Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Multidisciplinary Management of Fabry Disease: Current Perspectives
Authors Paim-Marques L, de Oliveira RJ, Appenzeller S ![]()
Received 19 October 2021
Accepted for publication 3 February 2022
Published 10 March 2022 Volume 2022:15 Pages 485—495
DOI https://doi.org/10.2147/JMDH.S290580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Luciana Paim-Marques,1 Rodrigo Joel de Oliveira,2 Simone Appenzeller2
1Department of Pediatrics, University of Florida, Gainesville, FL, USA; 2Department of Orthopedics, Rheumatology and Traumatology- School of Medical Sciences and University of Campinas (UNICAMP), São Paulo, Brazil
Correspondence: Simone Appenzeller, Department of Medicine, School of Medical Science, State University of Campinas, Cidade Universitária, Campinas, CEP 13083-970, SP, Brazil, Fax +55 19 3289-1818, Email [email protected]
Abstract: Fabry disease (FD) is a rare, recessive X-linked, multisystemic lysosomal storage disorder (LSD) that results from a deficiency in the hydrolase alpha-galactosidase A (α-GalA) caused by a GLA gene variant. The progressive accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in organs such as skin, kidney, brain, joints, vascular walls and eyes are responsible for the wide spectrum of clinical manifestations, often unspecific. In result, clinically relevant and life-threatening complications, such as malignant ventricular arrhythmia, sudden cardiac death, end stage kidney failure and stroke may occur. In this review, we will describe the clinical features and the current perspectives in the multidisciplinary management Of FD patients.
Keywords: Fabry disease, multidisciplinary care, alfa-galactosidase
Introduction
Fabry disease (FD) is a rare, recessive X-linked, multisystemic lysosomal storage disorder (LSD) that results from a deficiency in the hydrolase alpha-galactosidase A (α-GalA) caused by a GLA gene variant. Also recognized as Anderson-Fabry disease, it was initially described by doctors Johannes Fabry and William Anderson in 1898.1,2
FD is found in all races and regions worldwide, and its diagnosis frequency is difficult to obtain. Its birth prevalence was initially estimated at 1:40.000–170.000 for the general population.3 However, neonatal screenings have found a markedly higher incidence of FD than previously expected, with 1 to 3.000 males in Austria, 1 in 3.100 males reported in Northwestern Italy, and 1 in 1.500 among males in Taiwan.4,5 Despite increased frequency in newborn screening, they have mostly been suspected of having the later-onset phenotype. This review aims to describe the clinical features and the current perspectives in the multidisciplinary management of FD patients.
Physiopathology
Currently, more than 1000 GLA gene variants were identified in the chromosomal region Xq22.1. In addition, there are splicing alterations, deletions, translocations, complex gene rearrangement, and point missense variants, responsible for 60% of all changes with a single amino acid substitution in the α-GalA protein.6
The α-GalA deficiency in patient lysosomes with FD causes a progressive accumulation of the glycosphingolipid globotriaosylceramide (Gb3), that starts in the fetal stage of development, into the plasma and lysosomes of a variety of cells, particularly within the vascular endothelium and smooth muscle cells. Gb3 inclusion was detected in the fetal kidney cardiomyocytes, cornea, and placental tissue suggesting a prenatal Gb3 lysosomal accumulation.7,8 This process leads to significant cellular dysfunction, chronic inflammatory process, vessel occlusion, ischemia, and organ damage.
FD is characterized by a multidomain phenotype caused by GLA variants, missense, nonsense, small or large deletions.9 The amount of α-Gal A substrate accumulated on an individual patient, along with its residual enzymatic activity (< 1%) determines the disease severity in FD. Enzyme activity between 1 and 30%, leads to atypical phenotypes of FD.10,11
In addition, the correlations of genotype-phenotype in FD seems to suffer influence for several factors, such as the high proportion of private mutation, the large intra and inter-familial phenotype heterogeneity associated with the same mutation, and high prevalence disease-related complications.12 Therefore, variants associated with complete loss of enzyme function seem to be associated with the “classic” phenotype. In contrast, residual enzyme activity may lead to a slow progression of the disease, thought to be more related to the “late-onset” FD. Clinical symptoms may present at any age with a broad phenotypic variability, even among patients with the same variants.
Clinical Presentation
The disorder has described two major phenotypes: “classic” and the “later-onset.” In the classic phenotype, affected males have little or no residual α-GalA activity and present symptoms in early childhood, around 1 to 3 years old.8 In addition, there are acroparesthesias and angiokeratoma- a characteristic skin rash in Anderson-Fabry disease consisting of numerous tiny red lesions starting in the genital area and later becoming widespread; decreased sweating (hypohidrosis), and heat intolerance. The initial clinical symptoms are followed by gastrointestinal symptoms, particularly diarrhea, vomiting, abdominal pain, exercise intolerance, and ocular abnormalities (cornea verticillata), characteristic corneal dystrophy caused by Gb3 accumulation. With aging, there’s progressive involvement of multiple systems such as kidneys (renal insufficiency), heart (hypertrophic cardiomyopathy), and cerebrovascular disease such as transient ischemic attacks and strokes predominantly in the vertebrobasilar system.7
FD may also present with mild nonspecific symptoms frequently affecting the musculoskeletal system. Peripheral neuropathic pain, fever, arthritis, and elevated erythrocyte sedimentation rate (ESR) can also be observed.1–4. In the absence of specific enzyme replacement therapy (ERT), the mean survival of patients is about 55 years for males and nearly 70 years for females.13
The later-onset phenotype, previously called atypical variants of FD, is characterized by males with residual enzyme activity that develop the disease in adulthood, between the fourth and seventh decade of life, with end-stage renal, cardiovascular, or cerebrovascular diseases,14 but without acroparesthesias and angiokeratoma.15
Nonetheless, those patients may evolve with isolated cardiac or renal involvement due to specific GLA gene variants, making the diagnosis even more difficult. Furthermore, the correlation between genotype-phenotype is not fully established yet.15
As a recessive X-linked disease, it affects predominantly male patients. However, in the female carrier, clinical manifestations range from mild to severe symptoms, attributed to the pattern of X-chromosome randomized inactivation in each female cell.16 In addition, this process is a significant predictor of the natural history of the disease in female patients.16
Diagnosis
These varieties of clinical presentation may influence the diagnostic methods, clinical signs, life expectancies, and prognosis of FD patients. Nonetheless, heterozygous females have a better prognosis than affected males.
Although the α-Gal activity in plasma or leukocytes is the preferred method for diagnosis confirmation in male patients, this method is frequently inconclusive in female patients with low to normal enzyme levels. Therefore, male patients should have their diagnosis confirmed with enzyme titers, while female patients should have a direct genetic test.16
Early diagnosis is essential for adequate treatment. In addition to screen family members, physicians must be aware of the different phenotypes and clinical manifestations (Figure 1). Nonetheless, laboratory evaluation should be available worldwide to support clinical suspicion.
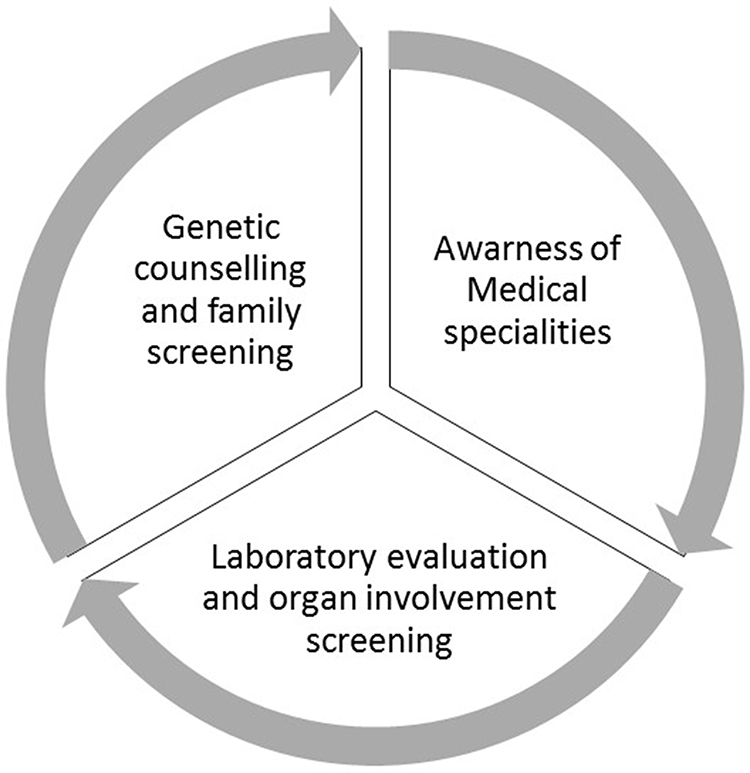 |
Figure 1 Early diagnosis of FD involves family screening and genetic counselling, physician awareness and availability of genetic testing. |
Clinical Manifestations of FD by Organ Systems
Cardiac Involvement
The heart is commonly affected in FD. Therefore, cardiac involvement represents one of the major causes of morbidity and mortality in adult patients with FD, and they are mainly related to the later-onset FD phenotype.17
The most frequent heart manifestation and the most specific one related to FD is the left ventricle hypertrophy (LVH), better defined initially as a concentric LVH caused by the accumulation of Gb3 leading to a ventricular wall thickening. This structural process is followed by cardiac remodeling, and finally, eccentric hypertrophy. In some cases, it may mimic hypertrophic cardiomyopathy.17 The prevalence of FD in hypertrophic cardiomyopathy patients ranges from 0.5–4%.18
FD also causes other cardiac abnormalities, including conduction abnormalities and valvular dysfunction.17 Conduction defects and arrhythmias are seen in childhood as early manifestations. They can be detected by ECG/Holter, including a shortening of the PR interval, likely due to accelerated AV nodal conduction. Another early manifestation of FD is diastolic dysfunction present throughout the disease and found in late stages.17 However, in adults, rhythm disturbances such as supraventricular tachyarrhythmia, conduction defects, bradycardia, ventricular arrhythmias, and sudden cardiac death are commonly associated with FD.19 In non-treated patients, they evolve to structural abnormalities 5. There are descriptions of a high incidence of pacemaker implantation mainly due to bradyarrhythmia.17 FD’s end cardiac stage is cardiac fibrosis, a severe and irreversible change associated with a poor prognosis. According to a Taiwan study in 2016, 38.1% of males and 16.7% of females with late-onset FD had cardiac fibrosis, even without LVH.20
Neurological, Cerebrovascular and Psychiatric Involvement
The pathophysiology underlying neurologic involvement in FD is still poorly delineated. The progressive accumulation of Gb-3 within the endothelium and vascular smooth muscles of intracranial blood vessels causes vascular regulation and endothelial dysfunctions, associated with multifocal involvement of small blood vessels, the proliferation of smooth muscles cells, and higher incidence of thrombosis.21 However, secondary factors, including abnormalities in cerebral blood flow velocity, prothrombotic state, and increased production of reactive oxygen species, have also been described as contributing to the development of stroke in FD.22–24
Neuropathic pain is, in general, the earliest FD manifestation in childhood, described as earlier as three years old of age or less.25 This pain is a distal, small fiber sensory peripheral neuropathy, characterized by acroparesthesias, burning pain, sensory loss starting in the palms of the hands or the soles of the feet. The patients describe two types of pain: a severe episodic burning pain that begins in the extremity and evolves to the entire body, lasting for hours or days and resistant to analgesics, and a chronic pain characterized by burning and tingling, and paresthesia.26 The acute attacks described are also called Fabry crises.27 In most patients, Fabry crises are triggered by sudden changes in temperature, cold or heat exposure, exercises, fatigue, stress.28 For that reason, even the difference in skin temperature after a vigorous workout is avoided by patients with the condition.29 In crises triggered or associated with fever, the patients may also have an elevated erythrocyte sedimentation rate.
This pain is very frequent among adults with FD (80%) and also highly prevalent in heterozygous females within their two first decades of life.27
The most frequent cerebrovascular abnormalities in FD are ischemic stroke and transient ischemic attacks, with up to 13–16% of the patients presenting those events over their life course.30 Although heterozygous females have shown to have a higher frequency of cerebrovascular events27 initially, a comprehensive study has shown the stroke prevalence is around 6.9% for FD males and 4.3% for FD females with the median age at the first stroke of 39 and 46 years, respectively,31 which is consistent with males exhibiting an earlier and severe FD. Nonetheless, among the females who had a stroke, 17% experienced their first stroke episode before 30 years old. Regardless, there are reports of early cerebral involvement in an 8-year-old boy with white matter lesions on the brain magnetic resonance.32
Among patients with cryptogenic stroke, the prevalence of FD ranges from 0.6% to 11.1%, with an average of 4.5% in men and 3.4% in women.33 Other cerebrovascular manifestations, ranging from mild to severe, were also reported in FD patients, such as headaches, vascular dementia, vertigo/dizziness, hearing loss, transient ischemic attacks, cervical dissection, thrombotic events;31 and hemorrhagic events with hemorrhagic strokes being described to be more common among men (16.9%) than women (6.9%).34 Nonetheless, neuropsychiatric symptoms are described, such as depression, learning difficulties, memory deficits, and despite remarkable structural alterations, individuals with FD only show mild cognitive deficits.31
Neuroimaging findings of FD patients, including brain magnetic resonance (MR), helped better understand the structural and cerebrovascular changes in FD. Although MR is the most used imaging, functional MR, MR spectroscopy, transcranial Doppler, diffusor tensor imaging, and positron emission tomography can also be used in FD.14 There have been reports of white matter lesions, periventricular, deep white matter changes, and small vascular infarctions, including in older and recent events. Additionally, radiological findings may include large vessels ectasia and tortuosity (dolichoectasia).14 Peculiar critical imaging related to neuroradiological aspects of FD is the pulvinar hyperintensities on T1-weighted images.14 The above-described neurological lesions are unspecific and can also be associated with other cardiovascular risk factors such as hypertension, coagulation cascade dysfunction, hyperlipidemia, or any aspect that enhances atherogenesis.
Studies described the presence of severe depression, excessive guilt, fatigue, occupational difficulty, suicidal ideation, and depressed mood in female patients with FD.35 In addition, cognitive decline and dementia were described in older patients.36
Renal Involvement
The deposition of Gb3 occur in all renal cells, such as endothelial cells, vascular smooth muscle cells, mesangial cells, podocytes, and distal tubular epithelial cells,37 and this accumulation has been noted as early as during fetal development.31 The first signs of renal disease in affected males with the classic phenotype usually manifest between 10 and 20 years old. The Gb3 in the tubular epithelial cells is associated with an early concentration defect. The deposition in podocytes, mesangial, and endothelial cells cause basal membrane thickening and glomerulosclerosis with microalbuminuria and subsequent proteinuria. Glomerular sclerosis exceeds 50% with tubulointerstitial damage progression between the third and fourth decades. With aging, between fourth- and fifth-decade males, patients develop chronic renal insufficiency and end-stage renal failure.38 In heterozygous females and late-onset phenotypes, the age of presentation of renal disease is variable, and the decline in filtering capacity is slower than in the classic phenotype. End-stage renal failure is less prevalent in females with FD, despite some reported cases.29
Gastrointestinal Manifestations
Gastrointestinal (GI) symptoms are considered early clinical manifestations of FD in childhood, which usually persist until adulthood. In some studies, GI problems were reported in 33% of males, and 20% of females with the classic phenotype of FD,5 while males with later-onset phenotypes reported GI symptoms much less frequently at baseline as compared with classic patients.39 Children’s frequent complaints are abdominal pain after meals, diarrhea, nausea, vomits, and which can be a significant cause of anorexia. Pediatric patients, in general, alternate diarrhea, constipation, bloating, and acute abdominal pain, frequently misdiagnosed with irritable bowel disease or appendicitis. Female carriers become symptomatic in early childhood or adolescence.40
Dermatologic Involvement
The most common skin manifestations are angiokeratomas, telangiectasis, and hypohidrosis. Angiokeratoma is a tiny red lesion, typically distributed on the posterior and anterior trunk, limbs, umbilicus, and genitals, but also can occur anywhere, including lips, oral mucosa, and conjunctiva.31 They become more numerous and more prominent with age. It is present in 66% of males, and 36% of female patients with the classic phenotype,14 generally beginning in adolescence. Histologically, these small superficial angiomas are caused by dermal vessel dilatation, secondary to cumulative damage of the vascular endothelial cells of the skin.41 The Fabry outcome Survey (FOS) reported telangiectasia and subcutaneous edema in pediatric patients.41 Hypohidrosis was the most prominent sweat abnormality described in the FOS study, more pronounced in adults. However, anhidrosis rarely occurred (25% of males and 4% of females) in younger patients. Both are associated with heat intolerance.41
Ocular Involvement
FD’s most typical ocular involvement is the cornea verticillata, present in affected boys and more than 70% of carrier females.7 These whorl-like, cream-colored opacities are in the corneal layers and distributed in a vortex pattern, explaining the “cornea verticillata” denomination. This ocular abnormality is caused by the presence of intra-epithelial deposits of dense laminated cytoplasmic inclusions, freely in the cytoplasm and membrane-bound.7
Retinal vessel tortuosity, conjunctival vascular changes, lenticular opacities, eyelid edema, optic atrophy, papilledema, peripapillary edema, nystagmus, internuclear ophthalmoplegia, and retinal vein occlusion were also described in FD patients.42 Preliminary FOS data suggested that vessel tortuosity correlates with the progression of the systemic disease, reflecting disease severity. However, there are too little available data to confirm this observation.7
Other Organs Involvement
Furthermore, as previously discussed, the α-Gal A deficiency results in multi-organ damage due to progressive accumulation of glycosphingolipids, mainly globotriaosylceramide (Gb3), in different organs.7
Auditory problems such as tinnitus, and hearing loss, are more prevalent in girls (55%) than in boys (39%) with FD and may present already in childhood.42
Pulmonary findings were also described in FD, such as dyspnea, wheezing, dry cough, sleep-disordered, considered secondary to accumulation of glycolipids in small-medium sized airway cells, causing obstructive airway disease (with or without restrictive changes). In the advanced stage of FD disease, patients may need additional chest X-ray/CT scan to monitor lung abnormalities such as pulmonary infiltrations, fibrosis, and air trapping.36
Lymphatic tissue can present with lymphedema in all or part of a limb, below the eyes, and pitting edema, resulting from an accumulation of glycolipids in the lymph vessels and fragmentation of the micro lymphatic network.41
Among the musculoskeletal symptoms described in FD, there are chronic inflammatory joint and bone diseases. In addition, there are descriptions of poly, oligo and monoarticular arthritis, gout, osteoporosis, and degenerative joint conditions, as well as neurologic arthropathies such as Charcot’s foot,43 Heberden-like nodules,44 and myositis.45
Currently, there has been shown the coexistence of FD and autoimmune disease, such as systemic lupus erythematosus,46 rheumatoid arthritis,47 autoimmune hypothyroidism,48 Ig A nephropathy,49 and granulomatosis with polyangiitis.50 Patients with FD and rheumatic manifestations have a significant delay in FD diagnosis that can last up to 16 years or more. The most common associated mutations observed in FD patient cohort in Brazil, presenting with rheumatic manifestations, were R118C and A143T.51 A recent study of our group observed a frequency of 1.12% of FD in a cohort of juvenile idiopathic arthritis (JIA) patients.52
Therefore, to the wide spectrum of clinical manifestations, FD patients should be followed by multidisciplinary team (Figure 2).
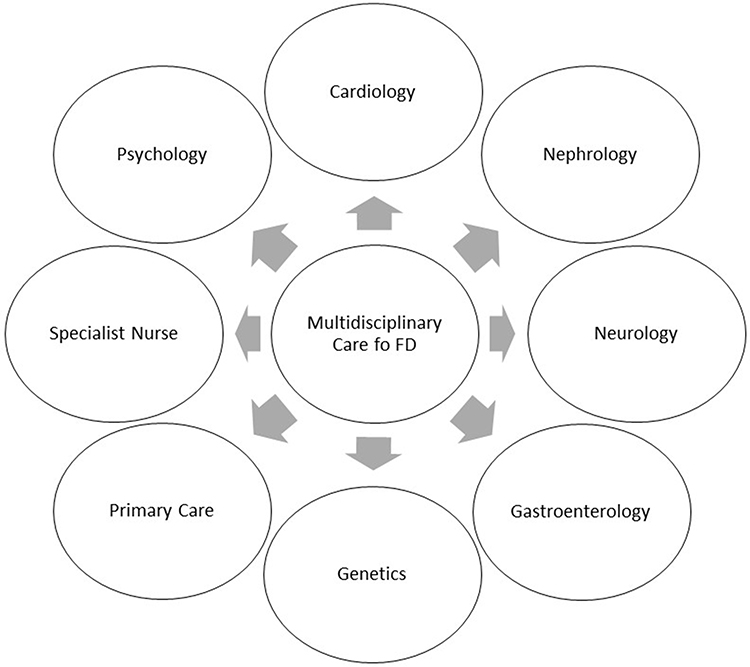 |
Figure 2 Multidisciplinary care in FD patients. |
Therapy and Multidisciplinary Care
As a lysosomal X-linked disease that requires the α-Gal-A enzyme activity measurement in affected females or a genetic study of the GLA gene in affected males, it is challenging to establish FD’s diagnosis, especially in early ages. A high index of suspicion is necessary at the early stages of the disease, and screening in high-risk patients is a cost-effective strategy for identifying FD patients.53
Once the diagnosis is established, the main goals of the FD young patient’s treatment must include eliminating Gb3 accumulation and alleviating the bothersome symptoms. Clinical trials have shown that Gb3 levels can be normalized in different cells with the enzyme replacement therapy (ERT) with agalsidase-α (Replagal; Shire Human Genetic Therapies, Cambridge, MA; 0.2 mg/kg per infusion) and agalsidase-β (Fabrazyme; Genzyme Corporation, Cam- bridge, MA, 1 mg/kg per infusion), such as multiple renal cell types, and vascular endothelial cells of kidney, heart, and skin. Although, still a point to be discussed, some clinical studies suggested ERT benefit and safety in the pediatric population, as early as 2.5 years old.32 Subsequently, patients have also reported significant improvements in gastrointestinal symptoms, and neuropathic pain, and normalization of heat intolerance/heat-pain, vibration detection thresholds, and sweat function also have been reported.54 However, the ERT does not seem to affect cerebrovascular damages, which different hypotheses could explain including small therapeutic effect of the ERT in the brain; or due to its size, the α-Gal A enzyme (>100kd) replaced cannot reach the entire thickness of the blood vessels; or even, the short half-life of the replaced enzyme (one to two days) and its intermittent replacement may be insufficient to control the Gb3 accumulation.55
In some trials, the ERT may slow the yearly decline in glomerular filtration rate and reduce the left ventricular mass. However, little evidence exists that ERT can change cardiac disease, including rhythm and conduction defects and the risk of cardiac death in adults. Once irreversible changes are established, as cardiac fibrosis, ERT may be no longer effective.56 In patients refractory to ERT, the treatment goal is to stabilize the existing complications or slow their progression to improve survival and quality of life.
In childhood, although there are no clear guidelines about when to start ERT, the newborn screening has brought the possible early ERT onset.57 The main objective is to prevent symptoms and to decrease unnecessary medications. This decision should be made in conjunction with the family and according to the clinical judgment of the physician specialist, taking into consideration the family history and patient’s symptoms, phenotype, and genotype. The family should also be aware that the disease causes an irreversible, progressive accumulation of Gb3 with end-organ damages and the possible improvement with early ERT.53
It is valid to remember that most FD end-organs damages are nonspecific, and diagnosis and treatment of prevalent diseases should be included in regular care (Figure 3).
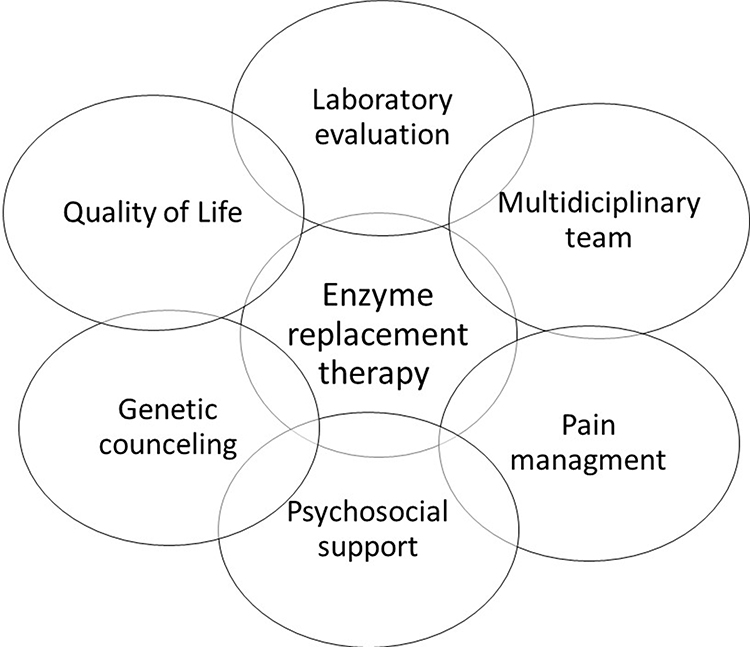 |
Figure 3 Treatment aspects in FD. |
Patients should receive general recommendations such as smoking cessation, increased aerobic exercises, a heart-healthy diet, and treatment of diabetes, hypertension, and dyslipidemia. However, no trials show substantial benefits of those measurements in morbidity/mortality specific for FD.58 According to the Fabry Registry, hypertension is a strong predictor of cardiovascular events such as myocardial infarction, heart failure, and cardiac-related death.59 When part of the patient’s symptoms, it should be treated with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for kidney protection. Another significant symptom is angina. Patients should receive calcium channel blockers, nitrates, and low-dose aspirin. FD can cause bradyarrhythmia and chronotropic incompetence, though beta-blockers can be used, however, with careful evaluation.60
FD’s proteinuria has been shown promising results when using the combination of angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blocker in association with ERT reducing proteinuria to less than 500mg/24h.61 Blood pressure control is also imperative for kidney function preservation and prevention of other vascular complications. For those with end-stage renal function, renal transplantation can be lifesaving, as in any other disorder. It is worth the comment that many FD patients are diagnosed with the pre-scanning workup for a live donor or cadaver renal transplant.54 Therefore, a nephrologist should be part of the multidisciplinary care of an FD patient.
Antiplatelet agents should be used to prevent strokes in patients with FD. Clopidogrel, aspirin, and long-acting dipyridamole have been effective, especially on those patients with a family history of stroke. As discussed above, this is a crucial point since the ERT does not reduce stroke incidence.54
Neuropathic pain decreases over time in adults and children, although no complete resolution of symptoms is observed. Neurologist or pain specialist follow these patients and in general, the treatment consists of low-dose anticonvulsants (Neurontin, carbamazepine, gabapentin, and lamotrigine), alpha-lipoic acid with vitamin B complex, and holistic therapy.62
A dermatology evaluation is valuable since FD’s skin manifestations are an essential feature of the disease.31 In addition to support diagnosis, dermatologists follow-up are important in the symptomatic control of peripheral edema and lymphedema and in the use of vascular lasers to improve the appearance and psychological impact of angiokeratomas. Yet unfortunately, there are little data in FOS on the benefits of ERT on the skin. However, in general, the initial and preliminary data on the effect of agalsidase alpha are encouraging.42
Pancreatic enzymes can prevent postprandial diarrhea in FD patients. The nonsteroidal anti-inflammatory drug seems to have lower effectiveness. In the case of associated chronic arthritis, methotrexate can be used with good results (personal observations). Narcotics should be avoided, despite being effective for musculoskeletal and neuropathic pain.54
The increased presence of depression in FD patients prompts the need for a multidisciplinary approach, including psychologists and psychiatrist. Therefore, further studies were recommended to establish the etiology, incidence, and response to pharmacotherapy and psychotherapy in FD.35
Quality of Life in FD
Patients who suffer from FD have a lower quality of life (QoL) compared to healthy individuals. Neuropathic pain, anhidrosis, ESRD and older age are predictors of decreased QoL.63 Studies analyzing ERT effect on QoL in FD are inconclusive.63 Depressive symptoms, pain and sleep disorders are frequently observed in FD, associated with disease severity, and interfere with QoL.64 Analysis of QoL and patient related outcomes (PRO) are essential in cost effectiveness analyses and should be included in research agenda to aid in reimbursement decisions.
Conclusion
FD is a multi-organ disease resulting in several organ clinical manifestations with progressive and irreversible damages. Therefore, a multidisciplinary team of experienced professionals should take care of FD patients. However, early diagnosis is essential for adequate treatment. High index of suspicion is necessary by physicians to suspect of FD. Laboratory tests have to widely available to support diagnosis.
Abbreviations
α-GalA, Alpha-galactosidase A; ANA, antinuclear antibodies; ECG, Electrocardiogram; Echo, Echocardiogram; FD, Fabry disease; Gb3, Globotriaosylceramide; JIA, juvenile idiopathic arthritis; PCR, Polymerase Chain Reaction.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Conselho Nacional Pesquisa Desenvolvimento-Brasil (CNPq 306723/2019-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) Finance Code 001.
Disclosure
All authors declare that they have no conflicts of interest related to the study.
References
1. Anderson WA. A case of angiokeratoma. Br J Dermatol. 1898;18:113–117.
2. Fabry J. Ein Beitrag zur Kenntnis der Purpura hammarrhagica nodularis. Arch Dermatol Syphilol. 1898;43:187–200.
3. Biegstraaten M, Arngrímsson R, Barbey F, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015;10:36. doi:10.1186/s13023-015-0253-6
4. Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40. doi:10.1086/504601
5. Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2(5):450–456. doi:10.1161/CIRCGENETICS.109.862920
6. Chien YH, Lee NC, Chiang SC, Desnick RJ, Hwu WL. Fabry disease: incidence of the common later-onset α-galactosidase A IVS4+919G→A mutation in Taiwanese newborns- superiority of DNA-based to enzyme-based newborn screening for common mutations. Mol Med. 2012;18(1):780–784. doi:10.2119/molmed.2012.00002
7. Sestito S, Ceravolo F, Concolino D. Anderson-Fabry disease in children. Curr Pharm Des. 2013;19(33):6037–6045. doi:10.2174/13816128113199990345
8. Vedder AC, Strijland A, Vd Bergh Weerman MA, Florquin S, Aerts JM, Hollak CE. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis. 2006;29(1):106–111. doi:10.1007/s10545-006-0196-0
9. Shimotori M, Maruyama H, Nakamura G, et al. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum Mutat. 2008;29(2):331. doi:10.1002/humu.9520
10. Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801–807. doi:10.1046/j.1523-1755.2003.00160.x
11. Yoshitama T, Nakao S, Takenaka T, et al. Molecular genetic, biochemical, and clinical studies in three families with cardiac Fabry’s disease. Am J Cardiol. 2001;87:71–75. doi:10.1016/S0002-9149(00)01275-3)
12. Viggiano E, Politano L. X chromosome inactivation in carriers of Fabry disease: review and meta-analysis. Int J Mol Sci. 2021;22(14):7663. doi:10.3390/ijms22147663)
13. Vedder AC, Linthorst GE, van Breemen MJ, et al. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis. 2007;30(1):68–78. doi:10.1007/s10545-006-0484-8
14. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi:10.1186/1750-1172-5-30
15. Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631–1641. doi:10.1681/ASN.2016090964
16. Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44–54. doi:10.1111/cge.12613
17. Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373.
18. Monserrat L, Gimeno-Blanes JR, Marín F, et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50(25):2399–2403. doi:10.1016/j.jacc.2007.06.062
19. Shah JS, Hughes DA, Sachdev B, et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. 2005;96(6):842–846. doi:10.1016/j.amjcard.2005.05.033
20. Hsu TR, Hung SC, Chang FP, et al. Later onset Fabry disease, cardiac damage progress in silence: experience with a highly prevalent mutation. J Am Coll Cardiol. 2016;68(23):2554–2563. doi:10.1016/j.jacc.2016.09.943
21. Moore DF, Kaneski CR, Askari H, Schiffmann R. The cerebral vasculopathy of Fabry disease. J Neurol Sci. 2007;257(1–2):258–263. doi:10.1016/j.jns.2007.01.053
22. DeGraba T, Azhar S, Dignat-George F, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. 2000;47(2):229–233.
23. Hilz MJ, Kolodny EH, Brys M, Stemper B, Haendl T, Marthol H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J Neurol. 2004;251(5):564–570. doi:10.1007/s00415-004-0364-9
24. Moore DF, Herscovitch P, Schiffmann R. Selective arterial distribution of cerebral hyperperfusion in Fabry disease. J Neuroimaging. 2001;11(3):303–307. doi:10.1111/j.1552-6569.2001.tb00051.x
25. Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006;95(1):86–92. doi:10.1080/08035250500275022
26. Charrow J. A 14-year-old boy with pain in hands and feet. Pediatr Ann. 2009;38(4):190–192. doi:10.3928/00904481-20090401-01
27. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242. doi:10.1111/j.1365-2362.2004.01309.x
28. Hilz MJ, Stemper B, Kolodny EH. Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. Pain. 2000;84(2–3):361–365. doi:10.1016/s0304-3959(99)00236-5
29. MacDermot J, MacDermot KD. Neuropathic pain in Anderson-Fabry disease: pathology and therapeutic options. Eur J Pharmacol. 2001;429(1–3):121–125. doi:10.1016/s0014-2999(01)01312-7
30. Buechner S, Moretti M, Burlina AP, et al. Central nervous system involvement in Anderson-Fabry disease: a clinical and MRI retrospective study. J Neurol Neurosurg Psychiatry. 2008;79(11):1249–1254. doi:10.1136/jnnp.2008.143693
31. Hsu TR, Niu DM. Fabry disease: review and experience during newborn screening. Trends Cardiovasc Med. 2018;28(4):274–281. doi:10.1016/j.tcm.2017.10.001
32. Laney DA, Peck DS, Atherton AM, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med. 2015;17(5):323–330. doi:10.1038/gim.2014.120
33. Shi Q, Chen J, Pongmoragot J, Lanthier S, Saposnik G. Prevalence of Fabry disease in stroke patients–a systematic review and meta-analysis. J Stroke Cerebrovasc Dis. 2014;23(5):985–992. doi:10.1016/j.jstrokecerebrovasdis.2013.08.010
34. Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40(3):788–794. doi:10.1161/STROKEAHA.108.526293
35. Sadek J, Shellhaas R, Camfield CS, Camfield PR, Burley J. Psychiatric findings in four female carriers of Fabry disease. Psychiatr Genet. 2004;14(4):199–201. doi:10.1097/00041444-200412000-00006
36. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–427. doi:10.1016/j.ymgme.2018.02.014
37. Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62(6):1933–1946. doi:10.1046/j.1523-1755.2002.00675.x
38. West M, Nicholls K, Mehta A, et al. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. 2009;20(5):1132–1139. doi:10.1681/ASN.2008080870
39. Hopkin RJ, Feldt-Rasmussen U, Germain DP, et al. Improvement of gastrointestinal symptoms in a significant proportion of male patients with classic Fabry disease treated with agalsidase beta: a Fabry Registry analysis stratified by phenotype. Mol Genet Metab Rep. 2020;25:100670. doi:10.1016/j.ymgmr.2020.100670
40. Hoffmann B, Schwarz M, Mehta A, Keshav S. Fabry Outcome Survey European Investigators. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5(12):1447–1453. doi:10.1016/j.cgh.2007.08.012
41. Orteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol. 2007;157(2):331–337. doi:10.1111/j.1365-2133.2007.08002.x
42. Keilmann A, Hajioff D, Ramaswami U, Investigators FOS. Ear symptoms in children with Fabry disease: data from the Fabry Outcome Survey. J Inherit Metab Dis. 2009;32(6):739. doi:10.1007/s10545-009-1290-x
43. Thévenot C, Crrouzet J, Villiaumey J, Avouac B, Le Charpentier Y. V. M. C. Les Manifestations Articulaires de la Maladie de Fabry. A Propos de Deux Observations. SEM.HÔP PARIS; 1992:486–493.
44. Lidove O, Zeller V, Chicheportiche V, et al. Musculoskeletal manifestations of Fabry disease: a retrospective study. Joint Bone Spine. 2016;83(4):421–426. doi:10.1016/j.jbspin.2015.11.001
45. Chimenti C, Padua L, Pazzaglia C, et al. Cardiac and skeletal myopathy in Fabry disease: a clinicopathologic correlative study. Hum Pathol. 2012;43(9):1444–1452. doi:10.1016/j.humpath.2011.09.020
46. Chatre C, Filippi N, Roubille F, Pers YM. Heart involvement in a woman treated with hydroxychloroquine for systemic lupus erythematosus revealing Fabry disease. J Rheumatol. 2016;43(5):997–998. doi:10.3899/jrheum.151357
47. Martinez P, Aggio M, Rozenfeld P. High incidence of autoantibodies in Fabry disease patients. J Inherit Metab Dis. 2007;30(3):365–369. doi:10.1007/s10545-007-0513-2
48. Katsumata N, Ishiguro A, Watanabe H. Fabry disease superimposed on overt autoimmune hypothyroidism. Clin Pediatr Endocrinol. 2011;20(4):95–98. doi:10.1297/cpe.20.95
49. Yin G, Wu Y, Zeng CH, Chen HP, Liu ZH. Coexistence of Fabry disease and IgA nephropathy: a report of two cases. Ir J Med Sci. 2014;183(4):671–675. doi:10.1007/s11845-014-1161-9
50. Hanaoka H, Hashiguchi A, Konishi K, Ishii T, Kuwana M. A rare association between Fabry’s disease and granulomatosis with polyangiitis: a potential pathogenic link. BMC Nephrol. 2014;15:157.
51. Rosa Neto NS, Bento JCB, Pereira RMR. Higher rate of rheumatic manifestations and delay in diagnosis in Brazilian Fabry disease patients. Adv Rheumatol. 2020;60(1):7. doi:10.1186/s42358-019-0111-7
52. Paim-Marques L, Cavalcante AV, Verçosa I, et al. Frequency of Fabry disease in a juvenile idiopathic arthritis cohort. Pediatr Rheumatol Online J. 2021;19(1):91. doi:10.1186/s12969-021-00563-9
53. Gonçalves MJ, Mourão AF, Martinho A, et al. Genetic screening of mutations associated with Fabry disease in a nationwide cohort of juvenile idiopathic arthritis patients. Front Med (Lausanne). 2017;4:12. doi:10.3389/fmed.2017.00012
54. Schiffmann R, Fuller M, Clarke LA, Aerts JM. Is it Fabry disease? Genet Med. 2016;18(12):1181–1185. doi:10.1038/gim.2016.55
55. Schiffmann R, Swift C, Wang X, Blankenship D, Ries M. A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J Inherit Metab Dis. 2015;38(6):1129–1136. doi:10.1007/s10545-015-9845-5
56. Rombach SM, Smid BE, Linthorst GE, Dijkgraaf MG, Hollak CE. Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: a systematic review and meta-analysis: effectiveness of ERT in different disease stages. J Inherit Metab Dis. 2014;37(3):341–352. doi:10.1007/s10545-014-9677-8
57. Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–555. doi:10.1203/PDR.0b013e318183f132
58. Acharya D, Doppalapudi H, Tallaj JA. Arrhythmias in Fabry cardiomyopathy. Card Electrophysiol Clin. 2015;7(2):283–291. doi:10.1016/j.ccep.2015.03.014
59. Patel MR, Cecchi F, Cizmarik M, et al. Cardiovascular events in patients with Fabry disease natural history data from the Fabry registry. J Am Coll Cardiol. 2011;57(9):1093–1099. doi:10.1016/j.jacc.2010.11.018
60. Hagège A, Réant P, Habib G, et al. Fabry disease in cardiology practice: literature review and expert point of view. Arch Cardiovasc Dis. 2019;112(4):278–287. doi:10.1016/j.acvd.2019.01.002
61. Warnock DG, Thomas CP, Vujkovac B, et al. Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy. J Med Genet. 2015;52(12):860–866. doi:10.1136/jmedgenet-2015-103471
62. Hoffmann B, Garcia de Lorenzo A, Mehta A, et al. Effects of enzyme replacement therapy on pain and health related quality of life in patients with Fabry disease: data from FOS (Fabry Outcome Survey). J Med Genet. 2005;42(3):247–252. doi:10.1136/jmg.2004.025791
63. Arends M, Hollak CEB, Biegstraaten M. Quality of life in patients with Fabry disease: a systematic review of the literature. Orphanet J Rare Dis. 2015;10:77. doi:10.1186/s13023-015-0296-8
64. Rosa-Neto NS, Bento JCB, Pereira RMR. Depression, sleep disturbances, pain, disability and quality of LIFE in Brazilian Fabry disease patients. Mol Genetics Metabol Rep. 2020;22:100547. doi:10.1016/j.ymgmr.2019.100547
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.