Back to Journals » Journal of Multidisciplinary Healthcare » Volume 14
Multidisciplinary Management of Ataxia Telangiectasia: Current Perspectives
Authors McGrath-Morrow SA ![]() , Rothblum-Oviatt CC
, Rothblum-Oviatt CC ![]() , Wright J, Schlechter H, Lefton-Greif MA, Natale VA, Crawford TO
, Wright J, Schlechter H, Lefton-Greif MA, Natale VA, Crawford TO ![]() , Lederman HM
, Lederman HM
Received 15 May 2021
Accepted for publication 10 June 2021
Published 28 June 2021 Volume 2021:14 Pages 1637—1643
DOI https://doi.org/10.2147/JMDH.S295486
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Sharon A McGrath-Morrow,1 Cynthia C Rothblum-Oviatt,2 Jennifer Wright,3 Haley Schlechter,4 Maureen A Lefton-Greif,5 Valerie A Natale,6 Thomas O Crawford,7 Howard M Lederman3
1Division of Pulmonary and Sleep, Children’s Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA; 2A-T Clinical Center, Johns Hopkins Hospital, Baltimore, MD, USA; 3Division of Pediatric Allergy and Immunology, The Johns Hopkins Medical Institutions, Baltimore, MD, USA; 4Institute for Clinical and Translational Research, Johns Hopkins School of Medicine, Baltimore, MD, USA; 5Eudowood Division of Pediatric Respiratory Sciences, Johns Hopkins School of Medicine, Baltimore, MD, USA; 6Forgotten Diseases Research Foundation, Santa Clara, CA, USA; 7Departments of Pediatrics and Neurology, Johns Hopkins School of Medicine, Baltimore, MD, USA
Correspondence: Sharon A McGrath-Morrow
Division of Pulmonary and Sleep, Children’s Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Email [email protected]
Abstract: Ataxia telangiectasia (A-T) is a rare autosomal recessive disease caused by mutations in the ataxia telangiectasia mutated (ATM) gene. In the absence of a family history, the diagnosis of A-T is usually not made until the child is older and symptomatic. Classic A-T is characterized by a constellation of clinical symptoms including progressive ataxia, oculocutaneous telangiectasias and sinopulmonary disease and is usually associated with absence of ATM protein. Other laboratory features associated with A-T include elevated serum levels of alpha-fetoprotein (AFP) and increased chromosomal breakage with in vitro exposure to ionizing radiation. Sinopulmonary symptoms can occur to varying degrees across the lifespan. Some children will also have hypogammaglobulinemia and impaired antibody responses requiring supplemental gamma globulin. People with hypomorphic ATM mutations are often considered to have mild A-T with onset of ataxia and neurological progression occurring later in life with less impairment of the immune system. The risk of malignancy, however, is significantly increased in people with either classic or mild A-T. While hematological malignancies are most common in the first two decades of life, solid organ malignancies become increasingly common during young adulthood. Deterioration of neurologic function with age is associated with dysphagia with aspiration, growth faltering, loss of ambulation and decline in pulmonary function, morbidities that contribute to shortened life expectancy and decreased quality of life. Premature death is often due to malignancies or chronic respiratory insufficiency. A-T is currently managed with supportive care and symptomatic treatment. Current clinical trials, however, represent progress and hope towards disease-modifying therapies for A-T.
Keywords: ataxia telangiectasia, ataxia telangiectasia mutated, DNA damage repair
Introduction
Ataxia telangiectasia (A-T) is an autosomal recessive disorder that affects between 1 in 40,000–300,000 live births worldwide.1 A-T occurs in all racial and ethnic backgrounds and is associated with premature death in most people afflicted with the disease. People with A-T have mutations in the ataxia telangiectasia mutated (ATM) gene; a gene first discovered in 1995.2 There is considerable heterogeneity of disease presentation and progression in people with A-T. The initial clinical description of the disease described a triad of symptoms including progressive ataxia, oculocutaneous telangiectasias and sinopulmonary disease.3 Premature death is common in A-T and current treatments are limited to supportive care. Although some of the heterogeneity in clinical presentation is due to ATM mutation-specific causes, other factors likely contribute to disease severity.
ATM Gene
Homozygosity for ATM mutations can occur; but most people diagnosed with A-T are found to have compound heterozygosity for mutations in the ATM gene; meaning that they inherit a different ATM mutation from each parent. The ATM gene is located on the long arm (q) of chromosome 11 (11q22-23). Genetic mutations in the ATM gene can include deletions, duplications, frameshift and nonsense mutations, which often result in the absence of ATM protein in the affected individuals. Individuals with ATM protein-null genotypes generally have a more severe disease phenotype termed “classic” A-T. Most children and adults with A-T have this more severe, “classic” form and manifest similar progression of clinical symptoms across their lifespan (Table 1). However, those with other complex ATM genotypes may manifest features across a range of severity. For instance, some individuals with A-T have hypomorphic ATM mutations that permit some protein production and function that is more directly relatable to their milder disease presentation.
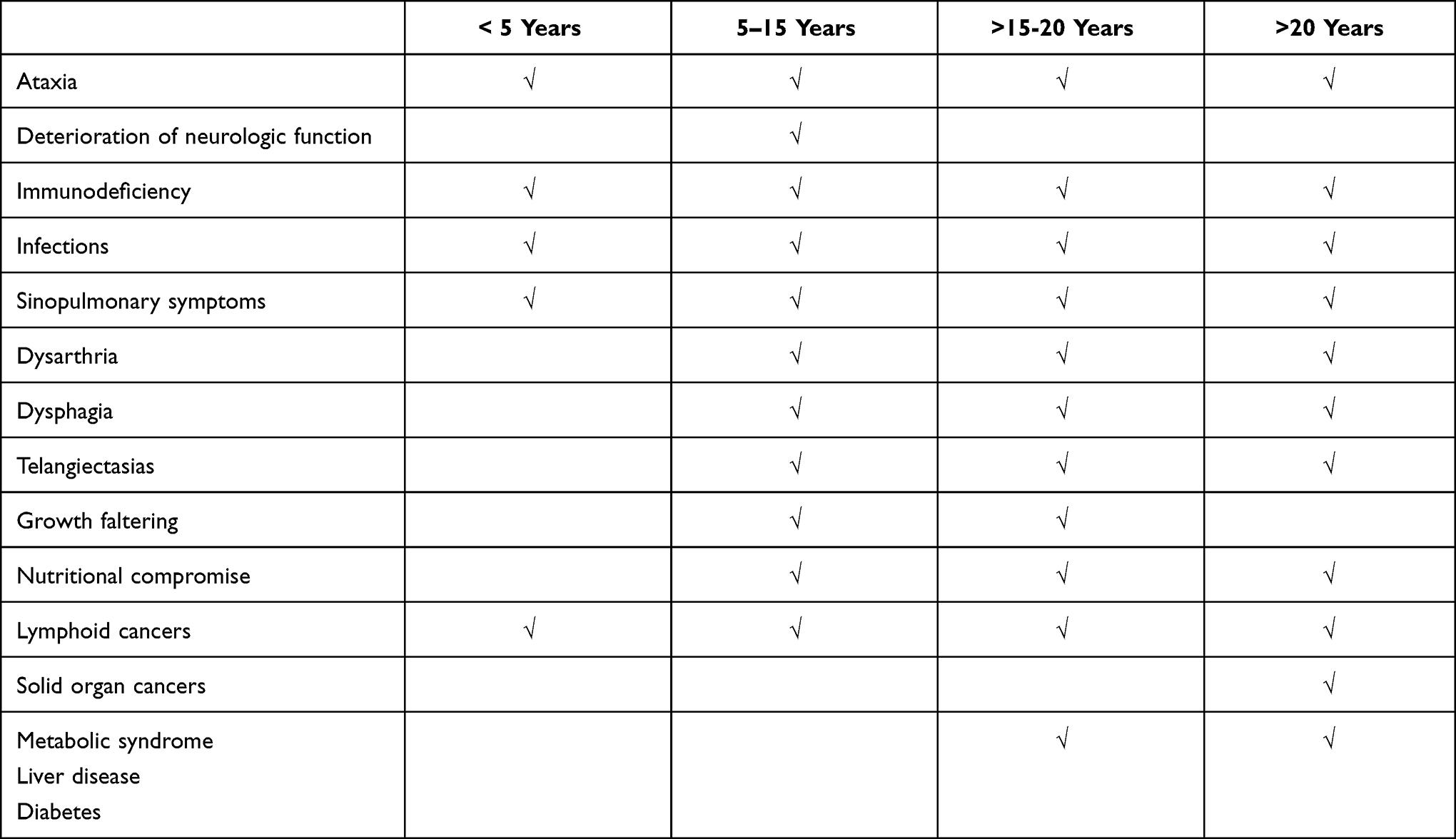 |
Table 1 Common Clinical Features of Classic A-T by Age |
ATM and Double-Stranded DNA Repair
The ATM protein is a 350 kDa serine/threonine protein kinase that is essential for double-stranded DNA repair. Double-stranded DNA breaks (DSBs) can occur following exposure to ionizing radiation. For this reason, individuals with A-T should avoid exposure to X-rays, unless radiographic imaging is clinically needed to direct therapy. Double-stranded DNA breaks also occur during V(D)J-recombination. V(D)J-recombination is integral to the adaptive immune response and is required for expansion of the antibodies and T cell receptor repertoire response to foreign pathogens. However, because there are functional redundancies in V(D)J recombination; total absence of ATM does not usually cause the severe immunodeficiency phenotype found in individuals with severe combined immunodeficiency (SCID).4
ATM and DNA Damage Response
In addition to DSBs, ATM is a critical constituent of the DNA damage response (DDR) pathway and is activated by reactive oxygen species and reactive nitrogen species.5 In the absence of ATM protein, both the DDR response is compromised; and cellular damage from oxidative stress may be amplified. Respiratory and systemic illnesses and indoor and outdoor environmental insults can induce localized and systemic oxidative stress. These exposures may have the potential to accelerate disease progression in people with A-T. Yeo et al reported that nasal epithelial cells from A-T individuals were more sensitive to the damaging effects of infection with S. pneumoniae than controls, and that this was partially due to production of H2O2 from the S. pneumoniae bacteria.6 Exposure to high levels of oxidative stress by this or other sources can be particularly injurious to people with A-T.
Clinical Characteristics of AT
Newborns
Children with classic A-T may be slightly smaller at birth than average but otherwise manifest no physical characteristics indicating the diagnosis.7 In the absence of a family history the early appearance does not enable diagnosis until later emergence of characteristic clinical features.
A recent new pathway to early diagnosis has emerged as a consequence of newborn screening for SCID, where at least some newborns with A-T trigger an abnormal result. This occurs because some infants with A-T have sufficiently low circulating copy numbers of T-cell receptor excision circles (TRECs) to trigger an abnormal report in that assay that detects SCID.8 In contrast to infants with SCID, infants with A-T seldom demonstrate severe deficiencies in T cell and B cell function and are unlikely to present with opportunistic infections.9 A positive SCID screen found not to be due to SCID, should lead to additional testing, and the diagnosis of A-T can be made when genetic studies are performed.
Preschool Years
Neurologic Symptoms and Laboratory Markers
The first clinical features of A-T are usually ataxic movements that most commonly develop in young children during the first two years of life. Young children often sway when seated, or have difficulty standing in one place without corrective steps even while they often walk at a normal age. With time the gait becomes more severely affected, and children may require the use of a wheelchair between 10 and 12 years of life. Children with hypomorphic mutations (and therefore some level of functional ATM) may be observed to have clumsiness with physical activities when compared to their peers. During the preschool years, children with A-T are often misdiagnosed with cerebral palsy, and the diagnosis is not suspected until there is more obvious decline in established functional abilities. It should be noted that telangiectasia may not appear until after the age of 5 years, so their absence is common during the earliest evaluations for ataxia, undermining the potential for early diagnosis.10 When A-T is suspected, genetic testing should be performed to rule out the presence of ATM mutations. Other laboratory features associated with the diagnosis of A-T include elevated serum levels of alpha-fetoprotein (AFP) and increased chromosomal breakage with in vitro exposure to ionizing radiation.
Sino-Pulmonary Symptoms in A-T
Some children with A-T will have frequent ear or respiratory infections during the preschool years often due to some degree of immunodeficiency. Reduced numbers of lymphocytes and abnormalities of T and B cell function are common in A-T. Many children with A-T are deficient in immunoglobulin A (IgA), and a smaller number will have hypogammaglobulinemia and impaired antibody responses.11 The latter group are at high risk for severe and progressive sinopulmonary disease if left untreated and can respond to gamma globulin supplementation. Many children with A-T have suboptimal antibody responses to pneumococcal vaccines, which can increase their risk for lower respiratory infections particularly following a viral respiratory infection. Children with A-T should be given pneumococcal vaccines at regular intervals throughout childhood to boost pneumococcal antibody responses along with annual influenza vaccines. COVID-19 vaccines also should be given to all age-appropriate individuals with A-T, and monoclonal antibodies should be considered in age-appropriate individuals with mild to moderate COVID symptoms. Antibiotics should be given when appropriate and chest physiotherapy and airway clearance techniques should be initiated in children with chronic or recurrent sinopulmonary disease to minimize development of bronchiectasis and chronic lung disease.
Other Common Clinical Findings in Preschool Children with A-T
Preschool children with A-T frequently present with dysarthria and drooling that can interfere with speech, communication and peer interactions. Eye movement abnormalities that are characterized by impaired initiation of volitional saccades and impaired eye/head coordination will affect most children with A-T; these deficits manifest with increasing difficulty reading.12 Brain imaging usually demonstrates cerebellar atrophy. Cognitive levels in children with A-T are usually normal, however learning difficulties are common. Ocular telangiectasias are unusual before 4–5 years of age.
School and Adolescent Years
Neurologic Problems
Children with A-T have progressive loss of independent ambulation as they age. Additionally, gait disturbances can worsen as sensory and motor neuropathies progress.13 Although children with A-T may benefit from orthopedic correction or therapies, loss of independent ambulation will occur in most individuals with classic A-T. Although physical and occupational therapies and exercise may help maintain overall physical well-being and quality of life, there are currently no treatments capable of slowing or halting the neurologic decline in A-T. Physical therapy and exercise should not be used to the point of fatigue. Certain drugs may be useful for managing the neurologic symptoms associated with A-T but should only be prescribed by a neurologist familiar with the treatment of movement disorders.
Growth Failure and AT
Although many children with classic A-T will have normal weight gain during the preschool years; most will start dropping growth percentiles by 5 years of age.7 Worsening dysphagia from neurological progression can contribute to the risk of growth faltering and suboptimal weight gain during childhood.14 Children often require longer mealtimes for several reasons, which can lead to less caloric intake. Reasons for longer mealtimes can include chewing difficulties, swallowing impairments, or self-feeding problems. Dysphagia from neurological progression can also increase the risk for aspiration and contribute to chronic lower respiratory tract symptoms, which can occur in the absence of fever.
Many people with classic A-T meet the World Health Organization criteria for wasting and stunting (defined as more than 2 standard deviations below mean weight-for-height and mean height for age) and this increases with age. In a cohort of children and young adults with A-T only 9% of children with A-T met the definition of wasting at 2 years of age but by 21 years 55% met that definition.7 Linear growth also usually falters in classic A-T. Approximately half of 2 year-olds with A-T were found to have heights at the 50th percentile; while adults with classic A-T had median heights at the 3rd percentile. Male percentile scores are slightly better than female scores. Problems with linear growth generally do not occur in people with mild A-T phenotypes.7,15
The early establishment of adequate nutrition for children and adults with A-T is essential for health and well-being. Feeding tubes are an option to improve nutritional status and to potentially mitigate respiratory complications from aspiration and respiratory muscle weakness. Placement is most beneficial before complications associated with nutritional compromise or dysphagia/aspiration are present.16 A study of feeding tube placement including optimal timing of placement in people with A-T would add considerable information about their utility in maintaining health.
Malignancy and A-T
All individuals with A-T are at higher risk for a malignancy. Loss of ATM is associated with an increased risk for developing leukemias and lymphomas particularly during the first two decades of life. Routine blood tests are not indicated, but any symptom characteristic of a hematologic malignancy such as unexplained fever, weight loss, or a persistently enlarged lymph node should be evaluated for the possibility of a malignancy. Individuals with A-T are also particularly susceptible to Epstein-Barr related malignancies that include diffuse large B cell and Hodgkin’s lymphomas.17 As individuals with A-T become adults, solid organ malignancies such as gastric, breast, pancreatic and skin cancers, become more common. Adults with A-T may benefit from annual whole-body MRIs to screen for malignancies. In patients found to have malignancies, treatment modifications and dose reductions are usually needed to minimize side effects and optimize outcomes.18,19
Lung Function and A-T
Declining lung function and emergence of respiratory symptoms often occurs in school and adolescent age groups.20 Impairment in lung function can occur as a result of multiple causes. Individuals with A-T can develop a progressive inability to initiate or take effective deep breaths which causes a decrease in their measured forced vital capacity (FVC). This can cause a functional restrictive lung phenotype, similar to individuals with neuromuscular weakness and associated with a decrease in pulmonary reserve. An ineffective or weak cough leading to impaired mucociliary clearance (MCC) can also contribute to respiratory decline in this age group. Decreased FVC and MCC can lead to recurrent respiratory infections; that can be further exacerbated by underlying immunodeficiency, aspiration and inadequate injury repair due to ATM deficiency. Bronchiectasis and lung fibrosis can develop in individuals with recurrent lower respiratory tract infections; particularly those in which early treatment of respiratory symptoms is delayed. Interstitial lung disease (ILD) has also been reported in people with A-T. Diagnosis of ILD is usually based on lung biopsy, although imaging and clinical features may suggest it. A-T specific ILD and may be responsive to systemic steroids.21
Children with A-T should undergo annual lung function studies starting at 6 years of age. Early screening for decline in lung function can allow for earlier interventions. These interventions may include the initiation of a cough assist device and chest physiotherapy to help improve MCC and lung inflation. They can be used as maintenance therapies on a daily basis. Some children with A-T may have airway reactivity that is responsive to bronchodilators. Use of antibiotics and gamma globulin supplementation should be considered for those with frequent lower respiratory tract infections and those with immunodeficiencies.
Skin Conditions and A-T
Cutaneous granulomas are the most common manifestation of granuloma formation in people with A-T, although systemic granulomas have also been reported.22 Recently, an association has been found between administration of live rubella vaccines and rubella-positive granuloma formation in people with A-T or other DNA repair disorders.23
Cutaneous granulomas can be persistent and difficult to treat. Treatments have included topical and systemic corticosteroids, anti-tumor necrosis factor therapy, and anti-virals. Cutaneous telangiectasia often emerge in sun-exposed areas of skin.
Cardiovascular and Liver and Metabolic Conditions
Individuals with A-T have been found to have cholesterol profiles associated with a higher risk for cardiovascular disease,24 diabetes with insulin resistance,25 and steatohepatitis.26,27 These findings suggest that ATM dysregulation is associated with the development of metabolic syndrome. People with A-T should undergo screening for these conditions during adolescence and early adult life so that timely treatments can be initiated.
Fatigue and A-T
Children and adolescents with A-T often complain of progressive daily fatigue which can be exacerbated by physical activities. The etiology for fatigue is not completely understood but its presence and severity may be associated with progressive neurological deterioration. Other causes of fatigue can include disruption or fragmentation of nighttime sleep, poor nutrition, depression or other medical causes.28 Quality of life may deteriorate in older children and adolescents due to their increasing disabilities, social isolation, and the development of depression and anxiety. Attention to mental health care needs should be given to all children and adults with A-T.
Adults with A-T
Life-expectancy for individuals with A-T is decreased. Malignancies and respiratory failure are the major contributing factors to premature death in people with A-T.29 Life expectancy for a person with A-T is determined in part by their ATM mutation with null mutations associated with lower survival. A 20-year survival rate of 53.4% was reported in a cohort of patients in the French national Reference Center for Primary Immunodeficiencies.29 Earlier diagnosis, supportive care and a multidisciplinary approach to patient follow-up can improve survival and quality of life.
Mild A-T
In people with hypomorphic ATM mutations, onset of ataxia and neurological progression may emerge later and progress more slowly, and impairment of the immune system be less marked.30 These individuals with hypomorphic ATM mutations, however, have similarly increased risk of malignancy as people with classic A-T.31 Isolated patients with predicted protein null mutations have been identified who manifest similar mild progression of symptoms, indicating that the relationship between genotype and phenotype is complex.32,33
Biomarkers and A-T
The majority of individuals with A-T have compound heterozygosity for mutations in their ATM gene. Predicted protein abrogating mutations occur throughout the gene, and while missense mutations cluster within the terminal exons encoding the kinase region, other apparent disease-causing missense mutation are identified throughout the gene. This complicates the search for therapeutic compounds and genetic approaches that could be used to treat the majority of afflicted individuals. Taken together the diverse heterogeneity of genetic and clinical symptoms in A-T makes it difficult to predict efficacy of a given treatment or intervention for this rare disease. Several serum biomarkers have been reported which could potentially be used as markers of response in people with A-T and to predict those who may be at risk for certain morbidities. Levels of neurofilament light chain from the blood has recently been shown to correlate with neurodegeneration in people with A-T.34,35 Elevated serum levels of IL-6 and IL-8 in people with A-T have also been associated with an increased risk of malignancies and worse lung function in people with A-T.36,37 Further work is needed to identify other biomarkers that can be used to assess disease progression and treatment effectiveness in clinical trials.
A-T Specific Neurologic and Functional Scales
A-T specific neurologic24 scales have been validated, and functional scales are in development to track neurologic function and daily activities in people with A-T. These could be used to assess progression and stability of people with A-T at baseline and for those are enrolled in clinical trials.
Current Clinical Trials for A-T
Although A-T is a complex disorder with symptoms varying from patient to patient, all individuals with A-T (mild and classic) suffer from neurologic abnormalities. These deficits arguably represent some of the most devastating symptoms of this disease, so therapies capable of halting disease progression as early as possible are highly desirable. Current intervention trials for A-T include triheptanoin, nicotinamide riboside, N-Acetyl-L-Leucine, and intra-erythrocyte dexamethasone sodium phosphate. Neurologic assessment is a primary or secondary clinical endpoint for each of these studies. The extraordinary progress in antisense oligonucleotide (ASO)-based treatments for other rare diseases, such as spinal muscular atrophy (SMA), have opened the door for exploration of the first mutation-targeted therapy for A-T. An ASO trial in A-T is now ongoing which is targeting an ATM mutation in a “n of 1 trial”. These types of trials are critically important to determine if personalized targeting of ATM mutations, will ultimately be useful in slowing or preventing disease progression and are important for the development of optimal clinical trial design. Although A-T is currently managed with supportive care and symptomatic treatment, these trials represent progress and hope towards disease-modifying therapies for A-T.
Future Directions
The highest priority is to develop therapies that delay or stop neurodegeneration. This will require a better understanding of why the loss of ATM causes the circuit-specific deficits that characterize the characteristic neurologic deficits. The lack of animal models with the human neurologic phenotype has made this research very difficult, but perhaps it can be solved with iPSC-derived Purkinje cells,38 or other in vitro systems for studying human cells.
Progress in the management of other aspects of A-T will have an equally important role in lengthening and improving the lives of affected individuals. Understanding the reasons for phenotypic variation may yield gains, not only to identify those individuals at highest risk for developing specific complications such as cancer, but also to learn of other therapeutic targets to mediate disease progression. We know how to manage the nutritional needs of people with A-T, but do not do an adequate job of communicating this to caregivers and convincing them to use gastrostomy tubes to deliver supplemental calories early enough in life to make a difference to their children. We need more effective and less toxic drugs to manage cancer and chronic inflammatory disorders when they develop. Finally, we need to think of A-T as a disease to live with and not to die from, and therefore be willing to take risks with aggressive therapies such as liver transplantation for people with end-stage liver disease who are otherwise healthy and enjoying what they consider to be a reasonable quality of life.
Funding
National Heart, Lung, and Blood Institute, Grant/Award Number: R01HL114800 (SMM), RFA-FD-19-001 (ML-G, HL, VN).
Disclosure
Professor Maureen A Lefton-Greif reports grants from National Institutes of Health (1R21TR003534), grants from Pediatric Clinical Research Center, grants from A-T Children’s Project, during the conduct of the study. Dr Howard M Lederman report grants from A-T Children’s Project, during the conduct of the study; contract to participate in a drug trial for A-T, and personal fees for serving on the medical advisory board from EryDel, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39:573–583.
2. Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi:10.1126/science.7792600
3. Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554.
4. Weitering TJ, Takada S, Weemaes CMR, van Schouwenburg PA, van der Burg M. ATM: translating the DNA damage response to adaptive immunity. Trends Immunol. 2021;42:350–365. doi:10.1016/j.it.2021.02.001
5. Blignaut M, Harries S, Lochner A, Huisamen B, Lane JD. ataxia telangiectasia mutated protein kinase: a potential master puppeteer of oxidative stress-induced metabolic recycling. Oxid Med Cell Longev. 2021;2021:8850708. doi:10.1155/2021/8850708
6. Yeo AJ, Fantino E, Czovek D, Wainwright CE, Sly PD, Lavin MF. Loss of ATM in airway epithelial cells is associated with susceptibility to oxidative stress. Am J Respir Crit Care Med. 2017;196:391–393. doi:10.1164/rccm.201611-2210LE
7. Natale VAI, Cole TJ, Rothblum-Oviatt C, et al. Growth in ataxia telangiectasia. Orphanet J Rare Dis. 2021;16:123. doi:10.1186/s13023-021-01716-5
8. Ravkov E, Slev P, Heikal N. Thymic output: assessment of CD4(+) recent thymic emigrants and T-cell receptor excision circles in infants. Cytometry B Clin Cytom. 2017;92:249–257. doi:10.1002/cyto.b.21341
9. Quinn J, Orange JS, Modell V, Modell F. The case for severe combined immunodeficiency (SCID) and T cell lymphopenia newborn screening: saving lives … one at a time. Immunol Res. 2020;68:48–53. doi:10.1007/s12026-020-09117-9
10. Cabana MD, Crawford TO, Winkelstein JA, Christensen JR, Lederman HM. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics. 1998;102:98–100. doi:10.1542/peds.102.1.98
11. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. 2016;11:159. doi:10.1186/s13023-016-0543-7
12. Tang SY, Shaikh AG. Past and present of eye movement abnormalities in ataxia-telangiectasia. Cerebellum. 2019;18:556–564. doi:10.1007/s12311-018-0990-x
13. Woods CG, Taylor AM. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med. 1992;82:169–179.
14. Lefton-Greif MA, Crawford TO, Winkelstein JA, et al. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J Pediatr. 2000;136:225–231. doi:10.1016/S0022-3476(00)70106-5
15. Hadchouel A, Rousseau J, Roze JC, et al. Association between asthma and lung function in adolescents born very preterm: results of the EPIPAGE cohort study. Thorax. 2018;73:1174–1176. doi:10.1136/thoraxjnl-2017-211115
16. Lefton-Greif MA, Crawford TO, McGrath-Morrow S, Carson KA, Lederman HM. Safety and caregiver satisfaction with gastrostomy in patients with Ataxia Telangiectasia. Orphanet J Rare Dis. 2011;6:23. doi:10.1186/1750-1172-6-23
17. Tatfi M, Hermine O, Suarez F. Epstein-Barr Virus (EBV)-related lymphoproliferative disorders in ataxia telangiectasia: does ATM regulate EBV life cycle? Front Immunol. 2018;9:3060. doi:10.3389/fimmu.2018.03060
18. Seidemann K, Henze G, Beck JD, et al. Non-Hodgkin’s lymphoma in pediatric patients with chromosomal breakage syndromes (AT and NBS): experience from the BFM trials. Ann Oncol. 2000;11(Suppl 1):141–145. doi:10.1093/annonc/11.suppl_1.S141
19. Machida S, Tomizawa D, Tamaichi H, et al. Successful treatment of diffuse large B-cell lymphoma in a patient with ataxia telangiectasia using rituximab. J Pediatr Hematol Oncol. 2013;35:482–485. doi:10.1097/MPH.0b013e3182804d59
20. McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, et al. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatr Pulmonol. 2010;45:847–859. doi:10.1002/ppul.21277
21. Schroeder SA, Swift M, Sandoval C, Langston C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr Pulmonol. 2005;39:537–543. doi:10.1002/ppul.20209
22. Szczawińska-Popłonyk A, Olejniczak K, Tąpolska-Jóźwiak K, et al. Cutaneous and systemic granulomatosis in ataxia-telangiectasia: a clinico-pathological study. Postepy Dermatol Alergol. 2020;37:760–765. doi:10.5114/ada.2020.100485
23. Buchbinder D, Hauck F, Albert MH, et al. Rubella virus-associated cutaneous granulomatous disease: a unique complication in immune-deficient patients, not limited to DNA repair disorders. J Clin Immunol. 2019;39:81–89. doi:10.1007/s10875-018-0581-0
24. Andrade IG, Costa-Carvalho BT, da Silva R, et al. Risk of atherosclerosis in patients with ataxia telangiectasia. Ann Nutr Metab. 2015;66:196–201. doi:10.1159/000430790
25. Donath H, Hess U, Kieslich M, et al. Diabetes in patients with ataxia telangiectasia: a national cohort study. Front Pediatr. 2020;8:317. doi:10.3389/fped.2020.00317
26. Viswanathan P, Sharma Y, Maisuradze L, Tchaikovskaya T, Gupta S. Ataxia telangiectasia mutated pathway disruption affects hepatic DNA and tissue damage in nonalcoholic fatty liver disease. Exp Mol Pathol. 2020;113:104369. doi:10.1016/j.yexmp.2020.104369
27. Paulino TL, Rafael MN, Hix S, et al. Is age a risk factor for liver disease and metabolic alterations in ataxia Telangiectasia patients? Orphanet J Rare Dis. 2017;12:136. doi:10.1186/s13023-017-0689-y
28. McGrath-Morrow SA, Sterni L, McGinley B, Lefton-Greif MA, Rosquist K, Lederman H. Polysomnographic values in adolescents with ataxia telangiectasia. Pediatr Pulmonol. 2008;43:674–679. doi:10.1002/ppul.20838
29. Micol R, Ben SL, Suarez F, et al. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J Allergy Clin Immunol. 2011;128:382–389. doi:10.1016/j.jaci.2011.03.052
30. Taylor AM, Lam Z, Last JI, Byrd PJ. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet. 2015;87:199–208. doi:10.1111/cge.12453
31. Roohi J, Crowe J, Loredan D, et al. New diagnosis of atypical ataxia-telangiectasia in a 17-year-old boy with T-cell acute lymphoblastic leukemia and a novel ATM mutation. J Hum Genet. 2017;62:581–584. doi:10.1038/jhg.2017.6
32. Alterman N, Fattal-Valevski A, Moyal L, et al. Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am J Med Genet A. 2007;143a:1827–1834. doi:10.1002/ajmg.a.31853
33. Worth PF, Srinivasan V, Smith A, et al. Very mild presentation in adult with classical cellular phenotype of ataxia telangiectasia. Mov Disord. 2013;28:524–528. doi:10.1002/mds.25236
34. Donath H, Woelke S, Schubert R, et al. Neurofilament light chain is a biomarker of neurodegeneration in ataxia telangiectasia. Cerebellum. 2021.
35. Veenhuis SJG, Gupta AS, de Gusmão CM, et al. Neurofilament light chain: a novel blood biomarker in patients with ataxia telangiectasia. Eur J Paediatr Neurol. 2021;32:93–97. doi:10.1016/j.ejpn.2021.04.002
36. McGrath-Morrow SA, Ndeh R, Collaco JM, et al. Inflammation and transcriptional responses of peripheral blood mononuclear cells in classic ataxia telangiectasia. PLoS One. 2018;13:e0209496. doi:10.1371/journal.pone.0209496
37. McGrath-Morrow SA, Collaco JM, Detrick B, Lederman HM. Serum interleukin-6 levels and pulmonary function in ataxia-telangiectasia. J Pediatr. 2016;171:256–261.e251. doi:10.1016/j.jpeds.2016.01.002
38. Muguruma K. Self-organized cerebellar tissue from human pluripotent stem cells and disease modeling with patient-derived iPSCs. Cerebellum. 2018;17:37–41. doi:10.1007/s12311-017-0905-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.