Back to Journals » International Journal of Women's Health » Volume 18
Multi-Omics Mendelian Randomization and Colocalization Reveal Key Glycolipid Metabolism-Related Genes in Gestational Diabetes Mellitus
Authors Lin X ![]() , Zheng J
, Zheng J ![]() , Qin N
, Qin N ![]() , Li Y
, Li Y ![]()
Received 17 December 2025
Accepted for publication 9 May 2026
Published 20 May 2026 Volume 2026:18 589782
DOI https://doi.org/10.2147/IJWH.S589782
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Everett Magann
Xiaoxiao Lin,* Jingjing Zheng,* Ningning Qin, Yimei Li
Department of Obstetrics and Gynecology, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325027, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yimei Li, Email [email protected]
Purpose: Gestational diabetes mellitus (GDM) poses significant health risks, yet the causal genetic and epigenetic mechanisms linking glycolipid metabolism dysregulation to GDM remain elusive. This study aimed to identify key causal genes and regulatory pathways by integrating multi-omics data with large-scale genetic association studies.
Patients and Methods: We leveraged GDM genome-wide association study (GWAS) data from the FinnGen consortium (18,581 cases/263,483 controls) as the discovery dataset and the UK Biobank (193 cases/219,789 controls) for replication. These were integrated with blood-based quantitative trait loci (QTL) for DNA methylation (mQTL), gene expression (eQTL), and protein abundance (pQTL) of glycolipid metabolism-related genes. Findings were rigorously validated using colocalization analysis (PP.H4 > 0.5), cross-QTL mediation analysis, and tissue-specific eQTL data (adipose, liver, pancreas) from GTEx. Protein-protein interaction (PPI) networks and drug druggability analyses were conducted to assess translational potential.
Results: The multi-omics SMR analysis identified 325 CpG sites, 58 gene expressions, and 6 protein abundances associated with GDM, which were refined by colocalization to 72 mQTLs, 6 eQTLs, and 3 pQTLs. Integrative analysis prioritized 6 core genes: HKDC1, CYP21A2, DCXR, GCDH, ACOT4, and GPX1. HKDC1 was identified as a core hub; its hypomethylation was associated with decreased expression and increased GDM risk, which was further validated in adipose, liver, and pancreatic tissues. CYP21A2 overexpression was confirmed as a risk factor, a finding substantiated by independent replication in the UK Biobank. Additionally, DCXR and GCDH were identified as novel susceptibility genes supported by significant gene-protein regulatory cascades. Finally, protein-protein interaction network and drug target analyses highlighted HKDC1 as a potential druggable target.
Conclusion: This study identifies glycolipid metabolism genes, particularly HKDC1, in GDM via a methylation-expression-phenotype axis, and nominates DCXR and GCDH as novel candidates. These findings reveal new molecular insights and therapeutic targets for GDM.
Keywords: gestational diabetes mellitus, glycolipid metabolism, Mendelian randomization, multi-omics, genetic regulation
Introduction
Gestational diabetes mellitus (GDM), hyperglycemia first detected during pregnancy, is a common complication affecting approximately 14% of pregnancies globally and posing significant health risks to both mother and offspring.1,2 In China, the prevalence of GDM has been reported to range from 14% to 20%, reflecting a trend similar to the global rise and contributing substantially to the national health burden.3 The rising GDM incidence, paralleling obesity and type 2 diabetes epidemics, underscores its substantial health and economic burden.1 While established risk factors like maternal age and obesity are known, the precise molecular mechanisms, particularly the causal roles of genetic and epigenetic factors in maternal metabolism, remain largely unclear.2,4 Therefore, elucidating these underlying molecular mechanisms is crucial for developing effective prevention and treatment strategies for GDM.
Glycolipid metabolism, encompassing the intricate pathways of glucose and lipid processing, is fundamental to maintaining maternal metabolic homeostasis, particularly during the physiological stresses of pregnancy.5 Pregnancy itself induces a state of progressive insulin resistance, primarily to ensure an adequate glucose supply to the developing fetus.6 Concurrently, maternal lipid profiles undergo significant alterations, including increased triglycerides and cholesterol levels, to support fetal growth and maternal energy demands.7 However, when these adaptive metabolic changes are dysregulated, or when pre-existing susceptibilities are unmasked by pregnancy, GDM can ensue. Observational studies have consistently linked abnormal maternal glucose levels and dyslipidemia, particularly elevated triglycerides and altered high-density lipoprotein cholesterol (HDL-C), to an increased risk of GDM.8,9 Despite these associations, the causal nature of specific alterations in glycolipid metabolic pathways and the key genetic regulators involved in GDM pathogenesis remain to be fully elucidated.
To overcome the limitations of traditional observational studies, such as confounding and reverse causation, Mendelian Randomization (MR) offers a robust approach for causal inference by utilizing genetic variants as unconfounded instrumental variables.10 Moreover, the increasing availability of large-scale genome-wide association study (GWAS) data and molecular quantitative trait loci (QTL) data has paved the way for powerful multi-omics MR studies.11–13 However, previous studies have largely examined these molecular layers in isolation, limiting the understanding of how genetic and epigenetic factors jointly regulate glycolipid metabolism to influence GDM risk. The lack of integrated multi-omics approaches hinders the identification of regulatory cascades spanning from DNA methylation to gene expression and protein abundance, and their collective impact on disease pathogenesis. This study, therefore, leverages a systematic multi-omics Summary-data-based MR (SMR) approach to investigate the causal effects of genetic variants influencing DNA methylation, gene expression, and protein levels of glycolipid metabolism-related genes on GDM risk. We aim to integrate blood-derived mQTL, eQTL, and pQTL data with large-scale GDM GWAS summary statistics to identify glycolipid metabolism-related genes potentially linked to GDM. The robustness of these findings will be further assessed through rigorous colocalization analysis to identify shared causal variants, cross-cohort replication in an independent GDM dataset, and tissue-specific eQTL validation in key metabolic organs. Additionally, the multi-omics cascade, defined as the sequential flow where genetic variants influence DNA methylation, which modulates gene expression and subsequent protein abundance to ultimately contribute to disease phenotype, will be explored to characterize regulatory relationships between omics layers, with protein-protein interaction network analysis and drug target prediction employed to elucidate the molecular mechanisms and translational value of identified GDM candidate genes. This comprehensive strategy is designed to systematically uncover novel causal genes in GDM, ultimately contributing to a deeper understanding of its pathogenesis and identifying new avenues for prevention and therapy.
Material and Methods
Study Design
This study employed SMR to systematically identify glycolipid metabolism-related genes causally associated with GDM risk. The overall analytical workflow is depicted in Figure 1. Briefly, we first collected summary statistics from large-scale GDM GWAS and QTL data (mQTLs, eQTLs, pQTLs) related to glycolipid metabolism genes. These molecular QTLs were then assessed for their causal associations with GDM. Significant associations were further scrutinized using colocalization analysis to identify shared causal genetic variants. Findings from the discovery GDM cohort (FinnGen) were then subjected to replication in an independent GDM cohort (UK Biobank). We also conducted cross-QTL SMR analyses to explore regulatory relationships between methylation and gene expression, and between gene expression and protein abundance. Tissue-specific eQTL validation was performed using data from the Genotype-Tissue Expression (GTEx) project for GDM-relevant tissues. Finally, genes showing robust evidence were carried forward for protein-protein interaction (PPI) network analysis and drug target prediction to identify core genes and translational applications.
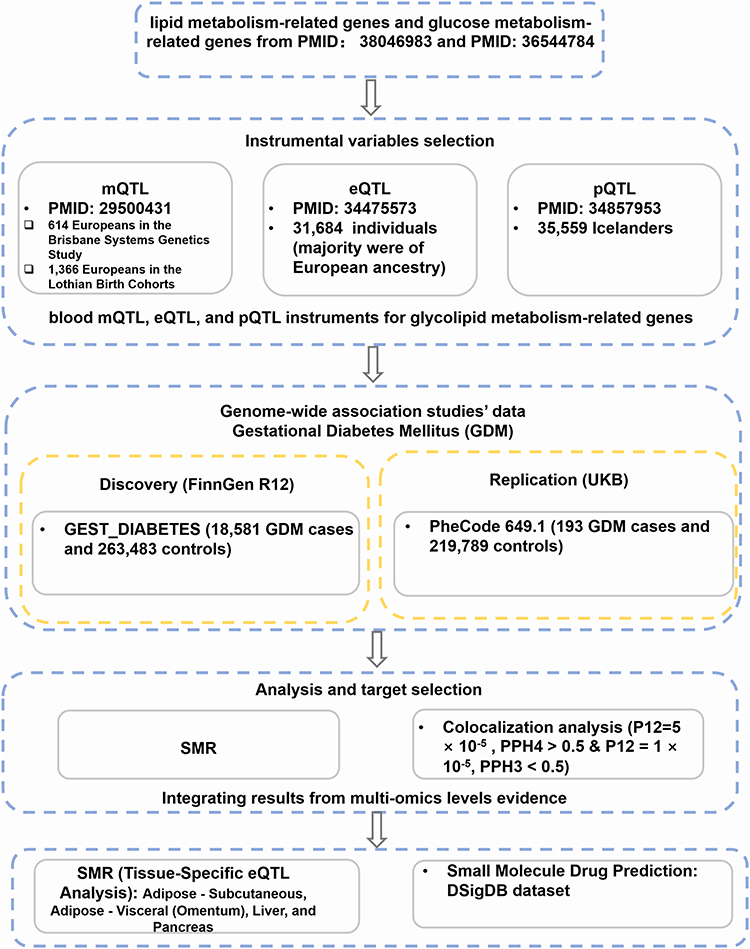 |
Figure 1 Methodological flowchart of the multi-omics Mendelian Randomization study. |
Glycolipid Metabolism-Related Gene Sets and Molecular QTL Data
The curated list of glycolipid metabolism-related genes was compiled based on previously published literature, with lipid metabolism-related genes identified from a 2023 publication utilizing KEGG, MSigDB, and GSEA databases,14 and glucose metabolism-related genes from a 2022 publication referencing the KEGG database15 (detailed in Box S1 and S2).
Summary-level data for blood-based molecular QTLs were obtained from several large-scale consortia and studies. Blood mQTL data were derived from a meta-analysis of 1,980 individuals of European ancestry from the Brisbane Systems Genetics Study and the Lothian Birth Cohorts, as reported by McRae et al.16 Blood eQTL summary statistics were sourced from the eQTLGen consortium, which analyzed genetic data from 31,684 individuals, predominantly of European descent.17 Blood pQTL data were obtained from a study by Pietzner et al, which included 10,708 European donors.18
GDM GWAS Summary Statistics
Summary statistics for GDM were primarily sourced from the FinnGen consortium, release R12 (https://www.finngen.fi/en), utilizing the GEST_DIABETES trait ID. This discovery dataset comprised 18,581 GDM cases and 263,483 controls of European ancestry, with analysis performed on 20,989,973 SNPs. For validation purposes, GDM GWAS summary statistics corresponding to PheCode 649.1 (Diabetes or abnormal glucose tolerance complicating pregnancy) were obtained from the UK Biobank (UKB) through the PheWeb portal (https://pheweb.org/). The UKB replication dataset included 193 GDM cases and 219,789 controls of European ancestry, covering 55,612,751 SNPs.
Tissue-Specific eQTL Data
To investigate the tissue-specific relevance of identified gene-GDM associations, eQTL summary statistics from GDM-relevant human tissues were retrieved from the GTEx project (https://gtexportal.org/home/), release v8. The GTEx v8 dataset encompasses data from 838 donors across 52 tissues and two cell lines. For the present study, eQTL data from four key metabolic tissues implicated in GDM pathophysiology were utilized: Adipose-Subcutaneous, Adipose-Visceral (Omentum), Liver, and Pancreas.
SMR Analyses
SMR analyses were conducted using the SMR software package (v1.3.1)12 to assess the causal relationships between molecular QTLs (CpG methylation, gene expression, protein abundance) and GDM risk, as well as to explore regulatory links between different omic layers (mQTLs to eQTLs, eQTLs to pQTLs). The SMR method leverages summary-level data from GWAS and QTL studies, providing increased statistical power when exposure and outcome data originate from large, independent cohorts. For cis-QTL selection, we considered SNPs located within ±1000 kb of the target gene/protein probe for eQTLs, pQTLs, and mQTL probes. Only top-associated cis-QTLs reaching genome-wide significance (p < 5.0 × 10−8) in the respective QTL studies were used as instrumental variables. Default SMR parameters were employed, including linkage disequilibrium (LD) estimation from the 1000 Genomes Project European reference panel. SNPs with an allele frequency difference greater than 0.2 between the QTL and GWAS datasets were excluded; if more than 5% of SNPs were excluded, the analysis for that probe was halted. The Heterogeneity in Dependent Instruments (HEIDI) test was utilized to distinguish causality from pleiotropy or linkage, with associations considered statistically significant if p SMR < 0.05 and p HEIDI > 0.05.
Colocalization Analysis
To determine whether an observed association between a molecular QTL and GDM was due to a shared causal variant rather than linkage disequilibrium, colocalization analysis was performed using the coloc R package.19 For each significant SMR signal, we assessed the posterior probability (PP) of five mutually exclusive hypotheses: H0 (no association with either trait), H1 (association with QTL only), H2 (association with GDM only), H3 (association with both traits, distinct causal variants), and H4 (association with both traits, shared causal variant). Following published guidelines,20–22 colocalization region windows were set to ±500 kb for mQTL-GWAS, and ±1000 kb for eQTL-GWAS and pQTL-GWAS analyses. A colocalization signal, indicative of a shared causal variant, was defined as PP.H4 > 0.5 (with prior p12 = 5 × 10−5) and PP.H3 < 0.5 (with prior p12 = 1 × 10−5).23
PPI Network Analysis
PPI network analysis was conducted for genes/proteins whose expression or abundance levels showed a significant SMR association with GDM risk in the blood eQTL-GWAS or pQTL-GWAS analyses. The STRING database (https://cn.string-db.org/) was used to construct the PPI network, with interactions based on Homo sapiens and a medium confidence score threshold (set at 0.4). The resulting network was visualized and further analyzed using Cytoscape software.24 Hub genes within the network were identified using the cytoHubba plugin, employing seven topological analysis algorithms: Degree, Stress, Radiality, Closeness, Maximal Clique Centrality (MCC), Maximum Neighborhood Component (MNC), and Edge Percolated Component (EPC). The top 20 nodes from each algorithm were selected, and genes appearing in the overlap of all seven algorithms were designated as key Hub Nodes.
Small Molecule Drug Prediction
To explore the druggability of the GDM-associated genes/proteins identified through SMR eQTL/pQTL analyses, we utilized the Drug Signatures Database (DSigDB v1.0, https://dsigdb.tanlab.org/DSigDBv1.0/). This database was queried to identify existing drugs or investigational compounds targeting these gene products, with each hit classified into one of the following categories: D1 (approved drugs); D2 (kinase inhibitors); D3 (compound-induced gene expression changes); D4 (database/literature-supported drug features).
Statistical Analysis
All statistical analyses were performed using R software (v4.3.0). Manhattan plots were generated using the ggplot2 and ggrepel packages. Forest plots were created using the forestplot package. Locus plots (SMRLocusPlot) and effect plots (SMREffectPlot) for SMR results were generated using scripts adapted from Zhu et al.12
Results
Identification of Glycolipid Metabolism-Related Molecular QTLs Associated with GDM via SMR and Colocalization
Multi-omics SMR analyses were performed using FinnGen GDM GWAS data as the outcome to identify causal associations with blood-based molecular QTLs. Associations meeting the criteria p SMR < 0.05 and p HEIDI > 0.05 were considered significant. A summary of all significant SMR associations identified in the FinnGen discovery cohort for mQTLs, eQTLs, and pQTLs is provided in Supplementary Table S1A–C.
Initially, SMR analysis identified 325 CpG sites (annotated to 160 genes) whose methylation levels (mQTLs) were significantly associated with GDM risk in the FinnGen cohort (details in Supplementary Table S2). For example, the methylation level of CpG site cg04586622, located in ADCY3, was positively associated with GDM risk (OR = 1.06, 95% CI [1.03–1.09]), while cg11023668 in the same gene showed a negative association (OR = 0.96, 95% CI [0.94–0.97]). To identify shared causal variants (PP.H4 > 0.5 and PP.H3 < 0.5), subsequent colocalization analysis refined these findings to 72 CpG sites (48 genes), demonstrating strong evidence of a shared genetic basis with GDM GWAS signals.
For gene expression (eQTLs), the SMR analysis revealed 58 genes whose expression levels were significantly associated with GDM risk (Supplementary Table S3). For instance, HKDC1 expression was negatively associated with GDM risk (OR = 0.88, 95% CI [0.83–0.94]). Among these, 6 genes (ACSL5, CYP21A2, GLTP, GPAM, HKDC1, RBKS) showed robust colocalization evidence.
Regarding protein abundance (pQTLs), 6 proteins (encoded by APOC1, CROT, DCXR, GCDH, MDH1, MINPP1) were found to be significantly associated with GDM risk (Supplementary Table S4). For example, DCXR protein abundance was positively associated with GDM risk (OR = 1.18, 95% CI [1.05–1.33]). 3 of these proteins (APOC1, DCXR, MINPP1) exhibited colocalization evidence. Representative forest plots summarizing these significant SMR associations for mQTLs, eQTLs, and pQTLs are presented in Figure 2, and Manhattan plots illustrating the genomic distribution of these key findings from the discovery cohort are shown in Figure 3. Representative colocalization plots for a key mQTL (cg05532178 for HKDC1), eQTL (HKDC1), and pQTL (DCXR) are provided in Supplementary Figure S1.
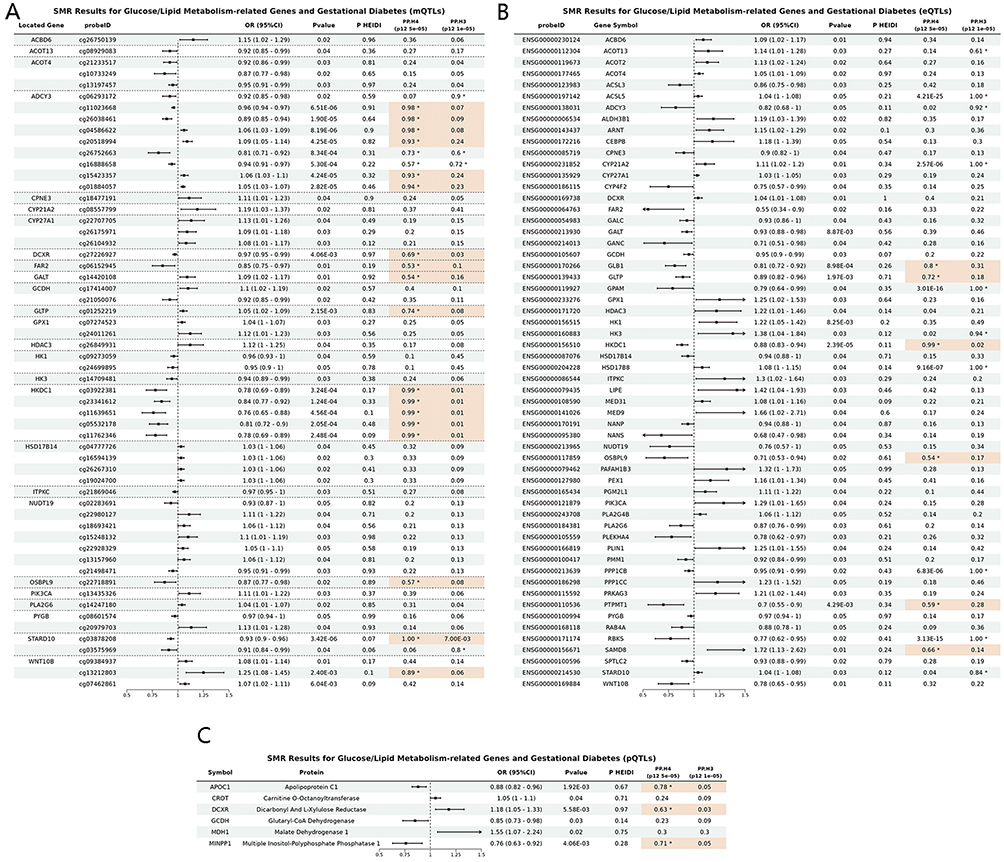 |
Figure 2 Forest plots of significant SMR associations between molecular QTLs and GDM risk in the FinnGen discovery cohort. Forest plots illustrating the estimated causal effect sizes (Odds Ratios, OR) and 95% confidence intervals (CI) for molecular QTLs significantly associated with GDM risk (p SMR < 0.05 and p HEIDI > 0.05). (A) For blood methylation QTLs (mQTLs), due to the large number of significant associations (325 CpG sites), this panel displays all 58 CpG sites located within or near the 26 unique genes that were identified as significant in the eQTL-GWAS SMR analysis, providing a focused overview of mQTLs potentially acting in conjunction with eQTLs. (B) Blood expression QTL (eQTL) SMR analysis results for significant genes. (C) Blood protein QTL (pQTL) SMR analysis results for significant proteins. The asterisk (*) indicates PPH4 > 0.5. Signals meeting colocalization criteria (PPH4 > 0.5 and PPH3 < 0.5) are highlighted in Orange. |
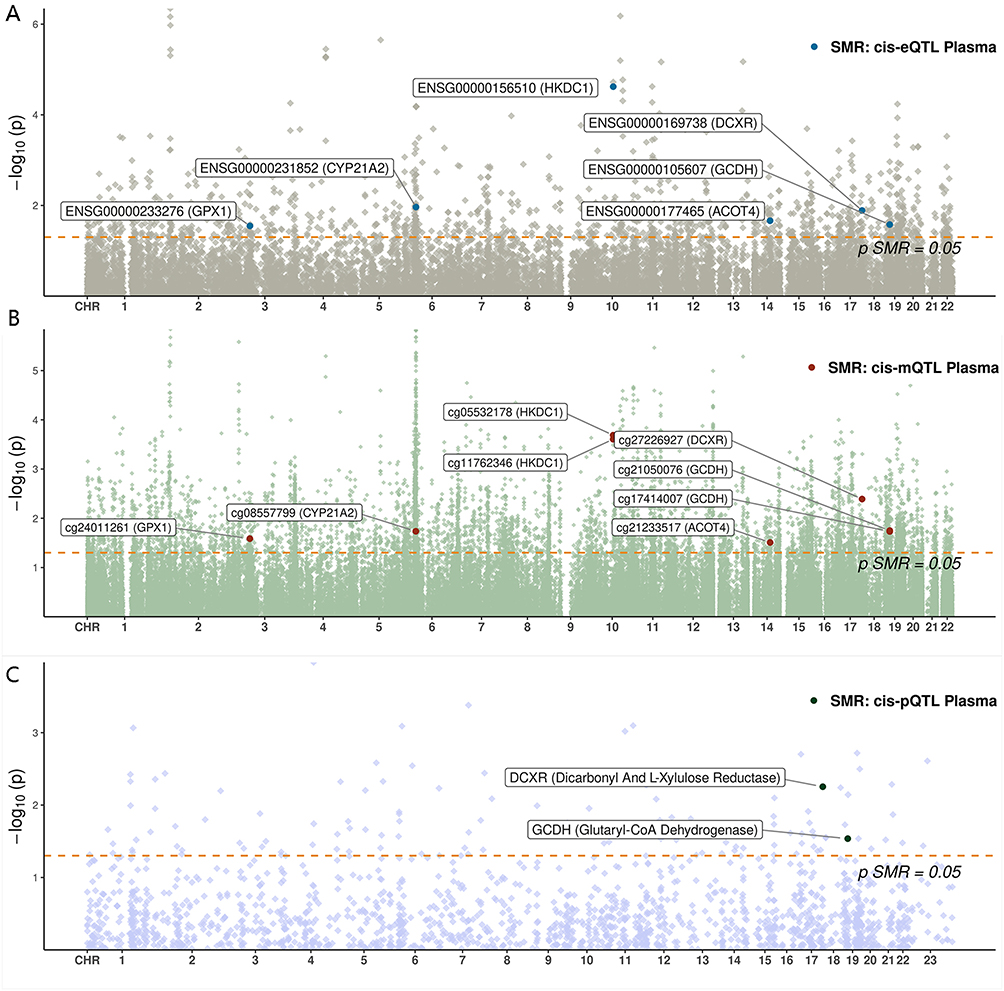 |
Figure 3 Manhattan plots illustrating the genomic distribution of GDM-associated molecular QTLs in the FinnGen discovery cohort. Manhattan plots showing the genomic positions of (A) CpG sites (mQTLs), (B) genes (eQTLs), and (C) proteins (pQTLs) significantly associated with GDM risk through SMR analysis. The red dashed line represents the significance threshold for SMR analysis at P = 0.05. |
Cross-Cohort Replication of SMR Findings in UK Biobank
To validate the robustness of the associations identified in the FinnGen discovery cohort, replication analyses were conducted in the independent UK Biobank GDM cohort (PheCode 649.1). For mQTLs, 13 CpG sites, associated with genes including ACSF3, CYP21A2, were successfully replicated (SMR < 0.05 and p HEIDI > 0.05) (Supplementary Table S5). Notably, the mQTL for CYP21A2 (cg08557799) was positively associated with GDM risk (OR=1.19, 95% CI [1.03–1.37]) in the discovery dataset, with validation in the replication dataset (OR=5.82, 95% CI [1.78–18.99]). At the eQTL level, the positive association between CYP21A2 gene expression and GDM risk was validated (discovery: OR = 1.11, 95% CI [1.02–1.20]; replication: OR = 2.84, 95% CI [1.51–5.37]) (Supplementary Table S6). However, none of the pQTL signals were replicated in the UK Biobank cohort (Supplementary Table S7).
Cross-Omics Regulatory Relationships
To explore potential causal regulatory cascades, two-step SMR analyses were performed, applying the same significance criteria. The analysis of mQTLs influencing eQTLs revealed that specific CpG site methylations significantly regulated the expression of nearby genes. Notably, methylation at cg21233517 was associated with ACOT4 expression, cg24011261 with GPX1 expression, and cg05532178/cg11762346 with HKDC1 expression (Table 1, full results in Supplementary Table S8). Furthermore, the eQTL to pQTL SMR analysis demonstrated that the gene expression levels of DCXR and GCDH significantly regulated the abundance of their respective encoded proteins (Table 1, full results in Supplementary Table S9).
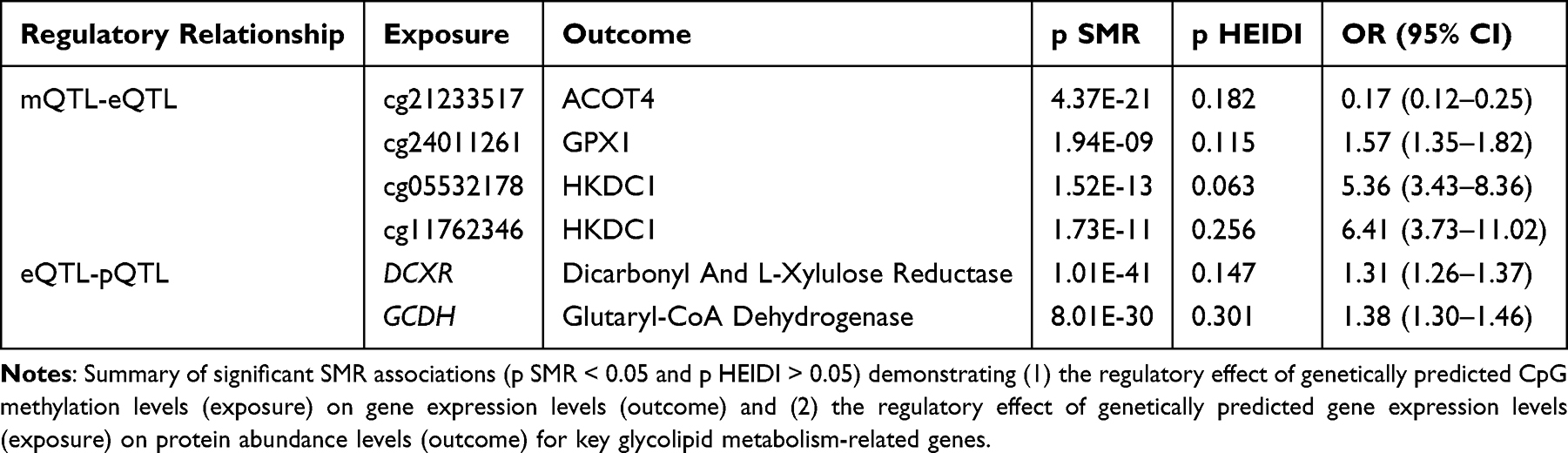 |
Table 1 Significant Cross-QTL SMR Analysis Results Indicating Potential Regulatory Relationships |
Tissue-Specific Validation of eQTL-GDM Associations
To assess the biological relevance of the identified eQTL-GDM associations in metabolically active tissues, SMR analyses were conducted using eQTL data from the GTEx project for adipose (subcutaneous and visceral omentum), liver, and pancreas. In subcutaneous adipose tissue, the expression of 17 genes, including ACOT4, CYP21A2, DCXR, GCDH, and HKDC1, showed validated associations with GDM risk. Similarly, in visceral omentum adipose tissue, 12 genes, including ACOT4, DCXR, GCDH,and HKDC1, were validated. Liver-specific validation confirmed associations for HKDC1, NUDT19, and STARD10, while pancreatic tissue validation supported associations for 6 genes, including ACOT4, GCDH, and HKDC1 (Table 2, full results in Supplementary Table S10A–D). These tissue-specific findings lend further support to the potential roles of ACOT4, HKDC1, CYP21A2, DCXR, and GCDH in GDM pathogenesis. GPX1 could not be assessed in these specific tissue eQTL datasets due to insufficient instrumental variables meeting the SMR analysis criteria.
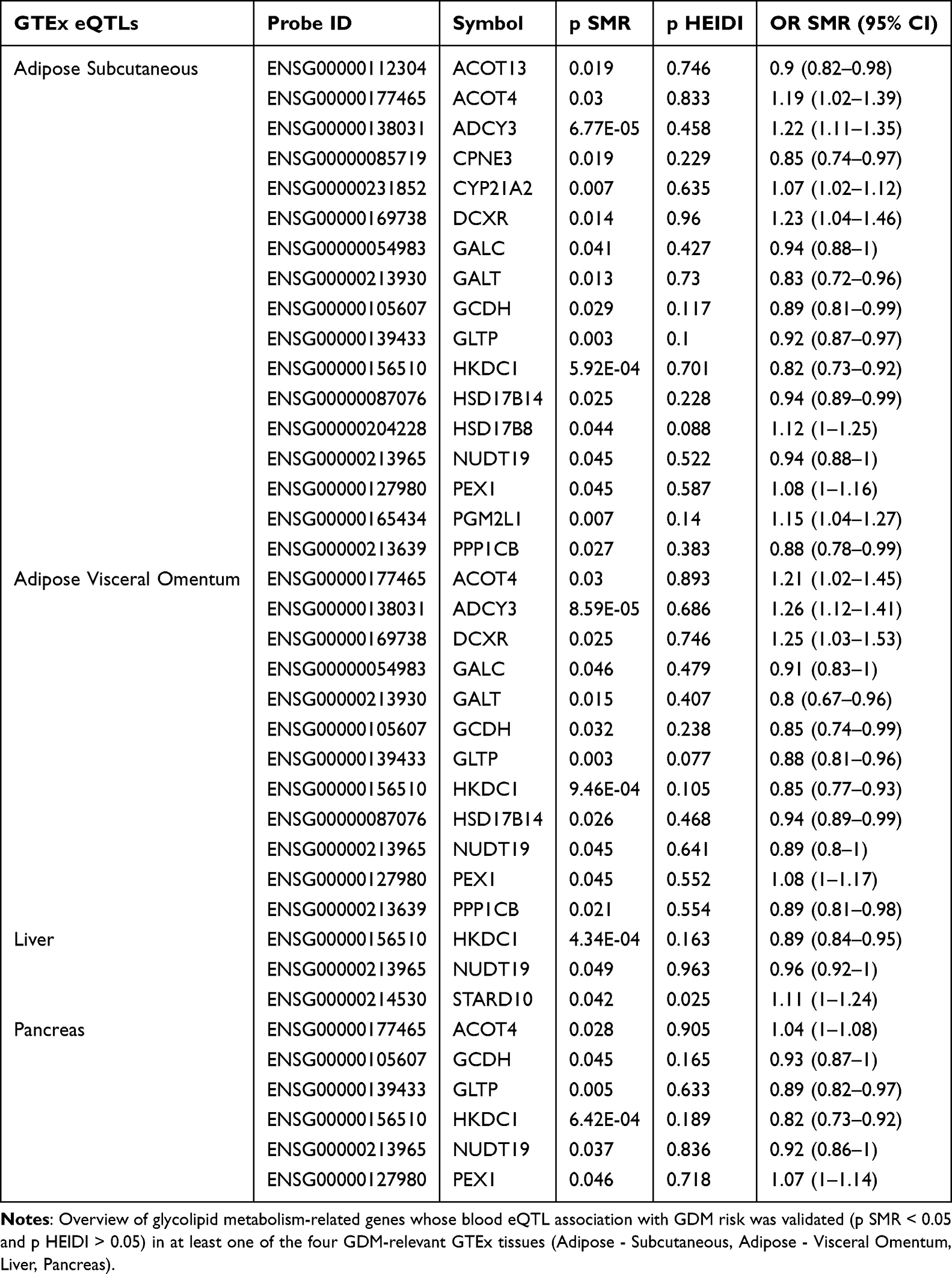 |
Table 2 Summary of Tissue-Specific eQTL Validation Results for GDM-Associated Genes in GTEx Tissues |
Integrated Multi-Omics Evidence Highlights Core Candidate Genes
Synthesizing evidence from multi-omics SMR, colocalization, cross-cohort replication, cross-QTL regulatory chains, and tissue-specific validations, several core genes emerged with strong indications of causal involvement in GDM.
HKDC1 displayed the most compelling evidence. Multiple blood mQTLs annotated to HKDC1 were significantly associated with GDM risk (cg05532178: OR = 0.81, 95% CI [0.72–0.90]; cg11762346: OR = 0.78, 95% CI [0.69–0.89]; both of which also showed strong colocalization). Similarly, HKDC1 gene expression was significantly associated and colocalized with GDM risk. A causal regulatory link from these CpG methylations (cg05532178 and cg11762346) to HKDC1 expression was established, and its eQTL-GDM association was validated across all four tested GDM-relevant tissues.
Novel candidates DCXR and GCDH also showed robust multi-omics support. For DCXR, blood mQTL (cg27226927: OR = 0.97, 95% CI [0.95–0.99]), eQTL (OR = 1.04, 95% CI [1.01–1.08]), and pQTL (as mentioned in section 2.1) levels were significantly associated with GDM risk, with colocalization reported for mQTL and pQTL. For GCDH, blood eQTL (OR = 0.95, 95% CI [0.90–0.99]) and pQTL levels were associated with GDM. Furthermore, eQTL-pQTL regulatory relationships were confirmed for both, and their eQTL-GDM associations were validated in adipose tissues, with GCDH also validated in pancreas. Representative locus plots and SMR effect plots for DCXR are provided in Supplementary Figures S2 and S3, respectively.
Other key genes with notable evidence include ACOT4, for which a significant mQTL-eQTL regulatory chain was identified (cg21233517 methylation negatively influencing ACOT4 expression). Its genetically predicted expression was positively associated with GDM risk, and this eQTL-GDM association was validated in subcutaneous adipose, visceral omentum adipose, and pancreatic tissues. GPX1 also featured a significant mQTL-eQTL regulatory link (cg24011261 methylation positively influencing GPX1 expression), with its genetically predicted expression being positively associated with GDM risk in blood. Lastly, CYP21A2 demonstrated robust evidence with both its mQTL and eQTL associations with GDM risk being replicated in the UK Biobank. Furthermore, the positive association of CYP21A2 expression with GDM risk was validated in subcutaneous adipose tissue.
PPI Network and Drug Target Analysis
A PPI network was constructed using the 62 genes/proteins identified as significantly associated with GDM risk from the primary SMR analyses (Figure 4). Topological analysis of this network identified 14 hub genes, including ACOT2, ACSL3, ACSL5, CYP27A1, GPAM, HK1, HK3, HKDC1, LIPE, MDH1, PLIN1, PPP1CB, PRKAG3, and PYGB (details in Supplementary Table S11), underscoring their central roles in the GDM-associated molecular network. Notably, HKDC1 was confirmed as a hub gene.
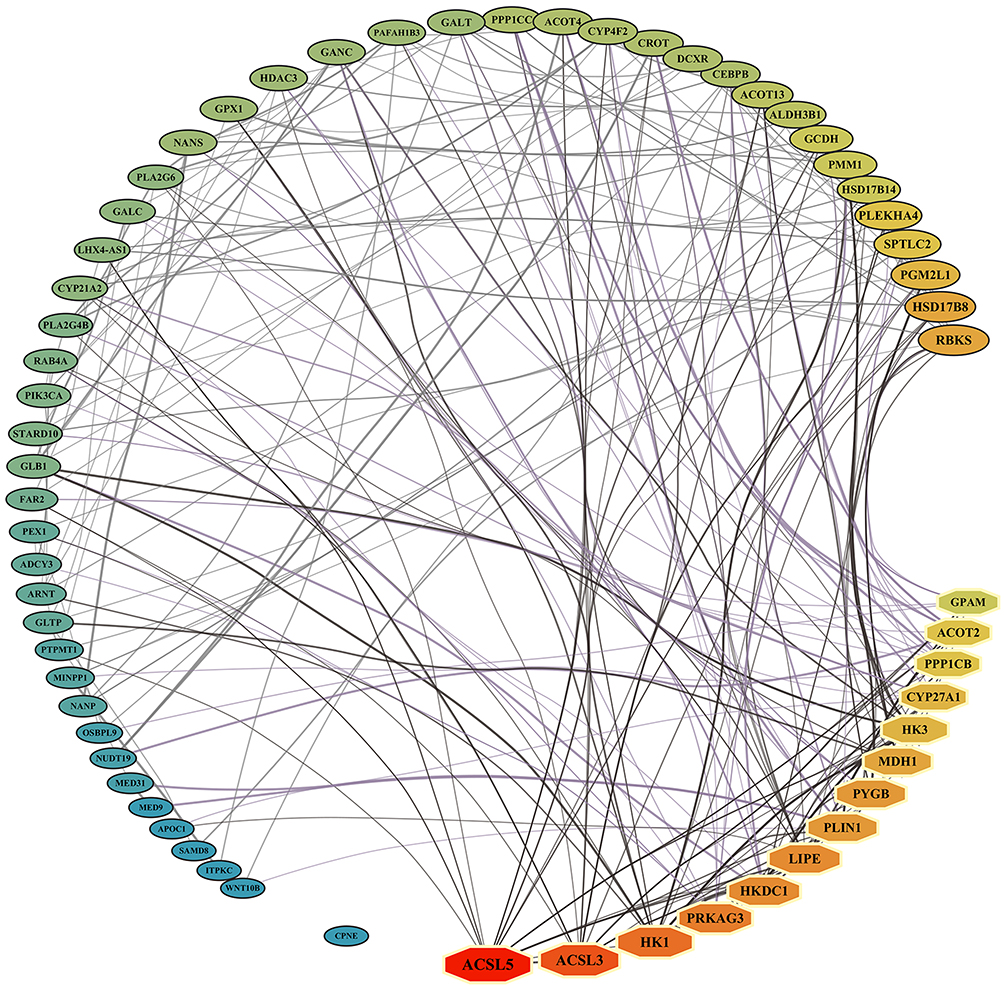 |
Figure 4 Protein-Protein Interaction (PPI) network of GDM-associated genes/proteins. PPI network constructed from the 62 genes/proteins identified through SMR eQTL/pQTL analyses as significantly associated with GDM risk. Key Hub Nodes within the interaction network are identified by octagonal shapes, while other candidate target nodes are represented by ellipses. Node size and color reflect the degree value (small to large, blue to green to red). |
Druggability analysis using the DSigDB database revealed that 42 of the 62 genes/proteins have potential interactions with existing drugs or compounds under investigation. Among the six core candidate genes highlighted in this study (HKDC1, CYP21A2, ACOT4, DCXR, GCDH, GPX1), HKDC1 was identified as a known target of existing compounds (eg., chlorhexidine). Additionally, other hub genes such as ACSL3, ACSL5, HK1, LIPE, PLIN1, PPP1CB, and PYGB were also identified as known targets of various drugs or compounds, suggesting avenues for therapeutic exploration in GDM (details in Supplementary Table S12).
Discussion
This comprehensive multi-omics MR study provides strong genetic evidence for the causal involvement of several glycolipid metabolism-related genes in GDM. By integrating genetic associations with methylation, gene expression, and protein abundance, and employing rigorous validation steps including colocalization, cross-cohort replication, tissue-specific eQTL analysis, PPI network construction, and drug target prediction, we have identified key molecular players relevant to GDM pathogenesis. Notably, our findings highlight HKDC1, CYP21A2, DCXR, GCDH, ACOT4, and GPX1 as core candidate genes with varying degrees of multi-layered evidence supporting their involvement in GDM.
Our study strongly reinforces the role of HKDC1, an enzyme with hexokinase activity involved in glucose metabolism,25 as a key candidate gene for GDM susceptibility. This aligns with GWAS that have consistently linked genetic variants in HKDC1 to gestational glucose homeostasis, particularly 2-hour glucose levels during pregnancy.26 Notably, the influence of HKDC1 on glucose regulation may be particularly pronounced or specific to the gestational period, distinguishing its role from many type 2 diabetes susceptibility loci.27 Functional studies in animal models further support its importance, demonstrating that hepatic HKDC1 can improve whole-body glucose tolerance during pregnancy,28 while intestinal HKDC1 may modulate dietary glucose absorption under metabolic stress.29 Our multi-omics MR results significantly extend these findings by establishing robust causal links from genetic variation, through specific CpG site methylations (cg05532178 and cg11762346) and subsequent gene expression changes, to GDM risk. We delineated a novel regulatory axis where lower methylation at these sites was associated with decreased HKDC1 expression, which in turn was associated with an increased risk of GDM. This eQTL-GDM association for HKDC1 was consistently validated across multiple metabolically active tissues (adipose, liver, and pancreas), underscoring its broad importance. Furthermore, its identification as a hub gene in our PPI network and as a known target of existing compounds (eg., chlorhexidine) elevates its clinical relevance for future GDM interventions.
The causal association of CYP21A2, an enzyme essential for adrenal steroidogenesis, particularly cortisol and aldosterone synthesis,30 with GDM was also supported by our findings. Mutations in CYP21A2 are the most common cause of congenital adrenal hyperplasia (CAH), an autosomal recessive disorder characterized by impaired steroidogenesis and subsequent androgen excess.30,31 Importantly, pregnant women with classic CAH are reported to be at an increased risk of developing GDM,30 and adults with classic CAH more frequently exhibit obesity, hyperinsulinemia, and insulin resistance compared to the general population, conditions that promote metabolic syndrome.31 Distinct from CAH-causing mutations, our findings suggest a broader role for CYP21A2 in GDM. Replicated mQTL and eQTL associations, with eQTL validation in adipose tissue, indicate that genetically predicted increased CYP21A2 expression is a risk factor for GDM. This implies that regulatory variations affecting CYP21A2, potentially by influencing steroid hormone balance and insulin sensitivity, contribute to GDM susceptibility beyond overt CAH.
A significant contribution of this study is the identification of DCXR and GCDH as novel candidate genes causally associated with GDM risk, for which direct links to GDM were previously scarce. DCXR, an enzyme active in L-xylulose reduction and involved in pentose and glucuronate interconversions as part of the polyol pathway, participates broadly in carbohydrate metabolism.32,33 Increased flux through the polyol pathway has been implicated in hyperglycemic conditions and the development of diabetic complications, partly via oxidative stress.34 Furthermore, DCXR expression has been linked to glycolysis in cancer cells34 and its dysregulation observed in type 2 diabetic rat hearts, where it is involved in clearing advanced glycation end-products.35 Our multi-omics findings, demonstrating that increased DCXR expression and protein abundance are associated with increased GDM risk, with an eQTL-pQTL regulatory chain and tissue-specific validation in adipose tissue, strongly suggest its involvement. Similarly, GCDH, a mitochondrial enzyme essential for the catabolism of L-lysine, L-hydroxylysine, and L-tryptophan,36 is traditionally linked to the rare metabolic disorder Glutaric Acidemia Type I (GA-I).37 However, emerging evidence points to the role of altered amino acid metabolism in insulin resistance and type 2 diabetes,38 and GCDH has been identified as a differentially expressed gene common to both obesity and hepatocellular carcinoma, potentially influencing immune infiltration.39 Our comprehensive findings for GCDH, including multi-omics associations (where decreased expression and protein abundance were associated with increased GDM risk), an eQTL-pQTL link, and validation in adipose and pancreatic tissues, suggest that dysregulation in specific amino acid metabolic pathways may contribute to GDM. These novel causal links for DCXR and GCDH warrant urgent functional validation to precisely elucidate their roles in GDM.
Our study also provides new insights into ACOT4, an enzyme that hydrolyzes acyl-CoAs and thus plays a significant role in fatty acid metabolism.40 Recent research has highlighted the involvement of ACOT4 in metabolic regulation. For instance, ACOT4 knockdown was shown to alleviate gluconeogenesis and lipid accumulation in hepatocytes, suggesting it as a potential target for non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes.40 Another study indicated that ACOT4 is a key gene regulated by Theabrownin in improving lipid metabolism in the skeletal muscle of GDM offspring.41 We identified a causal regulatory chain from CpG methylation (cg21233517) to ACOT4 expression, and increased ACOT4 expression to higher GDM risk. This finding, supported by consistent eQTL-GDM validation in adipose and pancreatic tissues, suggests a potential mechanism whereby dysregulation of fatty acid metabolism, mediated by ACOT4, contributes to GDM. GPX1 is a key antioxidant enzyme crucial in oxidative stress defense by reducing hydrogen peroxide and organic hydroperoxides.42 We identified a causal regulatory chain where methylation at cg24011261 increases GPX1 expression, which was associated with an increased risk of GDM. This observation seems counterintuitive, considering the generally protective function of GPX1 against oxidative stress, which is a recognized contributor to GDM.43 Indeed, some studies in GDM women have reported lower levels of the enzymatic antioxidant GPX compared to controls.43 However, the regulation and role of GPX1 can be complex. For instance, animal studies have shown that high dietary selenium intake, which can increase GPX1 expression and activity, induced gestational diabetes and insulin resistance in rats and their offspring.44 This suggests that an overabundance or dysregulation of GPX1 activity might not always be beneficial and could even contribute to metabolic disturbances. The observed positive association in our study warrants further investigation to understand the context-specific role of GPX1 in GDM.
The multi-omics MR design represents a major strength, allowing for the inference of causal relationships from genetic variation through molecular changes to GDM risk, thereby mitigating confounding and reverse causation inherent in observational studies. The integration of mQTL, eQTL, and pQTL data, coupled with the use of large-scale GWAS cohorts and comprehensive QTL datasets, provided substantial statistical power for discovery. Rigorous validation steps, including colocalization analysis, independent cohort replication, and tissue-specific eQTL validation in metabolically relevant organs, enhanced the biological plausibility of our findings. The identification of hub genes and their druggability assessment further provides a translational perspective.
Despite these strengths, several limitations should be acknowledged. First, the validity of MR relies on its core assumptions. While we applied HEIDI tests and colocalization analysis to mitigate pleiotropic effects, the possibility of residual or unmeasured confounding pathways influencing our results cannot be entirely ruled out. Second, the UK Biobank validation cohort, while independent, had a significantly smaller number of GDM cases (n=193) compared to the FinnGen discovery cohort (n=18,581). This limited replication sample size substantially reduced statistical power, which may partly explains the lack of replication of pQTL signals and the wide confidence intervals in the replication cohort. Therefore, further validation in larger, independent GDM cohorts with adequate power for all omics layers is crucial. Third, the QTL data used were primarily derived from blood samples of largely non-pregnant individuals. DNA methylation, gene expression, and protein levels can undergo dynamic changes during pregnancy to adapt to physiological demands. Thus, the QTLs utilized may not fully capture the specific regulatory landscape of gestation, potentially impacting the accuracy of GDM associations, highlighting an urgent need for future MR studies to leverage QTL data generated from samples collected during pregnancy and from pregnancy-specific tissues like the placenta. Fourth, our study population was predominantly of European ancestry, which may limit the generalizability of our findings to other ethnic groups where GDM prevalence and genetic architecture may differ; future studies in diverse populations are warranted. Fifth, while SMR infers directionality, complex molecular interplay require further functional validation, and the identification of GDM-associated genes as known targets of existing drugs or compounds via DSigDB is exploratory.
Future work should focus on functional characterization using CRISPR-Cas9-mediated gene editing in appropriate cellular or animal models, thereby confirming the causal involvement of candidate genes and dissecting the proposed regulatory cascades. Investigating cell-type specific effects within GDM-relevant tissues (eg., pancreatic beta cells, adipocytes, hepatocytes) could provide a more granular understanding of GDM mechanisms. The clinical implications of these findings represent crucial avenues for subsequent investigation. The identified genes may serve as early predictive biomarkers through polygenic risk scores constructed from their SNPs, enabling identification of high-risk populations. Additionally, the proteins encoded by these genes represent potential novel therapeutic targets for GDM intervention. Furthermore, understanding how these genetically influenced metabolic pathways interact with modifiable lifestyle factors, such as diet and physical activity, could open new avenues for personalized prevention strategies.
Conclusion
In conclusion, this study, through a comprehensive multi-omics MR analysis, identified a robust set of glycolipid metabolism-related genes (particularly HKDC1, CYP21A2, DCXR, GCDH, ACOT4, and GPX1 as key candidate genes) potentially associated with GDM. This study not only pinpoints HKDC1 as a central hub with a novel methylation-expression-GDM risk axis but also uncovers DCXR and GCDH as novel GDM-related genes, thereby expanding our understanding of GDM pathogenesis. These genetically validated targets offer promising avenues for future functional characterization, the development of novel diagnostic biomarkers, and targeted therapeutic strategies for GDM.
Data Sharing Statement
Summary statistics for the GWAS datasets used are publicly available from the FinnGen database (R12 version, GEST_DIABETES, https://www.finngen.fi/en/access_results) and UK Biobank (PheCode 649.1, https://www.ukbiobank.ac.uk/). Blood eQTL data are available from the eQTLGen consortium. Blood mQTL summary data are available from McRae et al (2018). Blood pQTL summary data are available from Pietzner et al (2021). Tissue-specific eQTL data are available from the GTEx Portal (https://gtexportal.org/home/). The SMR software (v1.3.1) is publicly available at https://cnsgenomics.com/software/smr/.
Ethics Approval and Consent to Participate
The study protocol was reviewed and approved by the Research Ethics Committee of the Second Affiliated Hospital of Wenzhou Medical University and Yuying Children’s Hospital of Wenzhou Medical University (Approval No. 2026-K-186-01). All methods were performed in accordance with the relevant guidelines and regulations.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Basic Public Welfare Scientific Research Project of Wenzhou Municipal Science and Technology Bureau (grant number Y2023059).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Sweeting A, Hannah W, Backman H. et al. Epidemiology and management of gestational diabetes. Lancet. 2024;404(10448):175–14. doi:10.1016/S0140-6736(24)00825-0
2. Kong D, Kowalik O, Garratt E, Godfrey KM, Chan SY, Teo AKK. Genetics and epigenetics in gestational diabetes contributing to type 2 diabetes. Trends Endocrinol Metab. 2025;36(10):929–942. doi:10.1016/j.tem.2025.03.014
3. Gao C, Sun X, Lu L, Liu F, Yuan J. Prevalence of gestational diabetes mellitus in mainland China: a systematic review and meta-analysis. J Diabetes Investig. 2019;10(1):154–162. doi:10.1111/jdi.12854
4. Atta N, Ezeoke A, Petry CJ, Kusinski LC, Meek CL. Associations of High BMI and Excessive Gestational Weight Gain With Pregnancy Outcomes in Women With Type 1 Diabetes: a Systematic Review and Meta-analysis. Diabetes Care. 2024;47(10):1855–1868. doi:10.2337/dc24-0725
5. Chandra M, Paray AA. Natural Physiological Changes During Pregnancy. Yale J Biol Med. 2024;97(1):85–92. doi:10.59249/JTIV4138
6. Kampmann U, Knorr S, Fuglsang J, Ovesen P. Determinants of Maternal Insulin Resistance during Pregnancy: an Updated Overview. J Diabetes Res. 2019;2019:5320156. doi:10.1155/2019/5320156
7. Mulder J, Kusters DM, Roeters van Lennep JE, Hutten BA. Lipid metabolism during pregnancy: consequences for mother and child. Curr Opin Lipidol. 2024;35(3):133–140. doi:10.1097/MOL.0000000000000927
8. Hivert MF, Backman H, Benhalima K, et al. Pathophysiology from preconception, during pregnancy, and beyond. Lancet. 2024;404(10448):158–174. doi:10.1016/S0140-6736(24)00827-4
9. Liu Y, Kuang A, Bain JR, et al. Maternal Metabolites Associated With Gestational Diabetes Mellitus and a Postpartum Disorder of Glucose Metabolism. J Clin Endocrinol Metab. 2021;106(11):3283–3294. doi:10.1210/clinem/dgab513
10. Lee S, Lee W. A Review of Mendelian Randomization: assumptions, Methods, and Application to Obesity-Related Diseases. J Obes Metab Syndr. 2025;34(1):14–26. doi:10.7570/jomes24031
11. Dong XX, Chen DL, Chen HM, et al. DNA methylation biomarkers and myopia: a multi-omics study integrating GWAS, mQTL and eQTL data. Clin Epigenetics. 2024;16(1):157. doi:10.1186/s13148-024-01772-1
12. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
13. Molendijk J, Parker BL. Proteome-wide Systems Genetics to Identify Functional Regulators of Complex Traits. Cell Syst. 2021;12(1):5–22. doi:10.1016/j.cels.2020.10.005
14. Jiang C, Peng M, Dai Z, Chen Q. Screening of Lipid Metabolism-Related Genes as Diagnostic Indicators in Chronic Obstructive Pulmonary Disease. Int J Chron Obstruct Pulmon Dis. 2023;18:2739–2754. doi:10.2147/COPD.S428984
15. Yang Y, Yang Y, Liu J, et al. Establishment and validation of a carbohydrate metabolism-related gene signature for prognostic model and immune response in acute myeloid leukemia. Front Immunol. 2022;13:1038570. doi:10.3389/fimmu.2022.1038570
16. McRae AF, Marioni RE, Shah S, et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci Rep. 2018;8(1):17605. doi:10.1038/s41598-018-35871-w
17. Võsa U, Claringbould A, Westra HJ, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–1310. doi:10.1038/s41588-021-00913-z
18. Pietzner M, Wheeler E, Carrasco-Zanini J, et al. Mapping the proteo-genomic convergence of human diseases. Science. 2021;374(6569):eabj1541. doi:10.1126/science.abj1541
19. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
20. Morrow JD, Glass K, Cho MH, et al. Human Lung DNA Methylation Quantitative Trait Loci Colocalize with Chronic Obstructive Pulmonary Disease Genome-Wide Association Loci. Am J Respir Crit Care Med. 2018;197(10):1275–1284. doi:10.1164/rccm.201707-1434OC
21. Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213.
22. Yoshiji S, Butler-Laporte G, Lu T, et al. Proteome-wide Mendelian randomization implicates nephronectin as an actionable mediator of the effect of obesity on COVID-19 severity. Nat Metab. 2023;5(2):248–264. doi:10.1038/s42255-023-00742-w
23. Pairo-Castineira E, Rawlik K, Bretherick AD, et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature. 2023;617(7962):764–768. doi:10.1038/s41586-023-06034-3
24. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
25. Zapater JL, Lednovich KR, Khan MW, Pusec CM, Layden BT. Hexokinase domain-containing protein-1 in metabolic diseases and beyond. Trends Endocrinol Metab. 2022;33(1):72–84. doi:10.1016/j.tem.2021.10.006
26. Pervjakova N, Moen GH, Borges MC, et al. Multi-ancestry genome-wide association study of gestational diabetes mellitus highlights genetic links with type 2 diabetes. Hum Mol Genet. 2022;31(19):3377–3391. doi:10.1093/hmg/ddac050
27. Lowe Jr WL. Genetics and Epigenetics: implications for the Life Course of Gestational Diabetes. Int J Mol Sci. 2023;24(7):6047.
28. Khan MW, Priyadarshini M, Cordoba-Chacon J, Becker TC, Layden BT. Hepatic hexokinase domain containing 1 (HKDC1) improves whole body glucose tolerance and insulin sensitivity in pregnant mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865(3):678–687. doi:10.1016/j.bbadis.2018.11.022
29. Zapater JL, Wicksteed B, Layden BT. Enterocyte HKDC1 Modulates Intestinal Glucose Absorption in Male Mice Fed a High-fat Diet. Endocrinology. 2022;163(6). doi:10.1210/endocr/bqac050
30. Maher JY, Gomez-Lobo V, Merke DP. The management of congenital adrenal hyperplasia during preconception, pregnancy, and postpartum. Rev Endocr Metab Disord. 2023;24(1):71–83. doi:10.1007/s11154-022-09770-5
31. Ambroziak U, Bednarczuk T, Ginalska-Malinowska M, et al. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency - management in adults. Endokrynol Pol. 2010;61(1):142–155.
32. Wang Q, Gao B, Yue X, et al. Weighted Gene Co-expression Network Analysis Identifies Specific Modules and Hub Genes Related to Subacute Ruminal Acidosis. Front Vet Sci. 2022;9:897714. doi:10.3389/fvets.2022.897714
33. Hara A, Nishinaka T, Abe N, El-Kabbani O, Matsunaga T, Endo S. Dimeric dihydrodiol dehydrogenase is an efficient primate 1,5-anhydro-D-fructose reductase. Biochem Biophys Res Commun. 2020;526(3):728–732. doi:10.1016/j.bbrc.2020.03.176
34. Garg SS, Gupta J. Polyol pathway and redox balance in diabetes. Pharmacol Res. 2022;182:106326. doi:10.1016/j.phrs.2022.106326
35. Edhager AV, Povlsen JA, Løfgren B, Bøtker HE, Palmfeldt J. Proteomics of the Rat Myocardium during Development of Type 2 Diabetes Mellitus Reveals Progressive Alterations in Major Metabolic Pathways. J Proteome Res. 2018;17(7):2521–2532. doi:10.1021/acs.jproteome.8b00276
36. Braissant O, Jafari P, Remacle N, Cudré-Cung HP, Do Vale Pereira S, Ballhausen D. Immunolocalization of glutaryl-CoA dehydrogenase (GCDH) in adult and embryonic rat brain and peripheral tissues. Neuroscience. 2017;343:355–363. doi:10.1016/j.neuroscience.2016.10.049
37. Yoldas Celik M, Canda E, Yazici H, et al. Glutaric aciduria type 1: insights into diagnosis and neurogenetic outcomes. Eur J Pediatr. 2024;184(1):72. doi:10.1007/s00431-024-05907-7
38. White PJ, McGarrah RW, Herman MA, Bain JR, Shah SH, Newgard CB. Insulin action, type 2 diabetes, and branched-chain amino acids: a two-way street. Mol Metab. 2021;52:101261. doi:10.1016/j.molmet.2021.101261
39. Chen Z, Ding C, Chen K, Gu Y, Qiu X, Li Q. Investigating the causal association between obesity and risk of hepatocellular carcinoma and underlying mechanisms. Sci Rep. 2024;14(1):15717. doi:10.1038/s41598-024-66414-1
40. Yuan Q, Zhang X, Yang X, et al. Knockdown of ACOT4 alleviates gluconeogenesis and lipid accumulation in hepatocytes. Heliyon. 2024;10(5):e27618. doi:10.1016/j.heliyon.2024.e27618
41. Liu R, She J, Li X, et al. Decoding the Impact of Theabrownin on Skeletal Muscle Function in Gestational Diabetic Offspring: insights from Integrated Metabolome and Transcriptome Analysis. J Agric Food Chem. 2025;73(20):12497–12512. doi:10.1021/acs.jafc.4c11857
42. Pei J, Pan X, Wei G, Hua Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front Pharmacol. 2023;14:1147414. doi:10.3389/fphar.2023.1147414
43. Saucedo R, Ortega-Camarillo C, Ferreira-Hermosillo A, Díaz-Velázquez MF, Meixueiro-Calderón C, Valencia-Ortega J. Role of Oxidative Stress and Inflammation in Gestational Diabetes Mellitus. Antioxidants (Basel). 2023;12(10). doi:10.3390/antiox12101812
44. Zeng MS, Li X, Liu Y, et al. A high-selenium diet induces insulin resistance in gestating rats and their offspring. Free Radic Biol Med. 2012;52(8):1335–1342. doi:10.1016/j.freeradbiomed.2012.01.017
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Impact of Immune Cells, Metabolites, Inflammatory Factors, and Circulating Proteins on Atopic Dermatitis: Insights from a Mendelian Randomization Study
Zhou D, Gan G, Song S, Zi C, Bao Y, Hao W, Chen Q
Clinical, Cosmetic and Investigational Dermatology 2024, 17:2999-3011
Published Date: 21 December 2024
Genetic Insights Into Lipid Traits and Lipid-Modifying Drug Targets in Pregnancy Complications: A Two-Sample Mendelian Randomization Study
Shao H, Xu C, Zhang C, Li L, Wu P, Chen Z, Guan R
International Journal of Women's Health 2025, 17:221-234
Published Date: 31 January 2025
Causal Associations Between Sarcopenia and Gestational Diabetes Mellitus

Huang Y, Zhao S, Hong J, Shen L, Wang Z, Wang D
International Journal of Women's Health 2025, 17:259-269
Published Date: 1 February 2025
A Monocyte-Driven Prognostic Model for Multiple Myeloma: Multi-Omics and Machine Learning Insights
Xie L, Gao M, Tan S, Zhou Y, Liu J, Wang L, Li X
Blood and Lymphatic Cancer: Targets and Therapy 2025, 15:21-37
Published Date: 16 June 2025
Causal Associations Between Oral Microbiota and Gestational Diabetes Mellitus: A Two-Sample Mendelian Randomization Study
Jin H, Wang Y, Li H, Cheng Y, Ma Y
International Journal of Women's Health 2025, 17:2777-2791
Published Date: 30 August 2025