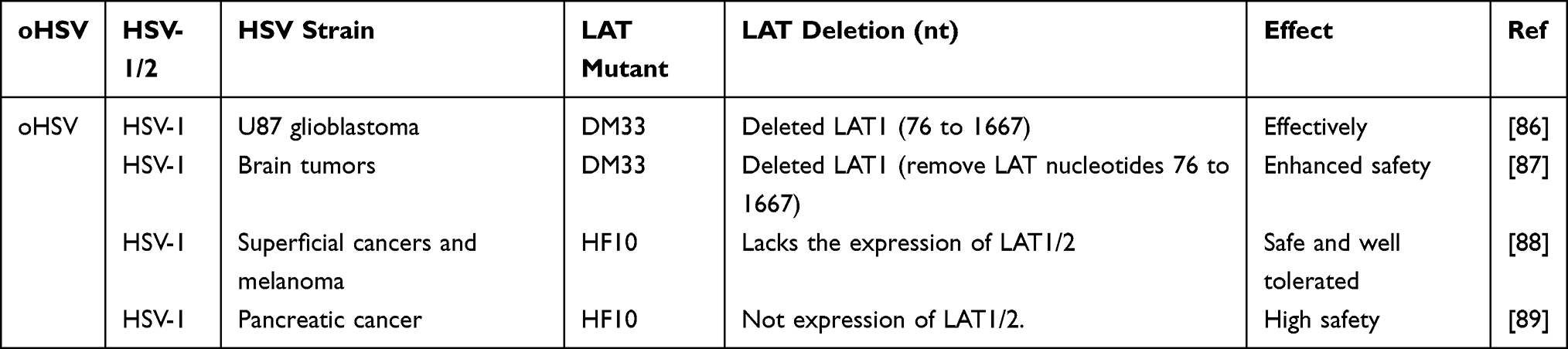 |
Table 2 Application of Deleted LAT in oHSV |
These effects have been confirmed in published literature (Table 1). In terms of the HSV-1, one experiment has made a LAT deletion via removing a 937–1074nt fragment from LAT to receive the M3 mutant. The M3 mutant was harvested from the infected human embryonic kidney cell line 293T cells, which showed that it is an attenuated strain with low pathogenicity during both acute and latent infections. The above result supports the use of the M3 mutant as a candidate for the development of an HSV-1 vaccine.6 Interestingly, another experiment built a CTRL2 deletion mutant virus by removing a 370-bp fragment from the 2.0-k bp LAT intron to form ΔCTRL2 mutant virus, which showed slightly higher levels of LAT expression during latent infection. However, this was indistinguishable during reactivation.31 Conversely, a CTRL2 deletion mutant virus (a 370-bp) resulted in the encroachment of heterochromatin onto the LAT region of the genome to disrupt the establishment of latency, and had lower survival rate.79 However, Lee et al31 showed that their ΔCTRL2 recombinant can establish an equivalent latent infection in the mouse TG and displayed no replication defect during the lytic infection.
Deletion of LAT in HSV affects not only HSV latency but also virus reactivation. One study has shown that deletion of LAT1 (76 to 1667) can effectively reactivate phenotype.80 Similarly, deletion of the core promoter of LAT and the first 1667 nts of LAT can increase the reactivation phenotype.56 Inversely, deletion of LAT (−161 to 1667) reduced the reactivation phenotype and enhanced apoptosis, resulting in the decrease in HSV reactivation rate.81 Other studies have shown that deletion of LAT can significantly reduce HSV reactivation.31,82,83 In addition, studies have also shown that deletion of LAT can significantly reduce reactivation to about 33% of the normal level.3 Similarly, partial LAT deletion mutation has been shown to lead to an approximately threefold increase of latency in the TG of wild-type mice infected with LAT(+) virus compared with mice infected with LAT(-) virus.29,49,84 On the contrary, another experiment has constructed a LAT deletion mutant virus by removing a −161 to +1667 fragment to form the dLAT2903 mutant virus, which showed increased neurovirulence and slightly more rapid reactivation.56 In HSV-2, removing LAT1/LAT2 significantly reduced LAT expression during productive infections and abolished LAT expression during latency.85
As the causal infectious virus, deletions of the LAT gene of HSVs will significantly reduce the latency and reactivation of the viral infection, which will alleviate the pain of clinical herpes patients. As oHSVs, there is growing interest about the genetic modification of LAT to make an effective and safe oHSVs for cancer treatment (Table 2). Recent studies have shown that modifying the LAT gene may significantly improve the targeting and safety of oHSVs for cancer therapy.86–89 For example, the oHSV DM33 was made by deleting γ34.5 and LAT genes, which removed LAT nucleotides 76 to 1667. DM33 was safe and effective in treating human U87 glioblastomas and brain tumors.86,87 Moreover, oHSV HF10 lacks the expression of LATs, showing a strong safe and well-tolerated effect in the treatment for superficial cancers, melanoma and pancreatic cancer.88,89
We look forward to developing a better therapeutic regimen based on the experimental application of HSV and oHSV by removing LAT through in-depth study, to give full play to its therapeutic potential. However, more clinical trials are needed to drive the development of HSV LAT-based diagnostic tests and therapeutic interventions to benefit patients with herpes and tumors.
Conclusion and Perspectives
In summary, latent infection of HSV is a multi-step complex process. Here, we have systematically reviewed the latest development of the multiple pathways to HSV latency, and the structure and expression, regulation, and function of the LAT, and the genetic analysis and engineering of LAT. LAT performs a wide range of function by different molecular mechanisms, including transcriptional and post-transcriptional regulation. Significantly, we first summarized the three HSV latent infection pathways, systematically analyzed the structure and expression of the LAT1 and LAT2 of HSV-1 and HSV-2, proposed the regulation of LAT expression by four pathways, and dissected the function of LAT mediated by five LAT products of miRNAs, sRNAs, lncRNAs, sncRNAs and ORFs. We further analyzed that application of HSV LAT deletion mutants in HSV vaccines and oHSVs. Deleting LAT significantly reduced the latency and reactivation of HSV, providing a new idea for the future development of HSV vaccines and safe oHSVs.
In different heterogeneous tissues, the translation level of LAT ORFs are strictly limited in time and space. Whether LAT can be translated into protein is uncertain. For example, studies have shown that LAT gene may encode a protein with a latency-related function, as has been demonstrated for bovine herpes virus.90 Currently, there has been one report of a LAT-encoded protein in latently infected primary neuron cultures,91 but it has not been confirmed. Fortunately, HSV-1 LAT is consistently detected in abundance during latency and in the neurons of infected humans, mice, and rabbits.92 In latency models, the abundance of the LAT RNAs increases during the establishment of latency and can be detected in neurons of ganglia from the sacral, thoracic, and lumbar regions of recently deceased humans.93 These reports provide us with the knowledge about the future application in establishing latency and reactivation models to further uncover the veil of HSV latency and reactivation.
It is well established that LAT controls apoptosis,94 which likely occurs via the type I interferon (IFN) pathway.95 The type I IFN pathway consisting mainly of IFN-α and -β is activated by several HSV-1 glycoproteins.95 However, the HSV-1 LAT transcript was shown to delay several IFN-α subtypes, including IFN-α1, IFN-α4, IFN-α5, IFN-α6, and IFN-α14, as well as IFN-β, during an acute infection in vitro, and IFN-α4 and IFN-β in vivo.96 In addition, LAT is responsible for CD8+ T cells immune exhaustion, which is also essential for latency establishment.97 Previous reports have shown a LAT-dependent elevation of the immune exhaustion markers CD4, CD8-α, CD8-β, PD-1, and Tim-3 as well as their associated cytokines IL-2, IL-21, IFN-γ, and TNF- α.98 Therefore, LAT interacting with immune system is considered to be related to HSV latency, which deserves more investigation in depth.
For restraining LAT latent infection, many promising vaccine approaches are being explored in animals and humans, such as modifying the LAT gene to make its latent period longer or reduce its reactivation for the appropriate vaccine type against HSVs.99,100 The M3 virus, which has a partial LAT deletion, was harvested from the infected human embryonic kidney cell line 293T cells and exhibited the lowest pathogenicity during acute and latent infection. Thus, LAT mutant is an ideal candidate for HSV vaccine development. In addition, the new CRISPR/Cas9 technology can now be used to make engineered HSVs for better vaccine and oHSV development.
In addition, RNA silencing and RNA interference have been discovered in plants, animals and human beings.101,102 Although it is shown that sRNA1-LAT and/or sRNA2-LAT inhibit the ICP0 gene expression by partial base complementation to maintain latency,61 its underlining mechanism is not known. We propose that RNA silencing or RNA interference may play an important role in HSV latency and reactivation, which is worth validating in future.
Acknowledgments
This work was partly supported by grants from the National Natural Science Foundation of China (81872412 to XHW). We thank Guangzhou Key Medical Discipline Construction Project (CSZ) and Yangtze University Fellowship to graduate student ZY.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ying-ying Wang Y-NL, Xin H-Y, Xin H-Y. Identification of putative UL54 (ICP27) transcription regulatory sequences binding to Oct-1, v-Myb, Pax-6 and hairy in herpes simplex viruses. J Cancer. 2019;10(2):430–440. doi:10.7150/jca.29787
2. Jones C. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res. 1998;51(1):81–133. doi:10.1016/s0065-3527(08)60784-8
3. BenMohamed L, Osorio N, Srivastava R, Khan AA, Simpson JL, Wechsler SL. Decreased reactivation of a herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) mutant using the in vivo mouse UV-B model of induced reactivation. J Neurovirol. 2015;21(5):508–517. doi:10.1007/s13365-015-0348-9
4. Kawamura Y, Bosch-Marce M, Tang S, Patel A, Krause PR. Herpes simplex virus 2 latency-associated transcript (LAT) region mutations do not identify a role for LAT-associated MicroRNAs in viral reactivation in guinea pig genital models. J Virol. 2018;92(14):89–181. doi:10.1128/JVI.00642-18
5. Yan C, Luo Z, Li W, et al. Disturbed Yin-Yang balance: stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta pharmaceutica Sinica B. 2020;10(3):383–398. doi:10.1016/j.apsb.2019.06.005
6. Xu X, Guo Y, Fan S, et al. Attenuated phenotypes and analysis of a herpes simplex virus 1 strain with partial deletion of the UL7, UL41 and LAT genes. Virol Sin. 2017;32(5):404–414. doi:10.1007/s12250-017-3947-1
7. Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog. 2014;10(7):e1004237. doi:10.1371/journal.ppat.1004237
8. Watanabe D, Goshima F. Oncolytic virotherapy by HSV. Adv Exp Med Biol. 2018;1045(4):63–84.
9. Liu XQ, Xin HY, Lyu YN, et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018;25(1):1950–1962. doi:10.1080/10717544.2018.1534895
10. Zhang Y, Xin Q, Zhang JY, et al. Transcriptional regulation of latency-associated transcripts (LATs) of herpes simplex viruses. J Cancer. 2020;11(11):3387–3399. doi:10.7150/jca.40186
11. Liu BM, Han ZQ, Branston RH, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. doi:10.1038/sj.gt.3301885
12. Andtbacka RHI, Kaufman HL, Frances C, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi:10.1200/JCO.2014.58.3377
13. Cabrera JR, Charron AJ, Leib DA. Neuronal subtype determines herpes simplex virus 1 latency-associated-transcript promoter activity during latency. J Virol. 2018;92(13):00430. doi:10.1128/JVI.00430-18
14. Raja P, Lee JS, Pan D, et al. Protein regulates the structure of latent viral chromatin. mBio. 2016;7(3):e00633–16. doi:10.1128/mBio.00633-16
15. Lou W, Ji F, Fu J, Han Z, Di W, Zhang N. Retracted Article: transcriptional retargeting of herpes simplex virus for cell-specific replication to control cancer. J Cancer Res Clin Oncol. 2018;144(2):1–11. doi:10.1007/s00432-017-2566-4
16. Guo Q, Weng W, Zeng F, Zheng R, Luo Y, Du J. Research of UL54-specific siRNA on herpes simplex virus type II replication. Int J Dermatol. 2011;50(3):362–366. doi:10.1111/j.1365-4632.2010.04732.x
17. Preston CM. Repression of viral transcription during herpes simplex virus latency. J General Virol. 2000;81(Pt 1):1–19.
18. Sturm RA, Das G, Herr W. The ubiquitous octamer-binding protein Oct- 1 contains a POU domain with a homeo box subdomain. Genes Dev. 1988;2(12):1582. doi:10.1101/gad.2.12a.1582
19. Ryan AK, Rosenfeld MG. POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev. 1997;11(10):1207. doi:10.1101/gad.11.10.1207
20. Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78(18):9689–9696. doi:10.1128/JVI.78.18.9689-9696.2004
21. Kenyon C, Colebunders R, Hens N. Determinants of generalized herpes simplex virus-2 epidemics: the role of sexual partner concurrency. Int J STD AIDS. 2013;24(5):375–382. doi:10.1177/0956462412472816
22. Margolis TP, Yumi I, Li Y, Vicky V, Krause PR. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol. 2007;81(4):1872. doi:10.1128/JVI.02110-06
23. Inman M, Perng GC, Henderson G, et al. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol. 2001;75(8):3636–3646. doi:10.1128/JVI.75.8.3636-3646.2001
24. Perng GC, Jones C, Ciacci-Zanella J, et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287(5457):1500–1503. doi:10.1126/science.287.5457.1500
25. Knipe DM, Lieberman PM, Jung JU, et al. Snapshots: chromatin control of viral infection. Virology. 2013;435(1):141–156. doi:10.1016/j.virol.2012.09.023
26. Katz JP, Bodin ET, Coen DM. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J Virol. 1990;64(9):4288–4295. doi:10.1128/jvi.64.9.4288-4295.1990
27. Preston CM. Repression of viral transcription during herpes simplex virus latency. J Gen Virol. 2000;81(1):1–19. doi:10.1099/0022-1317-81-1-1
28. Sawtell NM. The probability of in vivo reactivation of herpes simplex virus type 1 increases with the number of latently infected neurons in the ganglia. J Virol. 1998;72(8):6888. doi:10.1128/JVI.72.8.6888-6892.1998
29. Lee DH, Ghiasi H. An M2 Rather than a TH2 Response Contributes to Better Protection against Latency Reactivation following Ocular Infection of Naive Mice with a Recombinant Herpes Simplex Virus 1 Expressing Murine Interleukin-4. J Virol. 2018;92(10):e00051–18. doi:10.1128/JVI.00051-18
30. Lee JS, Raja P, Knipe DM. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio. 2016;7(1):e02007–15. doi:10.1128/mBio.02007-15
31. Lee JS, Raja P, Pan D, et al. CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. Mbio. 2018;9(1):e02372–17. doi:10.1128/mBio.02372-17
32. Henderson G, Jaber T, Carpenter D, Wechsler SL, Jones C. Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J Neurovirol. 2009;15(5–6):439. doi:10.3109/13550280903296353
33. Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76(2):717–729. doi:10.1128/JVI.76.2.717-729.2002
34. Doina A, Kent JR, Gartner JJ, Fraser NW. The stable 2-kb LAT intron of herpes simplex stimulates the expression of heat shock proteins and protects cells from stress. Virology. 2006;350(1):26–33. doi:10.1016/j.virol.2006.02.001
35. Sumit B, Wong AC, Steitz JA. Drosophila hnRNP A1 homologs Hrp36/Hrp38 enhance U2-type versus U12-type splicing to regulate alternative splicing of the prospero twintron. Proc Natl Acad Sci U S A. 2009;106(8):2577–2582. doi:10.1073/pnas.0812826106
36. Xu X, Fan S, Zhou J, et al. The mutated tegument protein UL7 attenuates the virulence of herpes simplex virus 1 by reducing the modulation of alpha-4 gene transcription. Virol J. 2016;13(1):152. doi:10.1186/s12985-016-0600-9
37. Liu X-Q, Xin H-Y, Lyu Y-N, et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018;25(1):1950–1962.
38. Diao L, Zhang B, Xuan C, et al. Activation of c-Jun N-terminal kinase (JNK) pathway by HSV-1 immediate early protein ICP0. Exp Cell Res. 2005;308(1):196–210. doi:10.1016/j.yexcr.2005.04.016
39. Sumiko KS, Ryuichi K, Tokiko O, Saya N, Kenichi N, Hiroshi K. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J Biol Chem. 2008;283(11):6988–6996. doi:10.1074/jbc.M707514200
40. Dziennis S, Alkayed NJ. Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev Neurosci. 2008;19(4–5):341–362. doi:10.1515/REVNEURO.2008.19.4-5.341
41. Hunt D, Raivich G, Anderson PN. Activating transcription factor 3 and the nervous system. Front Mol Neurosci. 2012;5(7):548. doi:10.3389/fnmol.2012.00007
42. Takaya A, Naohide O, Wataru Y, Masaru R. Rapid and preferential induction of ATF3 transcription in response to low doses of UVA light. Biochem Biophys Res Commun. 2003;310(4):1168–1174. doi:10.1016/j.bbrc.2003.09.143
43. Minfeng S, Te D, Grace Z, Bernard R. Role of activating transcription factor 3 in the synthesis of latency-associated transcript and maintenance of herpes simplex virus 1 in latent state in ganglia. Proc Natl Acad Sci U S A. 2015;112(39):E5420. doi:10.1073/pnas.1515369112
44. Millhouse S. ATF/CREB elements in the herpes simplex virus type 1 latency-associated transcript promoter interact with members of the ATF/CREB and AP-1 transcription factor families. J Biomed Sci. 1998;5(6):451–464. doi:10.1007/BF02255935
45. Washington SD, Edenfield SI, Lieux C, et al. Depletion of the insulator protein CTCF results in HSV-1 reactivation in vivo. J Virol. 2018;92(11):
46. Hafezi W, Lorentzen EU, Eing BR, et al. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012;8(5):e1002679. doi:10.1371/journal.ppat.1002679
47. Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83(16):8182. doi:10.1128/JVI.00712-09
48. Cliffe AR, Coen DM, Knipe DM. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio. 2013;4(1):169–172. doi:10.1128/mBio.00590-12
49. Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM. CTCF binding sites in the herpes simplex virus 1 genome display site-specific CTCF occupation, protein recruitment, and insulator function. J Virol. 2018;92(8):e00156–18. doi:10.1128/JVI.00156-18
50. Peng W, Vitvitskaia OD, Wechsler S, Jones C. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol. 2008;14(1):41–52. doi:10.1080/13550280701793957
51. Ambros V, microRNAs: tiny regulators with great potential. Cell. 2001;107(7):823–826. doi:10.1016/S0092-8674(01)00616-X
52. Liu Y, Yang HL, Zhong FF, Fan JY. Anti-apoptotic function of herpes simplex virus −2 latency-associated transcript RL1 sequence and screening of its encoded microRNAs. Clin Exp Dermatol. 2016;41(7):782–791. doi:10.1111/ced.12671
53. Jiang X, Brown D, Osorio N, et al. A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J Neurovirol. 2015;21(2):199–209. doi:10.1007/s13365-015-0319-1
54. Tang S, Patel Akrause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009;83(3):1433–1442. doi:10.1128/JVI.01723-08
55. Jennifer Lin U, Kramer MF, Igor J, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454(7205):780. doi:10.1038/nature07103
56. Jiang X, Brown D, Osorio N, Hsiang C, BenMohamed L, Wechsler SL. Increased neurovirulence and reactivation of the herpes simplex virus type 1 latency-associated transcript (LAT)-negative mutant dLAT2903 with a disrupted LAT miR-H2. J Neurovirol. 2016;22(1):38–49. doi:10.1007/s13365-015-0362-y
57. Duan F, Liao J, Huang Q, Nie Y, Wu K. HSV-1 miR-H6 inhibits HSV-1 replication and IL-6 expression in human corneal epithelial cells in vitro. Clin Dev Immunol. 2012;2012(6):192791. doi:10.1155/2012/192791
58. Shuang T, Bertke AS, Amita P, Kening W, Cohen JI, Krause PR. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci U S A. 2008;105(31):10931–10936. doi:10.1073/pnas.0801845105
59. Tang S, Bosch-Marce M, Patel A, Margolis TP, Krause PR. Characterization of HSV-2 primary miRNA transcript regulation. J Virol. 2015;85(2):261–265.
60. Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76(2):717–729.
61. Xianzhi J, Aziz Alami C, Chinhui H, et al. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol. 2011;85(5):2325. doi:10.1128/JVI.01791-10
62. Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26(1):389–420. doi:10.1146/annurev.immunol.26.021607.090404
63. Pardo J, Aguilo JI, Anel A, et al. The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes Infect. 2009;11(4):452–459. doi:10.1016/j.micinf.2009.02.004
64. Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2006;442(7098):82. doi:10.1038/nature04836
65. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–129. doi:10.1038/ng1001-117
66. Khvorova A, Kwak Y-G, Tamkun M, Majerfeld I, Yarus M. RNAs that bind and change the permeability of phospholipid membranes. Proc Natl Acad Sci U S A. 1999;96(19):10649–10654. doi:10.1073/pnas.96.19.10649
67. Wenwen S, Mariana SES, Tareq J, et al. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol. 2009;83(18):9131–9139. doi:10.1128/JVI.00871-09
68. Carpenter D, Henderson G, Hsiang C, et al. Introducing point mutations into the ATGs of the putative open reading frames of the HSV-1 gene encoding the latency associated transcript (LAT) reduces its anti-apoptosis activity. Microb Pathog. 2008;44(2):98–102. doi:10.1016/j.micpath.2007.07.001
69. Cheng JT, Wang L, Wang H, et al. Insights into biological role of LncRNAs in epithelial-mesenchymal transition. Cells. 2019;8(10):1178. doi:10.3390/cells8101178
70. Watson ZL, Washington SD, Phelan DM, et al. In vivo knock-down of the HSV-1 latency-associated transcript reduces reactivation from latency. J Virol. 2018;21:
71. Allen SJ, Antje RK, Mott KR, et al. Interactions between herpesvirus entry mediator (TNFRSF14) and latency-associated transcript during herpes simplex virus 1 latency. J Virol. 2014;88(4):1961. doi:10.1128/JVI.02467-13
72. Kubat NJ. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr Is hyperacetylated during latency independently of LAT transcription. J Virol. 2004;78(22):12508–12518. doi:10.1128/JVI.78.22.12508-12518.2004
73. Leticia Frizzo DS, Clinton J. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res. 2013;175(2):101–109. doi:10.1016/j.virusres.2013.04.005
74. Hobbs WE, Brough DE, Kovesdi I, Deluca NA. Efficient activation of viral genomes by levels of herpes simplex virus ICP0 insufficient to affect cellular gene expression or cell survival. J Virol. 2001;75(7):3391–3403. doi:10.1128/JVI.75.7.3391-3403.2001
75. Thomas SK, Lilley CE, Latchman DS, Coffin RS. A protein encoded by the herpes simplex virus (HSV) type 1 2-kilobase latency-associated transcript is phosphorylated, localized to the nucleus, and overcomes the repression of expression from exogenous promoters when inserted into the quiescent HSV genome. J Virol. 2002;76(8):4056–4067. doi:10.1128/JVI.76.8.4056-4067.2002
76. Perng GC, Ab GHN, Wechsler SL, Slanina SM. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J Virol. 1996;70(3):2014–2018. doi:10.1128/jvi.70.3.2014-2018.1996
77. Rajčáni J, Andrea V, Ingeborg R. Peculiarities of herpes simplex virus (HSV) transcription: an overview. Virus Genes. 2004;28(3):293. doi:10.1023/B:VIRU.0000025777.62826.92
78. Thomas S, Gough G, Coffin R, S. R. Herpes simplex virus latency-associated transcript encodes a protein which greatly enhances virus growth, can compensate for deficiencies in immediate-early gene expression, and is likely to function during reactivation from virus latency. J Virol. 1999;73(8):6618–6625. doi:10.1128/JVI.73.8.6618-6625.1999
79. Washington SD, Singh P, Johns RN, et al. The CCCTC binding factor, CTRL2, modulates heterochromatin deposition and the establishment of HSV-1 latency in vivo. J Virol. 2019;93(13):e00415–19. doi:10.1128/JVI.00415-19
80. Jin L, Perng G-C, Mott KR, et al. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol. 2005;79(19):12286–12295. doi:10.1128/JVI.79.19.12286-12295.2005
81. Mott KR, Osorio N, Jin L, et al. The bovine herpesvirus-1 LR ORF2 is critical for this gene’s ability to restore the high wild-type reactivation phenotype to a herpes simplex virus-1 LAT null mutant. J Gen Virol. 2003;84(11):2975–2985. doi:10.1099/vir.0.19421-0
82. Carpenter D, Singh S, Osorio N, et al. A speculated ribozyme site in the herpes simplex virus type 1 latency-associated transcript gene is not essential for a wild-type reactivation phenotype. J Neurovirol. 2008;14(6):558–562. doi:10.1080/13550280802275912
83. Harrison KS, Zhu L, Thunuguntla P, Jones C. Herpes simplex virus 1 regulates beta-catenin expression in TG neurons during the latency-reactivation cycle. PLoS One. 2020;15(3):e0230870. doi:10.1371/journal.pone.0230870
84. Watson ZL, Washington SD, Phelan DM, et al. In vivo knockdown of the herpes simplex virus 1 latency-associated transcript reduces reactivation from latency. J Virol. 2018;92(16):e00812–18. doi:10.1128/JVI.00812-18
85. Yoshikawa T, Stanberry LR, Bourne N, Krause PR. Downstream regulatory elements increase acute and latent herpes simplex virus type 2 latency-associated transcript expression but do not influence recurrence phenotype or establishment of latency. J Virol. 1996;70(3):1535–1541. doi:10.1128/jvi.70.3.1535-1541.1996
86. Samoto KEM. Perng GC. A Herpes simplex virus type 1 mutant with γ34.5 and LAT deletions effectively oncolyses human U87 glioblastomas in nude mice. Neurosurgery. 2002;50(3):599–606.
87. Samoto K, Perng GC, Ehtesham M, et al. A herpes simplex virus type 1 mutant deleted for γ34.5 and LAT kills glioma cells in vitro and is inhibited for in vivo reactivation. Cancer Gene Ther. 2001;8(4):269–277. doi:10.1038/sj.cgt.7700306
88. Ragab EI, Yoshinori N, Itzel B-V, et al. Genomic signature of the natural oncolytic herpes simplex virus HF10 and its therapeutic role in preclinical and clinical trials. Front Oncol. 2017;7(1):149–156. doi:10.3389/fonc.2017.00149
89. Hirooka Y, Kasuya H, Ishikawa T, et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. 2018;18(1):596. doi:10.1186/s12885-018-4453-z
90. Schang LM, Hossain A, Jones C. The latency-related gene of bovine herpesvirus 1 encodes a product which inhibits cell cycle progression. J Virol. 1996;70(6):3807–3814. doi:10.1128/jvi.70.6.3807-3814.1996
91. Doerig C, Pizer LI, Wilcox CL. An antigen encoded by the latency-associated transcript in neuronal cell cultures latently infected with herpes simplex virus type 1. J Virol. 1991;65(5):2724–2727. doi:10.1128/jvi.65.5.2724-2727.1991
92. Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol. 2018;92(24):01451. doi:10.1128/JVI.01451-18
93. Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens. 2017;6(2):2076–2817. doi:10.3390/pathogens6020024
94. Jiang X, Chentoufi AA, Hsiang C, et al. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol. 2011;85(5):2325.
95. Kati T, Sariah A, Kevin M, et al. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol. 2019;93(10):e00103–19. doi:10.1128/JVI.00103-19
96. Peng W, Benmohamed L, Perng GC, et al. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal Ganglia of acutely infected mice. J Virol. 2005;79(10):6162–6171. doi:10.1128/JVI.79.10.6162-6171.2005
97. Jaggi U, Matundan HH, Tormanen K, Wang S, Ghiasi H. Expression of murine CD80 by HSV-1 in place of LAT can compensate for latency-reactivation and anti-apoptotic functions of LAT. J Virol. 2019;94(6):e01798–19.
98. Mott KR, Bresee CJ, Allen SJ, Benmohamed L, Wechsler SL, Ghiasi H. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol. 2009;83(5):2246–2254. doi:10.1128/JVI.02234-08
99. Stanfield B, Kousoulas KG. Herpes simplex vaccines: prospects of live-attenuated HSV vaccines to combat genital and ocular infections. Curr Clin Microbiol Rep. 2015;2(3):125–136. doi:10.1007/s40588-015-0020-4
100. Johnston C, Gottlieb SL, Wald A. Status of vaccine research and development of vaccines for herpes simplex virus. Vaccine. 2016;34(26):2948–2952. doi:10.1016/j.vaccine.2015.12.076
101. Guo Z, Li Y, Ding SW, Small RN. A-based antimicrobial immunity. Nat Rev Immunol. 2019;19(1):31–44. doi:10.1038/s41577-018-0071-x
102. Li Y, Lu J, Han Y, Fan X, Ding SW. RNA interference functions as an antiviral immunity mechanism in mammals. Science. 2013;342(6155):231–234. doi:10.1126/science.1241911
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.