Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Multidisciplinary Management of Alagille Syndrome
Authors Menon J ![]() , Shanmugam N, Vij M
, Shanmugam N, Vij M ![]() , Rammohan A, Rela M
, Rammohan A, Rela M
Received 16 November 2021
Accepted for publication 14 January 2022
Published 23 February 2022 Volume 2022:15 Pages 353—364
DOI https://doi.org/10.2147/JMDH.S295441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jagadeesh Menon,1 Naresh Shanmugam,1 Mukul Vij,2 Ashwin Rammohan,3 Mohamed Rela3
1Department of Pediatric Gastroenterology & Hepatology, Dr Rela Institute & Medical Centre, Bharath Institute of Higher Education and Research, Chennai, India; 2Department of Histopathology, Dr Rela Institute & Medical Centre, Bharath Institute of Higher Education & Research, Chennai, India; 3Institute of Liver Disease & Transplantation, Dr Rela Institute & Medical centre, Bharath Institute of Higher Education and Research, Chennai, India
Correspondence: Jagadeesh Menon, Email [email protected]
Abstract: Alagille syndrome (ALGS) is an autosomal dominant disorder characterized by involvement of various organ systems. It predominantly affects the liver, skeleton, heart, kidneys, eyes and major blood vessels. With myriads of presentations across different age groups, ALGS is usually suspected in infants presenting with high gamma glutamyl transpeptidase cholestasis and/or congenital heart disease. In children it may present with decompensated cirrhosis, intellectual disability or short stature, and in adults vascular events like stroke or ruptured berry aneurysm are more commonly noted. Liver transplantation (LT) is indicated in children with cholestasis progressing to cirrhosis with decompensation. Other indications for LT include intractable pruritus, recurrent fractures, hepatocellular carcinoma and disfiguring xanthomas. Due to an increased risk of renal impairment noted in ALGS, these patients would require optimized renal sparing immunosuppression in the post-transplant period. As the systemic manifestations of ALGS are protean and a wider spectrum is being increasingly elucidated, a multidisciplinary team needs to be involved in managing these patients. Moreover, many basic-science and clinical questions especially with regard to its presentation and management remain unanswered. The aim of this review is to provide updated insights into the management of the multi-system involvement of ALGS.
Keywords: Alagille syndrome, chronic cholestasis, liver transplantation, congenital heart disease, vascular anomalies, multidisciplinary management
Introduction
Alagille syndrome (ALGS), also known as Watson–Alagille syndrome, is an autosomal dominant (AD) disorder with an incidence of 1 in 70,000 live births.1 It is caused by mutations involving the notch signaling pathway of either the JAG1 gene (encoding JAGGED) (94–99%) located on chromosome 20 or the NOTCH2 gene (1–4%) located on chromosome 1.1,2 Although traditionally labeled as arteriohepatic dysplasia confined to the liver, this disease affects multiple organs. With an appreciation of increasing numbers of phenotypes, a revised set of diagnostic criteria for ALGS has been proposed.1,3,4
The major diagnostic criteria for ALGS involve various organs, including the liver (cholestasis, bile duct paucity), heart, eyes, skeletal system, kidneys and structural vascular anomalies or characteristic facies (Table 1). ALGS has a varied presentation across different age groups, thereby making it a ubiquitous disorder presenting to any specialty care physician. The manifestations of ALGS may be byzantine and range from being a completely silent disease phenotype to severe life-threatening cardiovascular involvement and/or advanced liver disease (Table 1). Despite remarkable advances in the understanding of this disorder, many questions especially with regard to the presentation, diagnosis and management of ALGS remain unanswered..
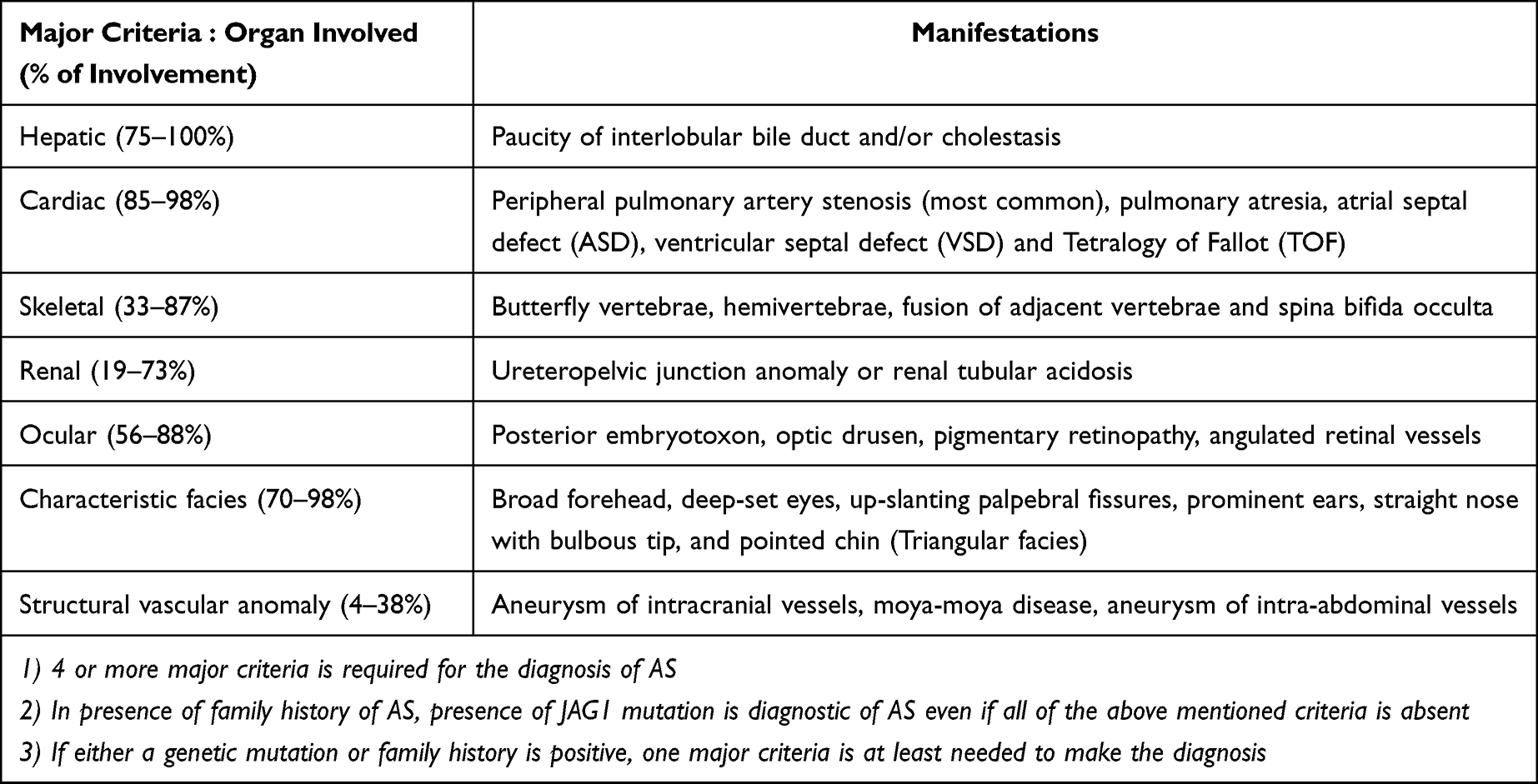 |
Table 1 Revised Diagnostic Criteria for Diagnosis of Alagille Syndrome |
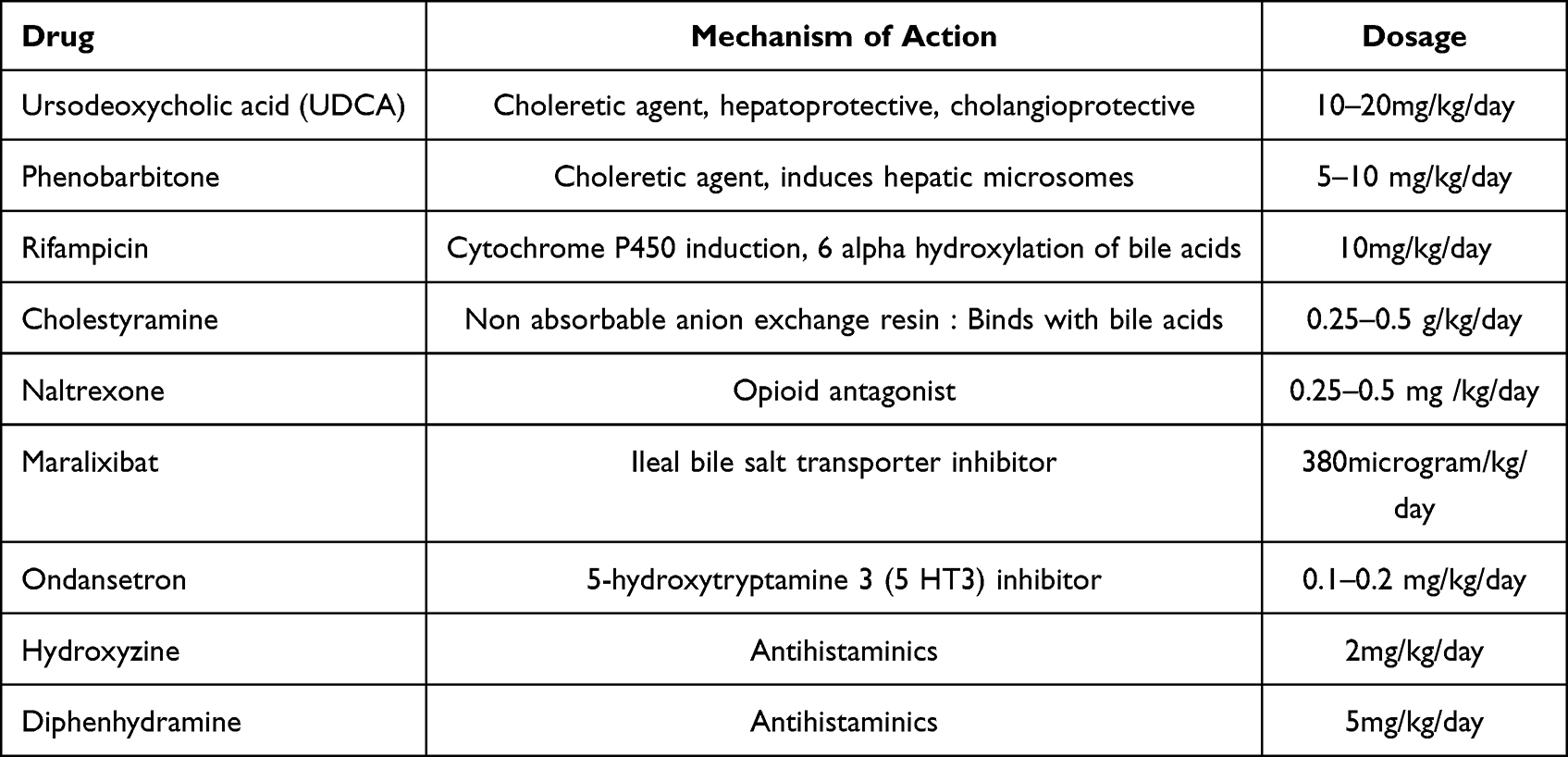 |
Table 2 Medical Therapy for Cholestasis in Alagille Syndrome |
This review appraises the current knowledge of ALGS with a focus on highlighting and appreciating the approaches in its treatment strategy and future challenges.
Genetics and Pathogenesis of ALGS
ALGS is caused by a mutation in either JAG1 or NOTCH2 gene, either of which produces single pass transmembrane proteins which are involved in the notch signaling pathway.5 While the NOTCH2 gene codes for the notch protein, JAG1 codes for cell surface protein Jagged1, a ligand for notch receptors. The signals originating from these receptors are involved in organogenesis. Mutations in these genes produce the multi-system clinical manifestations of ALGS. In a majority, the mutations in the JAG1 gene are protein truncations, others include gene deletions and missense mutations.2,6 Of all the organs involved, ALGS affects the liver and the heart the most. Jagged1 expression in the portal vein mesenchyme is necessary for bile duct formation. Notch2 signals are important for differentiation of hepatoblasts into biliary epithelial cells and their survival.7 Therefore, the classical hepatic manifestation of ALGS is the paucity of interlobular bile duct (PIBD) due to a defective morphogenesis of the bile ductules. Impaired notch2 signaling leads to defective embryogenesis in the heart. The mesodermal cells fail to differentiate into cardiomyocytes; endocardial cushion defects and valvular anomalies of the right heart may also occur.8 Jagged1 is required for angiogenesis, and a strong expression has been noted in all the major systemic arteries. Consequently, its defect leads to various vascular anomalies observed in ALGS. Interestingly, mutations in ALGS are inherited in only 30–50% cases, whereas de novo mutations account for the remaining 50%.5
It is possible to diagnose ALGS by pre-natal genetic tests and preimplantation diagnostic modalities. However, since there is no direct genotype–phenotype correlation, despite a positive mutation analysis, parents need to be cautioned regarding the unpredictable phenotypic expression and clinical course of ALGS.1
Hepatic Involvement
Liver involvement is observed in 89–100% of patients with ALGS.9 The spectrum of presentation of this hepatic affliction is varied. It presents in early infancy with cholestasis mimicking an obstructive cause like biliary atresia. Infants with ALGS develop pruritus around 6 months of age; with time this becomes intractable.10 The majority have hepatomegaly on examination. Splenomegaly is observed in 30–70% patients, usually occurring secondary to portal hypertension.11 Liver function tests are remarkable with elevated gamma glutamyl transpeptidase (GGT) and bilirubin.
The characteristic hepatic pathology in ALGS has been well described and consists of a paucity of interlobular bile duct with no significant bile ductular reaction (Figure 1A). A ratio of the number of bile ducts to the number of portal tracts of less than 0.5 was described in the initial report of this disease by Alagille.12,13 The presence and extent of bile duct paucity vary, largely as a result of the different ages of the patients at the time of biopsy.14 Cholangiodestructive lesions have also been reported in early infancy. Other histological findings observed in liver biopsies in patients with ALGS include hepatocanalicular bilirubinostasis with rosetting (Figure 1B), patchy giant cell transformation (Figure 1C), hemopoiesis and variable portal inflammation. Explanted liver specimen shows variable cholestasis (Figure 1D). Bile ductular proliferation is uncommon. Late pathologic features include portal fibrosis expansion with bridging fibrosis (Figure 2A) and even cirrhosis (Figure 2B) in some cases. Histochemical stains like orcein and rhodanine show copper deposition in the periportal hepatocytes (Figure 2C). Immunostaining with cytokeratin 7 is helpful to highlight the loss of bile duct and metaplastic biliary differentiation of hepatocytes (Figure 2D). Hepatocellular carcinoma has also been reported in ALGS (Figure 2E).
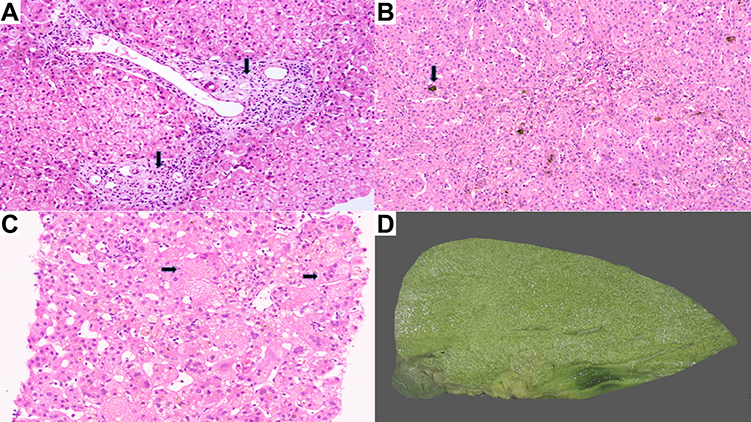 |
Figure 1 (A) Liver biopsy displaying paucity of interlobular bile ducts (arrow, H&E, ×40). (B) Hepatocanalicular bilirubinostasis (arrow, H&E, ×15). (C) Liver biopsy displaying giant cell formation (arrow, H&E, ×20). (D) Explant liver with cholestasis. |
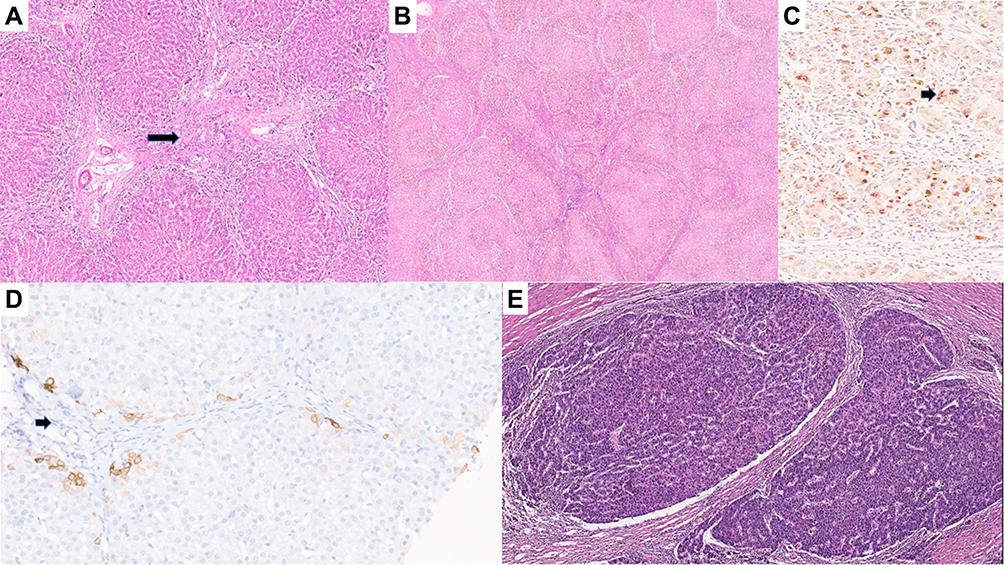 |
Figure 2 (A) Explant liver with bridging fibrosis (arrow, H&E, ×10). (B) Explant liver with cirrhotic transformation (H&E, ×5). (C) Rhodanine stain showing red copper granules in hepatocytes (arrow, H&E, ×25). (D) Immunostaining with cytokeratin 7 showing duct loss (arrow, H&E, ×10). (E) Hepatocellular carcinoma (arrow, H&E, ×8). |
It must be reiterated that it is difficult to predict the severity of liver involvement from the genotype of the patients. However, by 5 years of age, 15–30% of children with hepatic involvement will progress to advanced liver disease resulting in synthetic failure and portal hypertension, requiring a liver transplantation (LT).15 Apart from decompensated liver disease, other hepatic manifestations of ALGS like intractable pruritus have adverse implications on the patient’s quality of life. Even with a normal hepatic synthetic function, intractable pruritus may occur. This has other tumble-down effects like interruption of sleep, poor scholastics, lichenification of skin and secondary infections.9 Serum cholesterol levels remain high, and these children are likely to develop xanthomas causing dysmorphism/disfigurement and long-term cardiovascular adverse effects. Chronic cholestasis is also likely to lead to hepatic osteodystrophy resulting in fractures. Intracranial bleeds may occur secondary to vitamin K malabsorption.9
Management
Treatment of cholestasis is initially symptomatic with anti-pruritus medication (Table 2). The common agents include ursodeoxycholic acid, cholestyramine, rifampicin, ondansetron and naltrexone.15 A recent trial on the role of apical ileal bile salt transporter inhibitors (maralixibat) in patients with ALGS has shown a significant improvement in cholestasis evidenced by serum bile acid profiles and pruritus scores.16 Although there is symptomatic improvement, none of these medications are known to alter the natural history of ALGS. In intractable cases, plasmapheresis has shown to provide temporary relief from pruritus. Partial biliary diversion, either external or internal, is offered in patients with ALGS who present with isolated pruritus refractory to medical therapy.17 This surgical procedure should only be offered to non-cirrhotic patients. It is also found to reverse hypercholesterolemia and potentially reverse xanthoma formation. Nonetheless, even this surgical technique does not alter the natural history of liver disease in ALGS. Patients with bilirubin persistently above 6.5 mg/dL and cholesterol of more than 225 mg/dL have a higher propensity of progressing to end-stage liver disease.18
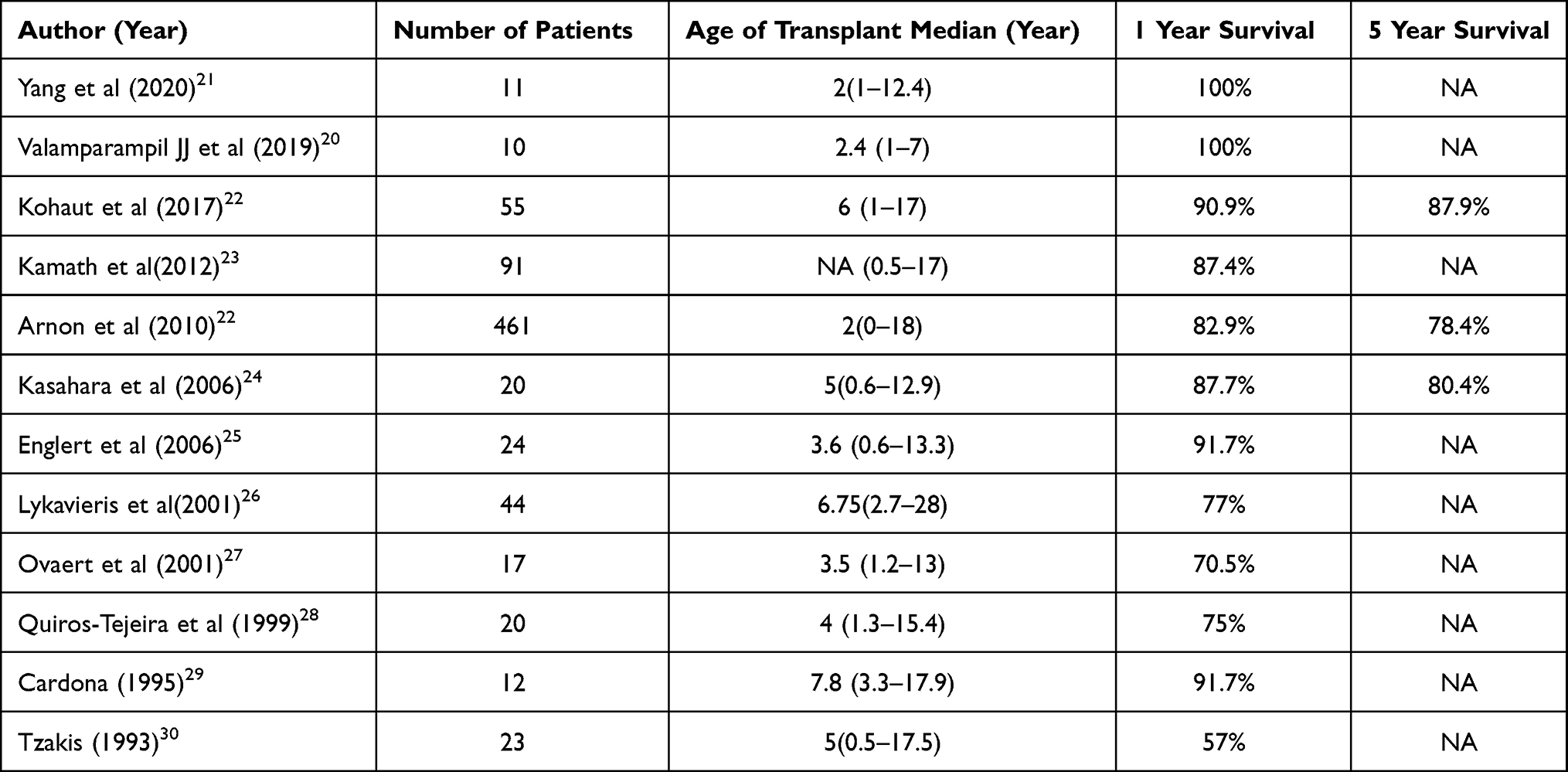 |
Table 3 Outcomes After Liver Transplantation in Patients with Alagille Syndrome |
Liver transplantation (LT) in ALGS is primarily indicated for end-stage liver disease leading to synthetic failure and/or portal hypertension.19 Other indications for LT include incidentally detected hepatocellular carcinoma (HCC) on routine screening. A persistently elevated alpha fetoprotein (AFP) provides a clue towards the occurrence of HCC. Patients with ALGS would also require LT when they have intractable pruritus, recurrent fractures or xanthomas at locations causing immobilization. LT in these patients have excellent long-term outcomes. (Table 3). Our experience of LT for ALGS concurs with that of other centers.
In our experience, the median age at LT was 28 months, with the commonest indication being intractable pruritus. On a median post-LT follow-up of 32 months, there has been 100% graft and patient survival.20 It is to be noted that the disease does not recur in the graft. Celiac artery stenosis and a hypoplastic hepatic artery are commonly noted in ALGS, and hence an extra-anatomical arterial inflow may be required. We did not encounter this issue in our series of patients.19,20
Due to inherent renal involvement as a part of the genetic defect, ALGS patients post-LT have a higher likelihood of developing calcineurin inhibitor (CNI)-induced nephrotoxicity.23 Targeting lower trough levels of CNI with renal sparing immunosuppression with anti-metabolites (mycophenolate mofetil) helps optimize immunosuppression in these patients. Using this protocol, our recipients have retained a normal estimated glomerular filtration rate (eGFR) in the post-LT period.20 There are isolated reports of combined liver–kidney transplantation in patients with ALGS.
An important consideration in living donor LT (LDLT) is that of the donor. Since ALGS is an AD disorder, either of the parents could carry the defective gene. The recommendations here is that the donors should be screened with liver function tests and abdominal imaging. The presence of any deviation indicates a pre-LT liver biopsy to rule out PIBD.20 If no stigmata are found, there is no contraindication for phenotypically normal first-degree relatives to be LDLT donors. Moreover, since there is no specific genotype–phenotype correlation in patients with ALGS, the role of genetic studies from the perspective of organ donation is not well defined. There are, however, certain centers which would universally exclude them as prospective donors.
Stunting (height for age) in ALGS is predominantly a genetic problem and should not be a sole indication for LT.28 Parents should be counseled that even though there is post-LT catch-up growth, the final height remains lower than the age-controlled population. As these patients are known to have an inherent growth hormone resistance, it is unclear if patients with ALGS suffering from stunting may benefit from insulin-like growth hormone (IGF-1) therapy.31
Both biliary atresia (BA) and ALGS have similar presentations, and it is important to differentiate these two common disorders which cause cholestasis in infancy. Both these diseases can have persistent pale stools, a high GGT and a hypoplastic gall bladder. Associated cardiac anomalies are also common in both these disorders. A liver biopsy also may not show classical features of BA in infants under 30 days of age, and those with ALGS may have PIBD in only 60% cases at 6 months of age. A percutaneous or intraoperative cholangiogram is the gold standard to diagnose BA, and these patients would require a timely Kasai portoenterostomy (KPE) surgery.32 It has been shown that accidentally done KPE for ALGS misdiagnosed as BA leads to poor outcomes. A previous analysis had shown higher mortality (31% vs 2.8%) and higher rate of LT (47.3% vs 13.9%) in patients undergoing a KPE for ALGS, when compared to patients who did not undergo the same surgery.33 Nevertheless, the outcomes of LT for ALGS does not depend on a previously done KPE.
Renal Involvement
NOTCH2 is expressed in renal tubular and glomerular epithelia, while JAGGED1 is expressed over the endothelium of glomeruli and collecting tubules in a fully formed nephron. NOTCH2 is present throughout the stages of formation of the kidneys and has a strong influence on the development of proximal tubule of the nephron and the podocyte. It is also important for the formation of the collecting tubule. Hence it is not surprising that patients with ALGS have various forms of renal involvement. Renal involvement is observed in 40–70% of patients with ALGS and is one among the revised diagnostic criteria in the diagnosis of ALGS9 (Table 1).
The spectrum of renal involvement in patients with ALGS can be divided into structural and functional disorders. Structural disorders include renal agenesis, hypoplasia, dysplasia, renal cysts, hydronephrosis, ureteropelvic obstruction, duplex collecting systems and renal vascular anomalies. The functional disorders include renal tubular acidosis, glomerulopathy and tubulointerstitial nephritis.34
In one of the largest series of ALGS from Canada, renal involvement was seen in 39% of patients, and the commonest anomaly was renal dysplasia.34 Independent of the effect of NOTCH2 mutation on the kidneys, patients may have a reduced eGFR despite a normal ultrasound of the kidneys. Purported reasons for this include hyperlipidemia causing a mesangiolipidosis leading to a focal or segmental glomerulopathy.34 Patients may present with persistent proteinuria.
Management
Patients need to be followed up under the care of a nephrologist. Those with structural renal anomalies require antibiotic prophylaxis followed by definite corrective procedures. As mentioned above, patients in the post-LT period should be commenced on renal sparing immunosuppression even if they have a normal eGFR prior to LT.20 In case of end-stage renal disease (ESRD), renal transplantation may be indicated, occasionally combined with a LT.
Cardiac Involvement
Cardiovascular manifestations are observed in 75–95% of patients with ALGS.35 The commonest anomalies involve the right ventricular outflow tract, notably that of the branched pulmonary artery (56% of patients).35 Peripheral pulmonary stenosis or valvular pulmonary stenosis is also common. Patients can also have complex structural heart diseases like tetralogy of Fallot (TOF) or double outlet right ventricle.36 Other lesions includes septal defects, right-sided aortic arch, coarctation of aorta, hypoplastic left heart syndrome and double superior vena cava. Involvement of heart in ALGS is important as it remains the primary cause of mortality, especially during infancy.37 The mortality of patients with TOF in ALGS is more than controls as the lesion is usually associated with co-existing pulmonary atresia (40% patients). It is noteworthy that the 6-year survival for patients of ALGS with or without cardiac anomalies is drastically different (40% vs 95%).11
Management
Corrective cardiac surgery needs to be done in a timely manner to prevent mortality during infancy. Patients with mild or focal pulmonary artery obstruction may be managed conservatively.35 Significant stenosis involving proximal, segmental or lobar branches of the pulmonary artery requires surgical or radiological arterioplasty followed by serial dilatations with or without stenting.38,39 Patients with ALGS having TOF need an early corrective surgery to prevent mortality. The outcomes nevertheless remain inferior.40
Consideration and management of co-existing cardiac illness are also important in the context of LT. Right-sided cardiac lesions which cause elevated pulmonary or right ventricular pressure could result in graft loss. Intraoperative hemodynamic changes including the need for clamping the inferior vena cava can lead to right ventricular overload and heart failure. The cardiac pathology can also result in a Budd–Chiari syndrome-like picture with hepatic congestion and decreased hepatic perfusion.19,41 Presence of tricuspid regurgitation indicates a higher likelihood of elevated right ventricular pressures. Right heart catheterization with pressure studies would be indicated in such cases. When the right ventricular pressures are over 50% of systemic pressures, a corrective procedure like balloon valvuloplasty or surgical correction is indicated prior to LT. Established pulmonary hypertension, especially when the mean pulmonary artery pressures (mPAP) is above 45–50 mmHg, remains an absolute contraindication to LT.42 When the mPAP is in the range 35–45 mmHg, targeted therapy to lower the pressures below 35 mmHg is instituted, following which an LT is performed. LT may still be performed in exceptional cases where the mPAP remains above 35 mmHg despite medical therapy. The caveat here remains that the patients should have a good cardiac reserve (right ventricular function) along with a normal pulmonary vascular resistance (<240 dynes/cm2).42 Rarely, patients can have a complex heart defect with coexisting decompensated liver disease, for which a combined heart and liver transplant may be offered.43
Vascular Involvement
JAGGED and NOTCH2 genes are crucial in normal angiogenesis, and any defect in these genetic proteins could result in multi-visceral vascular abnormalities. The important sites of involvement include cerebral vessels, mid-aorta, renal vessels and pulmonary arteries.36 The common manifestations in the cerebral vessels include aneurysms, moya-moya arteriopathy (MMA) and dolichoectasia. The incidence varies from 4% to 38% across various studies.44 Up to 25% of asymptomatic patients may have an abnormality in their blood vessels.44 Cerebral aneurysms are predominantly observed in the posterior circulation and may cause symptoms even in children. Hemorrhage from a cerebral aneurysm is an important cause of death in adults suffering from ALGS. MMA has a well-known association with ALGS. It is caused by a stenosis of the distal internal carotid artery with secondary collateral formation. MMA can cause ischemic stroke or transient ischemic attacks in children under 8 years of age and hemorrhagic stroke in adolescents and adults.45 MRI with MRA is diagnostic and shows the classical ivy sign and/or the brush signs. It is recommended that all patients with ALGS be screened with MRI of the brain. The mid-aortic syndrome (MAS) is characterised by stenosis of the proximal aorta along with stenosis of proximal renal and celiac arteries.47 Any patient with ALGS having unexplained hypertension needs to undergo detailed imaging for abdominal vasculature. An untreated MAS may lead to hypertensive encephalopathy and congestive heart failure. Patients with ALGS are also at high risk of systemic bleeds even in the presence of a normal liver function. The bleeding tendency in ALGS could be multifactorial, including inherent anomalies of blood vessels, JAGGED mutation causing deranged homeostasis, hypercholesterolemia and lastly due to coagulopathy caused by chronic cholestasis.48
Management
A corrective surgical procedure for cerebral aneurysm needs to be offered in symptomatic cases, or those prone to rupture. When detected on imaging, MMA needs corrective revascularization procedure including pial or durarterio synangiosis.45 If a child is planned for a major surgery like a LT, the underlying anomalies in the blood vessels of the central nervous system need to be treated prior to that.46 Patients with MAS should undergo percutaneous transluminal angioplasty or surgical correction to rectify the defect.47 The mode of therapy also needs to be decided based on status of liver function as it may have surgical implications (discussed above) during LT.
Skeletal Anomalies
The skeletal anomalies that occur in ALGS are secondary to either the genetic defect or the effect of chronic cholestasis. They may rarely be caused by chronic kidney disease afflicting these patients. Genetic defects in ALGS lead to facial dysmorphism; this diagnostic criterion is seen in up to 95% of the patients and is among the diagnostic criteria for this disorder.49 These characteristics include deep-set eyes, hypertelorism, triangular shaped face and bulbous nose tip, all of which may be noted starting from infancy (Figure 3A and B). The triangular shape of the face in children may disappear as they become adults. It is known that due to increasing awareness of ALGS, facial dysmorphism is being increasingly detected by pediatric hepatologists.50 The commonest skeletal manifestations of ALGS are the vertebral anomalies. These are noticed in at least 66–80% of the patients, of which butterfly vertebrae is the prototypical manifestation.51 It occurs due to a complete fusion defect of the anterior arches and is predominantly noted in the lower thoracic vertebrae (D6–D9).52 Incomplete fusion defects leading to mid-line cleft have also been reported. Even though these skeletal anomalies are also observed in conditions like Kabuki syndrome and 22q11 deletion, multiple vertebral involvement is specific to patients with ALGS. The other skeletal anomalies include spina bifida occulta, hemivertebrae, vertebrae plana, short ulna, absent 12th rib and hypoplastic phalanges.52
 |
Figure 3 Characteristic facies of Alagille syndrome. (A) In an infant with cholestasis of 2 months age. (B) In a 2-year-old child with Alagille syndrome. |
Chronic cholestasis in ALGS patients leads to osteopenia and fractures at multiple sites, of which the long bones are most affected.53 These are aggravated by the inherent genetic defects observed in ALGS. JAGGED1 and NOTCH genes are important for osteoblastic activity and inhibiting osteoclastic activity. The NOTCH gene defect affects endochondral ossification, resulting in a higher incidence of long bone fractures.
Recurrent fractures are an important indication for LT in patients with ALGS.54 With the bone mineral density falling in the first three months after LT, it is important to know that these patients might have an increased risk of fractures in this period.55 The risk is exacerbated by vitamin D deficiency and higher doses of steroids post-LT.
Management
Patients with chronic cholestasis should be started on vitamin D supplements with a dose of 800–1200 IU of cholecalciferol per day. Calcium 50–100 mg/kg/day and phosphorus of 25–50 mg/kg/day also need to supplemented.56 Bisphosphonates like alendronate have a role in improving bone mineral density and may prevent fractures in the pre- and post-LT period.57 Bisphosphonates should only be started in a patient with a normal vitamin D level along with calcium supplements, so as to prevent symptomatic hypocalcemia.
Neurocognitive Effects
The mean intelligence quotient (IQ) in patients with ALGS is lower than in patients who suffer from other cholestatic disorders. One analysis showed that 16% had gross motor delay and 2–10% suffered from intellectual disability.49,58 These children therefore require special education. Patients with ALGS are also known to have higher incidence of attention deficit hyperkinetic disorder, depression, anxiety and eating disorders. They may persist to suffer from cognitive defects in the post-LT period, and a delay in LT has a negative impact on their IQ.59 These children have a lower health-related quality of life (QOL) which is contributed to by intractable pruritus, dysmorphism, xanthomas, cardiac involvement and mental health derangement.60 A family-oriented therapy is required as the parents and their children are equally affected with the emotional burden in such cases. The outcome of patients with ALGS undergoing a LT in relation to QOL remains undefined.
Nutritional Rehabilitation
Patients with ALGS have malnutrition which may commence from infancy due to maldigestion associated with cholestasis.61 They are also malnourished due to the effects of chronic liver disease including poor intake, organomegaly or ascites, portal hypertensive enteropathy causing malabsorption, increasing resting energy expenditure, insulin resistance and abnormal growth hormone signaling.61 There can be renal losses of nutrients due to renal tubular acidosis. It is well known that sarcopenia associated with advanced chronic liver disease is an independent factor predicting mortality and even has a negative impact on outcomes in the post-LT period.62 Hence these patients needs a systematic approach including calculation of daily dietary intake, periodic anthropometric evaluation and personalized nutritional care. The recommendations of dietary constituents in a cholestatic child have been defined.61 Their energy requirements are 130% and protein needs to be given at 130-150% of the requirement for the age. Carbohydrates should be contributing to 40-60 % if the total calories, whereas fat should contribute to 30-50% of the calorie requirements. Of the calories derived from lipid, at least 30% should be contributed by medium chain triglycerides and the rest by long chain triglycerides. Vitamin A should be 5000IU per day in infants less than 10 kg and 10,000 IU for children more than 10 kg weight. Vitamin E need to be given at 15-25 U/kg/day, Vitamin D at 2000-5000 IU/day and Vitamin K at 2.5 – 5 mg /day. The water soluble vitamins needs to give at 2 times the recommended dietary allowance.
Ophthalmic Involvement
Both anterior and posterior chambers of the eye can be involved in patients with ALGS. Posterior embryotoxon, Axenfeld anomaly and microcornea are the predominant anomalies of anterior chamber, whereas pigmentary retinopathy, optic nerve head anomalies and abnormal vessels are the typical findings in the posterior segment of the eye.63 Ophthalmological manifestations are important for diagnosis of ALGS and are included among the major criteria.64 A majority of the anterior segmental anomalies do not impair the functional status of the patient. Elevated optic discs in ALGS are commonly caused by disc drusen, but rarely this can be caused by idiopathic intracranial hypertension.64 Hence a raised intracranial pressure should be considered in these patients having elevated optic discs, especially if there is a progressive loss of vision. There are reports where a lumboperitoneal shunt has been done for the aforementioned indication. Pigmentary retinopathy can be seen in up to 75% patients with ALGS.64 It is characterized by disruption of photoreceptor layer, atrophy or hyperplasia of pigment epithelium and hyperpigmentation of the nerve fiber layer. Rarely, maculopathy is also known to occur in patients with ALGS. Patients can have defects in their peripheral visual field along with nyctalopia. In the majority of cases, the electroretinograms are normal and there is no one-to-one correlation with the severity of pigmentary retinopathy. The natural history of progression of visual field defects in patients with ALGS remains uncertain. Chronic cholestasis in patients with ALGS could lead to vitamin A deficiency. This could lead to corneal ulceration, corneal scar and xerosis of the fundus, leading to loss of vision. Hence a prompt diagnosis is important, and early treatment with therapeutic doses of vitamin A should be offered to these patients.
Pregnancy and ALGS
With dramatic improvements in healthcare, and long-term post-LT outcomes, there are increasing numbers of reports of successful pregnancies in patients with ALGS.65 Those with advanced liver disease may have worsening portal hypertension during pregnancy.66 Also those having right heart involvement may have clinical deterioration. These patients require a closer follow-up during pregnancy. During labor, use of forceps or vacuum is encouraged to minimize maternal efforts. Immunosuppression needs to be modulated in LT recipients planning a family. Due to teratogenicity, antimetabolites need to be stopped at least 6 weeks prior to fertilization.67 Patients should be maintained on tacrolimus with more frequent monitoring of trough levels due to the changes in circulating blood volume.
Immune Dysregulation
The JAGGED–NOTCH pathway is closely associated with CD46 receptors, resulting in a higher risk of recurrent infections of the respiratory tract and the gut due to immune dysregulation in ALGS.68,69 However, the incidence of post-LT infections is similar to that of patients suffering from other etiologies leading to advanced liver disease.70 The defective Jagged gene would also increase T helper cells type 2, predisposing patients to asthma, food allergy and atopic dermatitis.
Conclusion
With dramatic improvements in medical science there is increasing knowledge and awareness of the byzantine presentations of ALGS. Patients who develop end-stage liver disease require a LT. Cardiovascular involvement is an important cause of morbidity and mortality not only in children but also in the adult patients suffering from ALGS and needs to be recognized and treated as early as possible. ALGS is a multi-system disorder requiring a multidisciplinary team of pediatric and adult subspecialists to effectively manage and ensure long-term survival along with a good QOL in these fragile patients.
Parental Consent
The parents have provided consent for the images to be published in the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Turnpenny P, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257. doi:10.1038/ejhg.2011.181
2. Kriegermeier A, Wehrman A, Kamath BM, Loomes KM. Liver disease in Alagille syndrome. In: Kamath B, Loomes K, editors. Alagille Syndrome. Cham: Springer; 2018. doi:10.1007/978-3-319-94571-2_4.
3. Ayoub MD, Kamath BM. Alagille syndrome: diagnostic challenges and advances in management. Diagnostics. 2020;10(11):907. doi:10.3390/diagnostics10110907
4. Kahn E, Daum F. Arteriohepatic dysplasia (Alagille’s syndrome): a common cause of conjugated hyperbilirubinemia. Ann Clin Lab Sci. 1984;14(6):480–486.
5. Krantz ID, Colliton RP, Genin A, et al. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62(6):1361–1369. doi:10.1086/301875
6. Warthen DM, Moore EC, Kamath BM, et al. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006;27:436–443. doi:10.1002/humu.20310
7. Lemaigre FP. Notch signaling in bile duct development: new insights raise new questions. Hepatology. 2008;48(2):358–360. doi:10.1002/hep.22480
8. Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res. 2008;102(10):1169–1181. doi:10.1161/CIRCRESAHA.108.174318
9. Kamath BM, Spinner NB, Piccoli DA, Bezerra JA, Mack CL, Shneider BL. Alagille Syndrome. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children.
10. Subramaniam P, Knisely A, Portmann B, et al. Diagnosis of Alagille syndrome-25 years of experience at King’s College Hospital. J Pediatr Gastroenterol Nutr. 2011;52(1):84–89. doi:10.1097/MPG.0b013e3181f1572d
11. Emerick K, Rand E, Goldmuntz E, Krantz I, Spinner N, Piccoli D. Features of Alagille syn- drome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822–829. doi:10.1002/hep.510290331
12. Quaglia A, Roberts EA, Torbenson M. Developmental and Inherited Liver Disease. In: Burt AD, Ferrell LD, Hubscher SG, editors. MacSween’s Pathology of the Liver.
13. Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110(2):195–200. doi:10.1016/S0022-3476(87)80153-1
14. Andrews AR, Putra J. Central hepatic regenerative nodules in Alagille syndrome: a clinicopathological review. Fetal Pediatr Pathol. 2021;40(1):69–79. doi:10.1080/15513815.2019.1675834
15. Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50(6):580–586. doi:10.1097/MPG.0b013e3181d98ea8
16. Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised Phase 2 study. Lancet. 2021;398(10311):1581–1592. doi:10.1016/S0140-6736(21)01256-3
17. van der Woerd WL, Kokke FT, van der Zee DC, et al. Total biliary diversion as a treatment option for patients with progressive familial intrahepatic cholestasis and Alagille syndrome. J Pediatr Surg. 2015;50(11):1846–1849. doi:10.1016/j.jpedsurg.2015.07.016
18. Kamath BM, Munoz PS, Bab N, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50(5):526–530. doi:10.1097/MPG.0b013e3181cea48d
19. Hsu E, Rand E. Transplant considerations in Alagille syndrome. In: Kamath B, Loomes K, editors. Alagille Syndrome. Cham: Springer; 2018. doi:10.1007/978-3-319-94571-2_5.
20. Valamparampil JJ, Reddy MS, Shanmugam N, Vij M, Kanagavelu RG, Rela M. Living donor liver transplantation in Alagille syndrome-Single center experience from south Asia. Pediatr Transplant. 2019;23(8):e13579. doi:10.1111/petr.13579
21. Yang WH, Zhang L, Xue FS, Riaz A, Zhu ZJ. Pediatric liver transplantation for Alagille syndrome: anesthetic evaluation and perioperative management. Ann Transplant. 2020;25:e924282. doi:10.12659/AOT.924282
22. Kohaut J, Pommier R, Guerin F, et al.Abdominal arterial anomalies in children with Alagille syndrome: Surgical aspects and outcomes of Liver transplantation. J Pediatr Gastroenterol Nutr.2017;64:888-891.
23. Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl. 2012;18(8):940–948. doi:10.1002/lt.23437
24. Kasahara M, Kiuchi T, Inomata Y, et al. Living-related liver transplantation for Alagille syndrome. Transplantation. 2003;75(12):2147–2150. doi:10.1097/01.TP.0000066804.33006.17
25. Englert C, Grabhorn E, Burdelski M, Ganschow R. Liver transplantation in children with Alagille syndrome: indications and outcome. Pediatr Transplant. 2006;10(2):154–158. doi:10.1111/j.1399-3046.2005.00432.x
26. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001;49(3):431–435. doi:10.1136/gut.49.3.431
27. Ovaert C, Germeau C, Barrea C, et al. Elevated right ventricular pressures are not a contraindication to liver transplantation in Alagille syndrome. Transplantation. 2001;72(2):345–347. doi:10.1097/00007890-200107270-00034
28. Quiros-Tejeira RE, Ament ME, Heyman MB, et al. Does liver transplantation affect growth pattern in Alagille syndrome? Liver Transpl. 2000;6(5):582–587. doi:10.1053/jlts.2000.9739
29. Cardona J, Houssin D, Gauthier F, et al. Liver transplantation in children with Alagille syndrome–a study of twelve cases. Transplantation. 1995;60(4):339–342. doi:10.1097/00007890-199508270-00007
30. Tzakis AG, Reyes J, Tepetes K, Tzoracoleftherakis V, Todo S, Starzl TE. Liver transplantation for Alagille’s syndrome. Arch Surg. 1993;128(3):337–339. doi:10.1001/archsurg.1993.01420150093017
31. Bucuvalas JC, Horn JA, Carlsson L, Balistreri WF, Chernausek SD. Growth hormone insensitivity associated with elevated circulating growth hormone-binding protein in children with Alagille syndrome and short stature. J Clin Endocrinol Metab. 1993;76(6):1477–1482. doi:10.1210/jcem.76.6.8501153
32. Ramachandran P, Safwan M, Reddy MS, Rela M. Recent trends in the diagnosis and management of biliary atresia in developing countries. Indian Pediatr. 2015;52(10):871–879. doi:10.1007/s13312-015-0735-6
33. Kaye AJ, Rand EB, Munoz PS, Spinner NB, Flake AW, Kamath BM. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51(3):319–321. doi:10.1097/MPG.0b013e3181df5fd8
34. Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A. 2012;158(1):85–89. doi:10.1002/ajmg.a.34369
35. McElhinney DB, Krantz ID, Bason L, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. doi:10.1161/01.CIR.0000037221.45902.69
36. Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354–1358. doi:10.1161/01.CIR.0000121361.01862.A4
37. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257. doi:10.1038/ejhg.2011.181
38. Trivedi KR, Benson LN. Interventional strategies in the management of peripheral pulmonary artery stenosis. J Interv Cardiol. 2003;16(2):171–188. doi:10.1046/j.1540-8183.2003.08031.x
39. Mainwaring RD, Hanley FL. Surgical techniques for repair of peripheral pulmonary artery stenosis. Semin Thorac Cardiovasc Surg. 2016;28(2):418–424. doi:10.1053/j.semtcvs.2016.07.003
40. Bauser-Heaton H, Borquez A, Han B, et al. Programmatic approach to management of tetralogy of Fallot with major aortopulmonary collateral arteries: a 15-year experience with 458 patients. Circ Cardiovasc Interv. 2017;10(4):e004952. doi:10.1161/CIRCINTERVENTIONS.116.004952
41. Png K, Veyckemans F, De Kock M, et al. Hemodynamic changes in patients with Alagille’s syndrome during orthotopic liver transplantation. Anesth Analg. 1999;89(5):1137–1142.
42. Krowka MJ, Fallon MB, Kawut SM, et al. International liver transplant society practice guidelines: diagnosis and management of hepatopulmonary syndrome and portopulmonary hypertension. Transplantation. 2016;100(7):1440–1452. doi:10.1097/TP.0000000000001229
43. Gandhi SK, Reyes J, Webber SA, Siewers RD, Pigula FA. Case report of combined pediatric heart-lung-liver transplantation. Transplantation. 2002;73(12):1968–1969. doi:10.1097/00007890-200206270-00024
44. Emerick KM, Krantz ID, Kamath BM, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41(1):99–107. doi:10.1097/01.MPG.0000162776.67758.2F
45. Baird LC, Smith ER, Ichord R, et al. Moyamoya syndrome associated with Alagille syndrome: outcome after surgical revascularization. J Pediatr. 2015;166(2):470–473. doi:10.1016/j.jpeds.2014.10.067
46. Pavanello M, Severino M, D’Antiga L, et al. Pretransplant management of basilar artery aneurysm and moyamoya disease in a child with Alagille syndrome. Liver Transpl. 2015;21(9):1227–1230. doi:10.1002/lt.24187
47. Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens. 2012;30(7):1300–1306. doi:10.1097/HJH.0b013e3283531e1f
48. Lykavieris P, Crosnier C, Trichet C, Meunier-Rotival M, Hadchouel M. Bleeding tendency in children with Alagille syndrome. Pediatrics. 2003;111(1):167–170. doi:10.1542/peds.111.1.167
49. Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822–829.
50. Kamath BM, Loomes KM, Oakey RJ, Krantz ID. Facial features in Alagille syndrome: specific or cholestasis facies? Am J Med Genet. 2002a;112(2):163–170. doi:10.1002/ajmg.10579
51. Sanderson E, Newman V, Haigh SF, et al. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002;32(2):114–119. doi:10.1007/s00247-001-0599-x
52. D’Amico A, Perillo T, Cuocolo R, et al. Neuroradiological findings in Alagille syndrome. Br J Radiol. 2021;95(1129):20201241. doi:10.1259/bjr.20201241
53. Bales CB, Kamath BM, Munoz PS, et al. Pathologic lower extremity fractures in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51(1):66–70. doi:10.1097/MPG.0b013e3181cb9629
54. Lee HP, Kang B, Choi SY, et al. Outcome of Alagille syndrome patients who had previously received kasai operation during infancy: a single center study. Pediatr Gastroenterol Hepatol Nutr. 2015;18(3):175–179. doi:10.5223/pghn.2015.18.3.175
55. Collier J. Bone disorders in chronic liver disease. Hepatology. 2007;46(4):1271–1278. doi:10.1002/hep.21852
56. Klein GL, Soriano H, Shulman RJ, et al. Hepatic osteodystrophy in chronic cholestasis: evidence for a multifactorial etiology. Pediatr Transplant. 2002;6(2):136–140. doi:10.1034/j.1399-3046.2002.01060.x
57. Kasturi KS, Chennareddygari S, Mummadi RR. Effect of bisphosphonates on bone mineral density in liver transplant patients: a meta-analysis and systematic review of randomized controlled trials. Transpl Int. 2010;23(2):200–207. doi:10.1111/j.1432-2277.2009.00976.x
58. Leung DH, Sorensen LG, Ye W, et al. Neurocognitive status in alagille syndrome: results of a multi-center prospective observational study. Hepatology. 2017;66(S1):647–648.
59. Lee JM, Jung YK, Bae J-H, et al. Delayed transplantation may affect intellectual ability in children. Pediatr Int. 2017;59(10):1080–1086. doi:10.1111/ped.13369
60. Elisofon SA, Emerick KM, Sinacore JM, et al. Health status of patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51(6):759–765. doi:10.1097/MPG.0b013e3181ef3771
61. Mouzaki M, Bronsky J, Gupte G, et al. Nutrition support of children with chronic liver diseases: a joint position paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2019;69(4):498–511. doi:10.1097/MPG.0000000000002443
62. Boster JM, Browne LP, Pan Z, et al. Higher mortality in pediatric liver transplant candidates with sarcopenia. Liver Transpl. 2021;27(6):808–817. doi:10.1002/lt.26027
63. Ho DK, Levin AV, Anninger WV, Piccoli DA, Eagle RC
64. Kim BJ, Fulton AB. The genetics and ocular findings of Alagille syndrome. Semin Ophthalmol. 2007;22(4):205–210. doi:10.1080/08820530701745108
65. Ferrarese A, Senzolo M, Burra P. Successful pregnancy in Alagille syndrome. Dig Liver Dis. 2015;47(1):86–87. doi:10.1016/j.dld.2014.08.047
66. Mikolasevic I, Filipec-Kanizaj T, Jakopcic I, et al. Liver disease during pregnancy: a challenging clinical issue. Med Sci Monit. 2018;24:4080–4090. doi:10.12659/MSM.907723
67. Parhar KS, Gibson PS, Coffin CS. Pregnancy following liver transplantation: review of outcomes and recommendations for management. Can J Gastroenterol. 2012;26(9):621–626. doi:10.1155/2012/137129
68. Le Friec G, Sheppard D, Whiteman P, et al. The CD46- Jagged1 interaction is critical for human T(H)1 immunity. Nat Immunol. 2012;13(12):1213–1221. doi:10.1038/ni.2454
69. Tilib Shamoun S, Le Friec G, Spinner N, et al. Immune dysregulation in Alagille syndrome: a new feature of the evolving phenotype. Clin Res Hepatol Gastroenterol. 2015;39(5):566–569. doi:10.1016/j.clinre.2015.02.003
70. Baker A. Immune dysregulation in Alagille syndrome: a feature of the evolving phenotype. In: Kamath B, Loomes K, editors. Alagille Syndrome. Cham: Springer; 2018. doi:10.1007/978-3-319-94571-2_10.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.