Back to Journals » The Application of Clinical Genetics » Volume 16
Multicenter Study of Diagnostic Tool for Patients with Hemophilia: From Bedside to Comprehensive Investigations
Authors Chuansumrit A ![]() , Natesirinilkul R, Sirachainan N
, Natesirinilkul R, Sirachainan N ![]() , Kadegasem P, Surapolchai P, Tangbubpha N, Kempka K, Khlangtan T
, Kadegasem P, Surapolchai P, Tangbubpha N, Kempka K, Khlangtan T
Received 8 August 2023
Accepted for publication 5 November 2023
Published 1 December 2023 Volume 2023:16 Pages 215—223
DOI https://doi.org/10.2147/TACG.S434470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Ampaiwan Chuansumrit,1 Rungrote Natesirinilkul,2 Nongnuch Sirachainan,1 Praguywan Kadegasem,1 Pacharapan Surapolchai,3 Noppawan Tangbubpha,1 Ketsuda Kempka,1 Tanyanee Khlangtan1
1Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 2Department of Pediatrics, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand; 3Department of Pediatrics, Faculty of Medicine, Thammasat University, Pathum Thani, Thailand
Correspondence: Ampaiwan Chuansumrit, Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Rachathewi District, Bangkok, 10400, Thailand, Tel +66 2 2011749, Fax +66 2 2011748, Email [email protected]
Background: Hemophilia cannot be diagnosed in most laboratories of economically less-developed countries leading to high mortality and morbidity rates.
Aim: A diagnostic tool was established ranging from bleeding assessment and a simple bedside test of mixing venous clotting time (VCT) to comprehensive DNA analysis for patients with hemophilia.
Methods: Patients with known (n=80) and suspected hemophilia (n=14) were included. Their bleeding symptoms were initially evaluated using verified translated-Thai ISTH bleeding assessment tool. Then, blood samples were drawn using a two-syringe technique, 2 mL each was placed in three tubes, for the mixing VCT and citrate blood was kept for coagulogram and coagulation factor assay. Finally, DNA analysis was determined.
Results: A total of 94 patients with hemophilia (A68, B26) defined as severe (A 57, B 17), moderate (A 7, B 5), and mild degrees (A 4, B 4) with the mean (SD) age of 14.0 (11.7) years and 24 normal controls aged 25.5 (4.5), were enrolled in the study. The mean (SD) bleeding score of patients with hemophilia was 13.5 (5.5), which did not significantly differ between patients with hemophilia A and B. The mixing venous clotting time offered the presumptive diagnosis of hemophilia A and B, which were subsequently confirmed by the prolonged APTT, low FVIII:C and FIX:C and mutations on the factor VIII and IX genes.
Conclusion: A diagnostic tool for bleeding assessment, mixing venous clotting time, coagulogram, coagulation factor assay, and DNA analysis for patients with hemophilia has been established in the existing health-care system.
Keywords: diagnostic tool, hemophilia A & B, bleeding score, mixing VCT, F8 & F9 gene mutation
Introduction
Hemophilia is a hereditary bleeding disorder found in 29.6 cases of 100,000 males at birth1 and is inherited by a sex-linked recessive pattern and found among males, while females are carriers. However, 30% of patients were sporadic cases where the origin of mutations were found in the last two generations of mother and grandmother in 40 of 45 studied families (88.8%). Most carrier mothers had a de novo mutation on the X-chromosome of paternal origin.2 Moreover, inversion of intron 22 has been commonly found in patients with severe hemophilia A.3,4 Patients with hemophilia often present bleeding in the muscles and joints, inducing severe pain. Cases of inadequate replacement therapy of factor VIII or IX concentrates result in deformities of the musculoskeletal system and patients can even succumb to excessive bleeding in the vital organs and central nervous system during childhood and seldom reach adulthood.5 The specific investigation cannot be performed in most laboratories in economically less-developed countries. As a consequence of delayed diagnosis, patients with hemophilia face unfavorable outcomes from avoidable complications. However, the existing health-care system in Thailand involving health stations and district hospitals, provincial hospitals, regional hospitals, and university hospitals act as primary, secondary, tertiary, and comprehensive care centers, respectively. The tertiary and comprehensive care centers already have the capacity of diagnosing hemophilia, while it remains challenging to establish access to hemophilia diagnosis at the primary and secondary care centers.
In 2006, the National Health Security Office in Thailand initiated the nationwide treatment program for Thai patients with hemophilia. Initially, a limited amount of factor VIII and factor IX concentrate was prescribed for patients with hemophilia A and B, respectively. Due to the incompleteness of local medical record and referral system, several patients identified themselves as presenting hemophilia without indicating a specific type of hemophilia A or B and asking for their allowance of factor concentrate at the assigned hemophilia treatment centers. This created constraint among medical personnel, patients, and family members in prescribing proper factor concentrate. The laboratory investigation of factor VIII and factor IX clotting activity (FVIII:C and FIX:C) is not immediately available at the hemophilia treatment centers. Therefore, a diagnostic kit of mixing venous clotting time (VCT) to determine the status of hemophilia A and B at bedside followed by the subsequent coagulation factor assay was invented6,7 to ease the confronted crisis.
The study aimed to establish a complete diagnostic tool for patients with hemophilia ranging from simple bedside tests, including mixing VCT, to comprehensive investigation of mutation identification. It can be applied in rural areas without sophisticated equipment and extended to other low and lower-middle income countries. An accurate diagnosis would be helpful to appropriately manage and lessen the pain of patients and family members. Importantly, morbidity and mortality rates will decrease.
Materials and Methods
An observational study was conducted at the Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, the Faculty of Medicine, Chiang Mai University Hospital, Chiang Mai, and the Faculty of Thammasat University Hospital, Pathum Thani, Thailand, from November 2020 to October 2022. The severity of hemophilia is based on FVIII:C and FIX:C levels: severe <0.01 IU/mL, moderate 0.01 to 0.05 IU/mL and mild >0.05 to <0.40 IU/mL.8 The study was approved by the Faculty Ethics Committee, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand (COA.MURA 2020/1863), and conducted following the Declaration of Helsinki. Written informed consent was obtained from patients and parents.
The study included patients with a known or suspected bleeding disorder of hemophilia, aged 2 years old or older. They might have or not have a family history of bleeding disorder. Patients with known hemophilia abstained from factor concentrate replacement at least 72 hours before participating in the study. The suspected patients were those exhibiting frequent bruise, bleeding at the muscles and joints spontaneously or related to minor trauma. Also, post-dental or surgical-related bleeding was noted. The study excluded patients with liver or kidney diseases and those taking medications or herbs interfering with hemostasis. Also, healthy volunteers without personal and family history of bleeding disorders and not taking any medication were included as normal controls.
All studied patients and normal controls had their bleeding symptoms assessed, their blood drawn for the mixing VCT, coagulogram, and FVIII:C/FIX:C. Inhibitor to FVIII:C or FIX:C was determined in indicated cases. Only patients with a diagnosis of hemophilia had DNA analysis for factor VIII and factor IX gene mutations.
Procedure
Bedside Bleeding Assessment
The bleeding symptoms were evaluated using the verified translated-Thai ISTH bleeding assessment tool9 by a hematologist or well-trained nurse at the outpatient clinic. It contained 14 items of bleeding manifestations with scores ranging from 0 (absence or trivial symptom) to 4 points (symptom requiring medical intervention) for each item. The bleeding score was obtained from the summation of scores from all items ranging from a minimum of 0 to a maximum of 56. The higher scores represented a greater bleeding risk. The bleeding score ≥4 among adult males, ≥6 among adult females and ≥3 among children was considered abnormal as published.10
Bedside Mixing Venous Clotting Time (VCT) Test
The blood samples were drawn using a two-syringe technique. The first one milliliter of blood in the first syringe was placed in an EDTA tube for complete blood count. Then 11 mL of blood was drawn in the second syringe and the first stopwatch was started; 4.5 mL was placed in a citrate tube for coagulogram and coagulation factor determination and 2 mL each was placed in three tubes for the mixing VCT test. The first stopwatch recorded the VCT in the first tube as control. The second and third tubes contained reconstituted factor VIII and factor IX concentrate, respectively. After mixing the patient’s blood with factor concentrate by titling the tubes up and down five times and the second and third stopwatches were started, the clotting time in the second and third tubes was recorded by the respective stopwatches. Patients with hemophilia A will have shortened clotting time in the second tube, while patients with hemophilia B will have shortened clotting time in the third tube compared with the remaining tubes.6,7 The presumptive diagnosis of hemophilia A and B was obtained at this stage.
Further Investigation
The patients’ presumptive diagnosis of hemophilia A and B was confirmed by the prolonged activated partial thromboplastin time (APTT) and low levels of FVIII:C and FIX:C in the citrate plasma. Then the buffy coat from the EDTA and citrate blood was collected for DNA extraction and analyzed for the mutation on the factor VIII and factor IX genes accordingly.
Preparing the Mixing VCT Test
Each set of mixing VCT test contained three glass tubes of 13×100 mm. The clean test tubes were placed in acid dichromate overnight, cleaned with ultrapure water the next day and dried at 37°C until completely dry before use. The plasma-derived factor VIII and factor IX concentrates were reconstituted with sterile water to obtain 100 units/1000 microliter (mcL). Then 2.4 units of factor VIII and factor IX concentrate equal to 24 mcL were aliquoted and added to the bottom of the second and third tubes, respectively. The tubes were properly sealed with parafilm and stored at −20°C before lyophilization. On the day of lyophilization, the parafilm on top of each reconstituted factor concentrate tube was punctured with a small needle and maintained at −20°C for 2 hours, followed by lyophilization using a Thermo Model Supermodulyo −230 for 3 hours. The lyophilized factor VIII and factor IX concentrate tubes were resealed with parafilm and kept at 4°C before use. The lyophilized factor VIII and factor IX concentrate was reconstituted with 30 mcL of sterile water immediately before use.
Coagulation Factor Assay
Whole blood was mixed with 3.2% sodium citrate at the ratio of 9:1, immediately placed on ice and centrifuged at 3000 g for 15 minutes to obtain platelet poor plasma. Coagulogram, FVIII:C, and FIX:C were assayed within 4 hours or in the plasma stored at −20°C for less than 3 months before testing. The FVIII:C and FIX:C assays were based on one-stage APTT11 using the ACL 200 Coagulometer with commercial factor VIII and IX deficient plasma purchased from Instrumentation Laboratories, Lexington, MA, USA. The inhibitor was determined using the Nijmegen-Bethesda assay.12
DNA Analysis
DNA was extracted using an in-house salting out method.13 The intron 22 and intron 1 inversion was the initial mutation determination for patients with severe and moderate hemophilia A using the inverse-shifting PCR technique.14 For patients with hemophilia A without intron 22 and intron 1 inversion and all patients with hemophilia B, all exons were sequenced using direct Sanger sequencing.15
Statistical Analysis
The Student’s paired t-test was used for paired samples, the Chi-Square or Fisher exact test was used for discrete data, while the Mann–Whitney U or Wilcoxon Signed rank test was used for continuous data. The level of significance was set at p≤0.05.
Results
Totally, 94 patients (90 males, 4 females) included 80 cases of known hemophilia and 14 cases of suspected hemophilia. Altogether, 68 patients with hemophilia A and 26 patients with hemophilia B defined as severe (A 57, B 17), moderate (A 7, B 5), and mild degrees (A 4, B 4) with a mean (SD) age of 14.0 (11.7) years were enrolled in the study. However, 7 and 4 patients with severe hemophilia A possessed low inhibitor (0.8 to 4.4 Bethesda unit, BU) and high inhibitor (42.0 to 58.4 BU), respectively. Also, 24 healthy volunteers (12 males, 12 females) with a mean (SD) age of 25.5 (4.5) years were included. The mean (SD) bleeding score of normal controls was very low at 0 (0.2) and that of patients with hemophilia was 13.5 (5.5) which did not significantly differ between patients with hemophilia A and B [13.6 (5.5) vs 13.4 (5.5), p=0.970]. Also, no significant difference of bleeding score was found between known and suspected patients with hemophilia. However, the scores related to muscle and joint bleeding among patients with hemophilia A were significantly higher than those of patients with hemophilia B [2.7 (1.7) vs 1.8 (1.7); 2.9 (1.6) vs 2.1 (1.7)] with p-values of 0.017 and 0.016, respectively.
The mean (SD) bleeding scores among patients with hemophilia A without inhibitor in severe, moderate, and mild degrees did not significantly differ as shown in Table 1. However, patients with severe hemophilia A exhibited significantly higher bleeding scores at the muscle and joint compared with those of patients with mild degree [3.0 (1.6) vs 1.0 (1.4), 2.9 (1.7) vs 0.75 (1.5)] with p-values of 0.033 and 0.026, respectively. Furthermore, patients with moderate hemophilia A had significantly higher bleeding scores at the joint compared with those with mild degree [3.1 (1.5) vs 0.75 (1.45)] with a p-value of 0.024. In addition, the mean (SD) bleeding scores among patients with severe hemophilia A without inhibitor (n=46) of 13.6 (5.4) did not significantly differ from those with inhibitor either low inhibitor (n=7) of 18.3 (5.7) or high inhibitor (n=4) of 12.5 (5.3) with p-values of 0.081 and 0.796, respectively, as well as between patients with high and low inhibitor (p=0.315). Patients with low inhibitor had a slightly higher age of 10.8 (4.9) years than those with high inhibitor of 9.6 (5.8) years but no significant difference (p=0.788). On the contrary, no significant difference was observed of bleeding score at various sites among patients with hemophilia B.
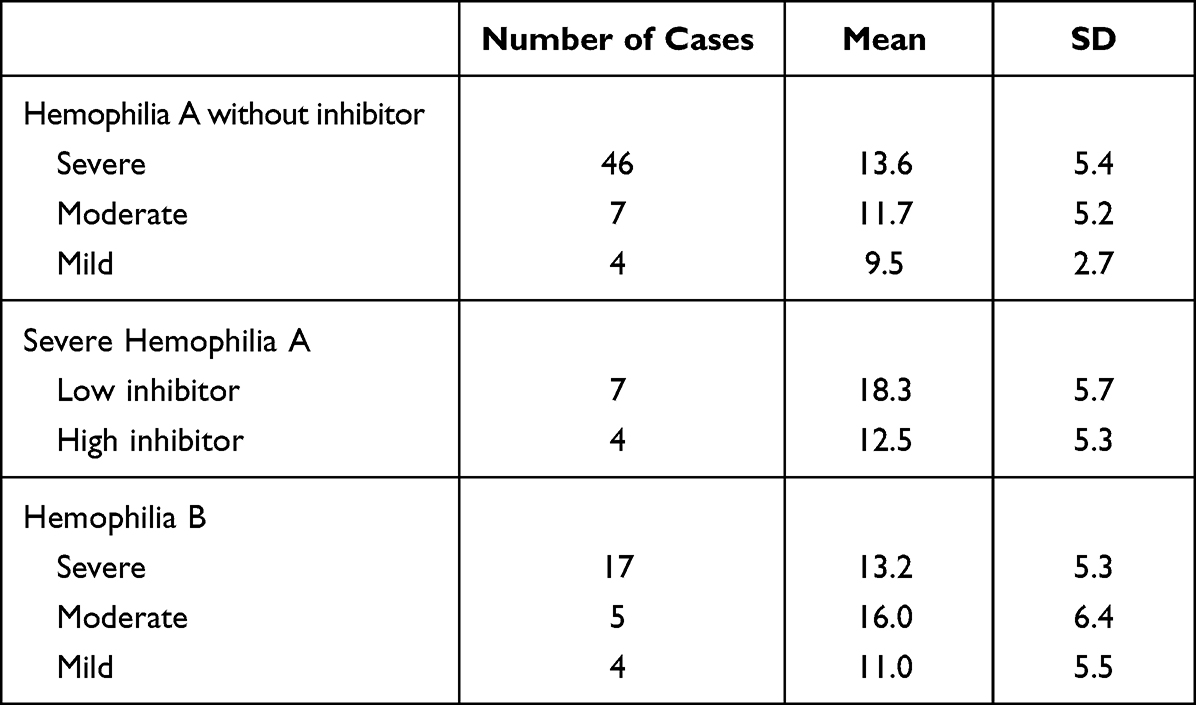 |
Table 1 Bleeding Scores Expressed as Mean and SD Among Patients with Hemophilia A and B |
Interestingly, the mean (SD) bleeding scores among patients older than 10 years (n=32) of 15.6 (5.3) were significantly higher than those ≤10 years (n=62) of 12.4 (5.3) with a p-value of 0.009. They had a higher mean (SD) post-dental procedure bleeding score of 1.7 (1.6) compared with those ≤10 years of 1.1 (1.5) with a p-value of 0.032. In addition, two and one adult females in reproductive life with mild hemophilia A and B, respectively, exhibited heavy menstrual bleeding, postpartum hemorrhage, and excessive postoperative bleeding, while another girl with severe hemophilia A and Turner’s syndrome exhibited frequent bleeding at the muscle and joint similar to those of a boy with severe hemophilia A. The foreseen heavy menstrual bleeding is expected to be more serious than that of females with mild hemophilia and appropriate intervention at the university hospital setting should be planned.
The results of mixing VCT test among the studied patients are shown in Table 2. The median (IQR) VCT of whole blood alone among patients with hemophilia A of 60.0 (24.6, 60.0) minutes (min) was significantly prolonged compared to those with hemophilia B of 17.3 (12.9, 42.8) min with a p-value <0.0001. Some patients with hemophilia B with severe and moderate degrees had a normalized VCT, while some had a prolonged VCT similar to those of patients with hemophilia A. However, both hemophilia A and B with mild degree had a normalized or slightly prolonged VCT, which were significantly shortened than those with respective severe degree with p-values of 0.049 and 0.009. Subsequently, the patients’ VCT were significantly shortened in the second and third tubes with reconstituted factor VIII and factor IX concentrate accordingly, no matter whether the VCT of whole blood alone was initially prolonged or normalized. Interestingly, the percentage of VCT correction among patients with initially normalized VCT was significantly less than those with initially prolonged VCT especially patients with hemophilia B.
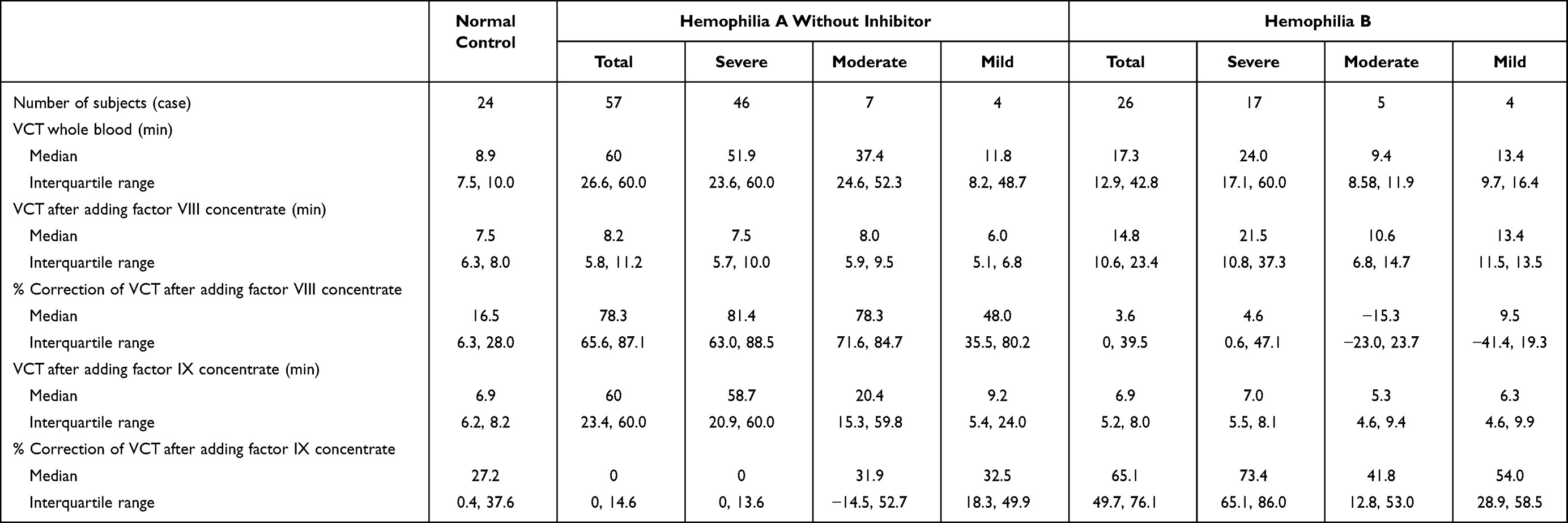 |
Table 2 Venous Clotting Time (VCT) of Whole Blood Alone and the Correction of VCT After Adding Factor VIII and Factor IX Concentrates Among Patients with Hemophilia and Normal Controls |
In addition, the VCT of whole blood alone of patients with severe hemophilia without inhibitor did not significantly differ from those with low and high inhibitor with p-values of 0.083 and 0.096, respectively, as shown in Table 3. The median VCT and the percentage of correction after adding factor VIII concentrate among patients with low inhibitor did not significantly differ from those without inhibitor. On the contrary, the median VCT and the percentage of correction after adding factor VIII concentrate among patients with high inhibitor significantly differed from those without inhibitor (55.5 vs 7.5 min, p<0.0001; 12.0 vs 81.4%, p<0.0001) and low inhibitor (55.5 vs 10.9 min, p=0.006; 12.0 vs 80.4%, p=0.006), respectively. It reflected the uncorrectable and undiagnostic hemophilia A with high inhibitor at bedside. As a result, the mixing VCT had the presumptive diagnosis of hemophilia A and B among 90 of 94 patients (95.7%) except four patients with hemophilia A and high inhibitors.
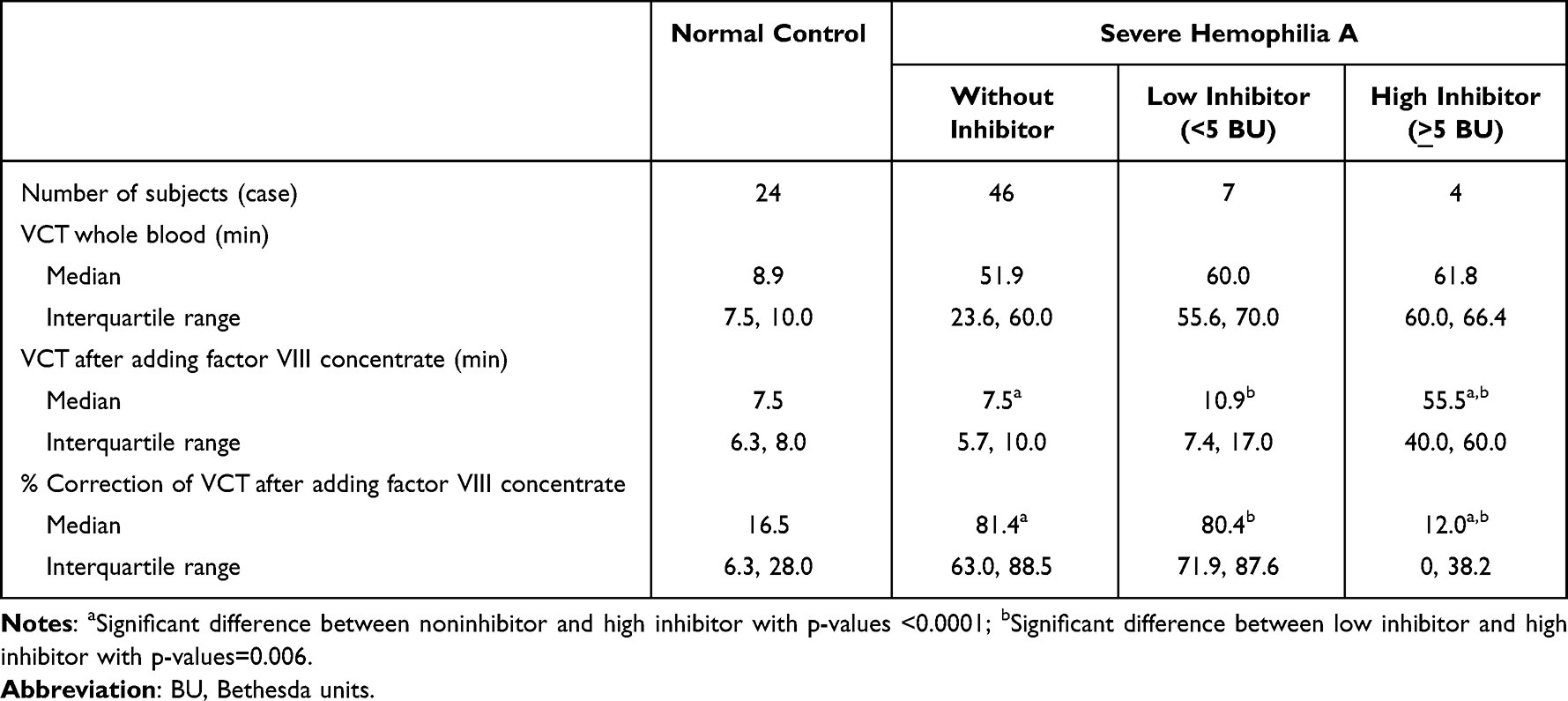 |
Table 3 Venous Clotting Time (VCT) of Whole Blood Alone and the Correction of VCT After Adding Factor VIII Concentrates Among Patients with Severe Hemophilia A with and without Inhibitor |
The coagulogram showed prolonged APTT among all patients except two females with mild hemophilia A with FVIII:C at 0.34 IU/mL exhibiting a slightly prolonged APTT. Further FVIII:C and FIX:C assays provided the definite diagnosis of hemophilia A and B with different severity accordingly. As a result, all patients with the presumptive diagnosis of hemophilia A and B had their diagnosis confirmed. In addition, all healthy volunteers had normalized coagulogram and levels of FVIII:C and FIX:C.
Further DNA analysis revealed mutations on the factor VIII and factor IX genes in all studied patients as shown in Table 4. Inversion intron 22 was commonly found among patients with hemophilia A while the missense mutation was commonly found among patients with hemophilia B. Mutation of A>G transition at position +1157 downstream of the stop codon (c.2545 A>G) in the 3’untranslated region was found in one patient with moderate hemophilia B.16 Three novel mutations were found in 3 patients with severe hemophilia A (c.578G>C, c.2385_2390 del AACACC ins G, c.5642 del C).
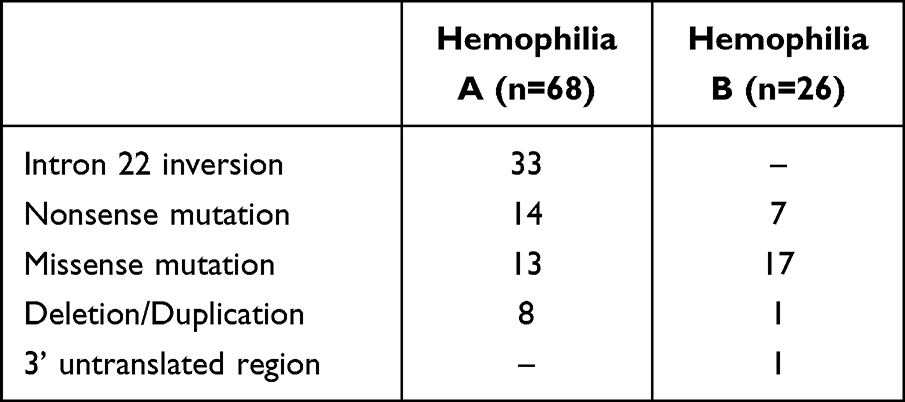 |
Table 4 Mutations on the Factor VIII and IX Genes Among Studied Patients with Hemophilia A and B |
Discussion
Hemophilia is rare and under-recognized in low and lower-middle income countries.17 The definite diagnosis is often delayed in economically less-developed countries, so patients often experience several bleeding episodes during infancy and childhood period resulting in high morbidity and mortality rates. The diagnostic tool for patients with hemophilia in the current study has been designed to apply to the existing health-care system of district, provincial, regional, and university hospitals acting as primary, secondary, tertiary, and comprehensive hemophilia treatment centers, respectively. The initial bleeding assessment and mixing VCT test could be performed at bedside at district hospitals without using sophisticated technology or equipment. They could provide a presumptive diagnosis of hemophilia A and B, which was helpful for prescribing appropriate factor concentrate to control and prevent bleeding episodes.
Patients with hemophilia in the current study exhibited a high bleeding score reflecting the frequent bleeding experience, similar to a related report from Pakistan.18 Similarly, patients at older age exhibited a high bleeding score due to the risk of more bleeding episodes especially post-dental procedure bleeding. Patients received inadequate replacement and improper care resulting in poor dental hygiene. The unawareness and unrecognition of hemophilia among health-care providers leads to avoidable bleeding complications.
The origin of the mixing VCT strategy was the percentage of VCT correction ranging from 40 to 80% after adding the factor VIII or IX concentrate among hemophiliac patients with severe, moderate, and mild degrees even though they had prolonged, slightly prolonged and normalized VCT of their whole blood alone, while healthy controls with normalized level of FVIII:C and FIX:C had percentage of VCT correction ranging from 16.5 to 27.2%. Patients with severe degree had a higher percentage of VCT correction than those with moderate or mild degrees.
Moreover, patients with hemophilia A with high inhibitor ≥5 BU had a lower percentage of their venous clotting time correction after adding factor VIII concentrate in the second tube explained by the insufficient factor VIII concentrate to overcome the high inhibitor. On the contrary, the added factor VIII concentrate was sufficient to neutralize the low inhibitor <5 BU to obtain the correct presumptive diagnosis for patients with hemophilia A.
The in-house mixing VCT test was proved an accurate bedside presumptive diagnostic tool after obtaining patient’s blood for 15 minutes. The diagnostic kit can be kept at 4°C for one year. However, identifying the severity of severe, moderate, and mild degrees is one limitation of the test, and it cannot provide the presumptive diagnosis for patients with hemophilia A and high inhibitors ≥5 BU. Moreover, the current study was not tested in the intended use population nor did their test population include non-hemophilia diagnoses.
The mixing VCT is relevant for centers and countries with limited resources in need of a quick way to identify patients at risk of hemophilia and high inhibitor ≥5 BU. Inspite of an availability of coagulogram, a prolonged APTT cannot distinguish the status of hemophilia A and B, while the mixing VCT has its capacity in determining the status of hemophilia A and B at bedside. To our knowledge, the current mixing VCT to distinguish hemophilia A and B is novel. This could easily be adopted across other geographies especially low and lower-middle income countries and would not require specific hemophilia expertise.
However, the pre-analytical and analytical issues should not be overlooked. The pre-analytical issue of VCT involved four steps. First, the cleaned test tube without reusing process; second, the complete reconstitution of the lyophilized factor VIII and factor IX concentrate from the bottom of the test tubes; third, obtaining freely flow blood sample without partial clot; and finally, the thorough mixing of patient’s blood and the reconstituted factor concentrate. Furthermore, the analytical issue involved the accuracy of starting and stopping the stopwatches for recording the clotting time of each test tube. At least two personnel were required for the mixing VCT.
Therefore, the initial bleeding assessment using the ISTH-bleeding assessment tool could provide information of bleeding risk and the subsequent mixing VCT test could provide the presumptive diagnosis of hemophilia A and B at bedside in different health-care systems such as district hospitals. Further confirmation of prolonged APTT, FVIII:C, and FIX:C assays could be performed on the citrate blood sent to nearby provincial and regional hospitals. For cost saving, the citrate plasma could be stored at −20°C up to 3 months before a monthly or 3-month coagulation factor assay schedule.
Finally, DNA analysis could be performed at the university hospital acting as comprehensive hemophilia treatment centers on EDTA blood or buffy coat sent by mail without ice.19,20 The advanced technology can offer the specific mutations on the factor VIII and factor IX genes among all studied patients in the current study, similar to previous reports.21,22 Genetic testing in routine clinical practice is relevant for complete genetic counseling.23 The definite diagnosis of carriers among family members could be further determined. Alternatively, the informative markers using the linkage analysis of restriction fragment length polymorphisms on the factor VIII and factor IX genes could be provided for regional hospitals to use in both carrier and prenatal diagnosis.
These know-hows could be applied to other countries with limited resources. However, the DNA analysis for the mutation identification requires further planning. The facility for DNA analysis should be shared among common hereditary disease services in the country such as thalassemia and sickle cell anemia.24 The World Federation of Hemophilia may be involved through twinning or laboratory upgrading programs.
In conclusion, a diagnostic tool of bleeding assessment, mixing VCT test, coagulogram and coagulation factor assays, and DNA analysis for patients with hemophilia has been established using the existing health-care resources.
Data Sharing Statement
The data from the findings of the study are available from the corresponding author upon reasonable request.
Acknowledgments
This research was funded by the Grifols Hemophilia Awareness Global Award. Grifols did not have any role in the design, execution, analysis, or interpretation of the study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content, agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
Ampaiwan Chuansumrit acts as advising board of Novo Nordisk and has received honoraria from Novo Nordisk, Grifols, Takeda, and Roche, while the other authors state that they have no interests which might be perceived as posing a conflict or bias in this work.
References
1. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registry. Ann Intern Med. 2019;17(8):540–546. doi:10.7326/M19-1208
2. Mårtensson A, Ivarsson S, Letelier A, Manderstedt E, Halldén C, Ljung R. Origin of mutation in sporadic cases of severe haemophilia A in Sweden. Clin Genet. 2016;90(1):63–68. doi:10.1111/cge.12709
3. Lu Y, Xin Y, Dai J, et al. Spectrum and origin of mutations in sporadic cases of haemophilia A in China. Haemophilia. 2018;24(2):291–298. doi:10.1111/hae.13402
4. Sasanakul W, Chuansumrit A, Sirachainan N, Kadegasem P. Prominent mutation of intron 22 inversion in sporadic hemophilia: is it worth the antenatal screening? Appl Clin Genet. 2022;19(15):49–54. doi:10.2147/TACG.S363132
5. Evatt BL. Demographics of hemophilia in developing countries. Semin Thromb Hemost. 2005;31(5):489–494. doi:10.1055/s-2005-922218
6. Chuansumrit A, Pongtanakul B, Kadegasem P, et al. Accurate bedside diagnostic kit for determining haemophilia A and B. Haemophilia. 2009;15(1):361–364. doi:10.1111/j.1365-2516.2008.01783.x
7. Sasanakul W, Kadegasem P, Chaiyaratana W, Wongwerawattanakoon P, Sirachainan N, Chuansumrit A. Simple and accurate bedside diagnostic kit for determining haemophilia A and B: a revised version. Haemophilia. 2013;19(1):e48–e49. doi:10.1111/hae.12047
8. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
9. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065. doi:10.1111/j.1538-7836.2010.03975.x
10. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost. 2012;10(11):2223–2229. doi:10.1111/j.1538-7836.2012.04923.x
11. Hardisty RM, Macpherson JC. A one-stage factor VIII (antihaemophilic globulin) assay and its use on venous and capillary plasma. Thromb Diath Haemorrh. 1962;7:215–228.
12. Duncan E, Collecutt M, Street A. Nijmegen-Bethesda assay to measure factor VIII inhibitors. Methods Mol Biol. 2013;992:321–333. doi:10.1007/978-1-62703-339-8_24
13. Sasanakul W, Chuansumrit A, Kadegasem P, Hathirat P. A comparison of DNA extraction between conventional phenol-chloroform and in-house modified method. Rama Med J. 1997;20:119–124.
14. Rossetti LC, Radic CP, Larripa IB, De Brasi CD. Developing a new generation of tests for genotyping hemophilia-causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. J Thromb Haemost. 2008;6(5):830–836. doi:10.1111/j.1538-7836.2008.02926.x
15. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74(12):5463–5467. doi:10.1073/pnas.74.12.5463
16. Krooss S, Werwitzke S, Kopp J, et al. Pathological mechanism and antisense oligonucleotide-mediated rescue of a non-coding variant suppressing factor 9 RNA biogenesis leading to hemophilia B. PLoS Genet. 2020;16(4):e1008690. doi:10.1371/journal.pgen.1008690
17. Pierce GF, Adediran M, Diop S, et al. Achieving access to haemophilia care in low-income and lower-middle-income countries: expanded Humanitarian Aid Program of the World Federation of Hemophilia after 5 years. Lancet Hematol. 2022;9(9):e689–e697. doi:10.1016/S2352-3026(22)00209-5
18. Borhany M, Fatima N, Abid M, Shamsi T, Othman M. Application of the ISTH bleeding score in hemophilia. Transfus Apher Sci. 2018;57(4):556–560. doi:10.1016/j.transci.2018.06.003
19. Sasanakul W, Chuansumrit A, Rurgkhum S, Udomsubpayakul U, Hathirat P. DNA extraction and amplification of 10-day, room-temperature blood samples. J Med Assoc Thai. 1999;82(Suppl 1):S186–S189.
20. Kadegasem P, Rurgkhum S, Sasanakul W, Chuansumrit A. DNA extraction from buffy coat sent by mail without ice. Thai J Hematol Transfus Med. 2001;11:167–171.
21. Kulkarni S, Hegde R, Hegde S, et al. Mutation analysis and characterisation of F9 gene in haemophilia- B population of India. Blood Res. 2021;56(4):252–258. doi:10.5045/br.2021.2021016
22. Andersson NG, Labarque V, Letelier A, et al. Novel F8 and F9 gene variants from the PedNet hemophilia registry classified according to ACMG/AMPes. Hum Mutat. 2020;41(12):2058–2072. doi:10.1002/humu.24117
23. Pezeshkpoor B, Oldenburg J, Pavlova A. Insights into the molecular genetic of hemophilia A and hemophilia B: the relevance of genetic testing in routine clinical practice. Hamostaseologie. 2022;42(6):390–399. doi:10.1055/a-1945-9429
24. Chuansumrit A. Treatment of haemophilia in the developing countries. Haemophilia. 2003;9(4):387–390. doi:10.1046/j.1365-2516.2003.00763.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.