")
Back to Journals » The Application of Clinical Genetics » Volume 8
Moyamoya disease and syndromes: from genetics to clinical management
Authors Guey S, Tournier-Lasserve E, Herve D, Kossorotoff M
Received 19 June 2014
Accepted for publication 28 August 2014
Published 16 February 2015 Volume 2015:8 Pages 49—68
DOI https://doi.org/10.2147/TACG.S42772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Stéphanie Guey,1,3 Elisabeth Tournier-Lasserve,1,2 Dominique Hervé,1,3 Manoelle Kossorotoff4
1Inserm UMR-S1161, Université Paris 7 Denis Diderot, Sorbonne Paris Cité, Paris, France; 2AP-HP, Groupe hospitalier Lariboisière-Saint-Louis, Service de génétique neurovasculaire, Paris, France; 3Service de Neurologie, Centre de Référence des maladies Vasculaires Rares du Cerveau et de l'Œil (CERVCO), Groupe Hospitalier Saint-Louis Lariboisière-Fernand Widal, Assistance Publique-Hôpitaux de Paris, Paris, France; 4Pediatric Neurology Department, French Center for Pediatric Stroke, University Hospital Necker-Enfants Malades, AP-HP Assistance publique-Hôpitaux de Paris, Paris, France
Abstract: Moyamoya angiopathy is characterized by a progressive stenosis of the terminal portion of the internal carotid arteries and the development of a network of abnormal collateral vessels. This chronic cerebral angiopathy is observed in children and adults. It mainly leads to brain ischemic events in children, and to ischemic and hemorrhagic events in adults. This is a rare condition, with a marked prevalence gradient between Asian countries and Western countries. Two main nosological entities are identified. On the one hand, moyamoya disease corresponds to isolated moyamoya angiopathy, defined as being “idiopathic” according to the Guidelines of the Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis. This entity is probably multifactorial and polygenic in most patients. On the other hand, moyamoya syndrome is a moyamoya angiopathy associated with an underlying condition and forms a very heterogeneous group with various clinical presentations, various modes of inheritance, and a variable penetrance of the cerebrovascular phenotype. Diagnostic and evaluation techniques rely on magnetic resonance imaging (MRI), magnetic resonance angiography (MRA) conventional angiography, and cerebral hemodynamics measurements. Revascularization surgery can be indicated, with several techniques. Characteristics of genetic moyamoya syndromes are presented, with a focus on recently reported mutations in BRCC3/MTCP1 and GUCY1A3 genes. Identification of the genes involved in moyamoya disease and several monogenic moyamoya syndromes unraveled different pathways involved in the development of this angiopathy. Studying genes and pathways involved in monogenic moyamoya syndromes may help to give insights into pathophysiological models and discover potential candidates for medical treatment strategies.
Keywords: moyamoya disease, moyamoya syndrome, stroke, surgical revascularization, genetics
Introduction
Moyamoya cerebral angiopathy is characterized by a progressive stenosis or occlusion of the intracranial internal carotid artery (ICA) and/or the proximal portion of the anterior cerebral artery (ACA) and middle cerebral artery (MCA). This steno-occlusive pattern is associated with a compensatory development of a collateral network of vessels at the base of the brain, appearing as a “puff of smoke” on conventional angiography (“moyamoya” in Japanese).
This angiopathy was first described in Japan in 1957,1 and was called “moyamoya disease” for the first time by Suzuki and Takaku in 1969.2
When used alone, the term “moyamoya phenomenon” refers to this cerebral angiopathy regardless of its cause, although this angiographic pattern can have multiple causes. Moyamoya disease (MMD) refers to isolated and primary moyamoya angiopathy, usually bilateral. Moyamoya syndromes (MMS) correspond to moyamoya angiopathy associated with other neurological or extra-neurological manifestations, with or without a well-known associated inherited or acquired condition. Hereditary MMS constitute a heterogeneous group that tends to be clarified with advances in genetic research. It includes Mendelian and non-Mendelian genetic causes that will be detailed below.
Whatever the cause, moyamoya angiopathy increases the risk of ischemic and hemorrhagic brain damage. The specific treatment to prevent such complications is usually revascularization surgery, associated or not with therapeutics relating to the underlying disease.
Pathophysiology of moyamoya remains unknown to date. The identification of genes involved in MMD/MMS provides clues to understand the pathophysiology and gives hope for the development of treatments that could limit vascular damage.
Moyamoya disease
Epidemiological data
Epidemiological studies on MMD are heterogeneous, using various methodologies, making valid comparisons between these studies difficult. In Japan, where MMD is frequent, numerous epidemiological nationwide surveys were performed.3 In other countries, data were derived from smaller studies which sometimes mixed MMD and MMS and are therefore more heterogeneous.
MMD is found in all ethnicities, but there is a marked East-West gradient: if MMD is a rare condition in Western countries, its occurrence is about ten times higher in East Asian countries.4–6 The highest prevalence of MMD is found in Japan.7–10 High standard epidemiological studies found an MMD prevalence of 3.16–10.5/100,000 and an incidence of 0.35–1.13/100,000/year in this country.7–10 All studies showed a female preponderance with twice as many females as males.
Age of MMD onset follows a bimodal distribution with a first peak in childhood between 5 and 10 years of age and a second peak during the fourth decade.11 A familial history is noted in 10% to 15% of cases in Japan.
Incidence does not seem to be uniform across East Asian countries. The People’s Republic of China or Korea have high incidence and prevalence rates, close to those observed in Japan. In contrast, the incidence rate in Taiwan is similar to that found in Western countries.12–15
In Western countries, incidence and prevalence of MMD are much lower than those in East Asian countries. A study conducted in California and Washington State showed that the annual incidence rate of MMD/MMS was 0.086/100,000 persons, ie, 4–13 times lower than incidence of MMD in Japan.5 However, the observation of an ethnicity-related pattern suggests a strong genetic determinism in MMD: the incidence in Californian people originating from Asia was similar to the Japanese incidence. In line with this observation, in Hawaiian patients of Japanese descent a significantly higher prevalence was observed than in those of Caucasian origin.14–16
In Europe, MMD incidence was estimated 1/10 of MMD incidence in Japan.6 In French pediatric patients, the prevalence of pediatric MMD/MMS was approximately 1/20 of that estimated in Asia, with a less frequent familial history, around 7.5%.17
Data about MMD for African and African American population are scarce. A study conducted in California showed that ethnicity-specific incidence of moyamoya was higher among African American people (0.13/100,000 person-years) than among Caucasian people (0.06/100,000 person-years), but these two incidences became similar when patients with a diagnosis of sickle cell disease were removed.5
Clinical presentation
Clinical presentation of MMD differs between adults and children. Adults present with transient or permanent cerebral infarction and intracranial hemorrhage, whereas children present mainly with ischemic events.18–20 However, in occidental countries, adults seem to present with a lower rate of hemorrhage than in Asian countries.21–23
Ischemic manifestations, due to steno-occlusive lesions, are usually multiple and recurrent. Patients present with acute transient or permanent symptoms related with cerebral ischemia in the carotid branches territories or in watershed regions: mono or hemiparesis, sensory impairment, or aphasia/dysarthria. Rarely, patients may develop atypical symptoms such as syncope, visual symptoms, or pseudo-psychiatric symptoms. Ischemic events can be triggered by hyperventilation due to physical or intellectual activity, dehydration, fever, and crying in babies.24 In these patients, cortical vessels are most often maximally dilated at steady state to compensate for chronic cerebral ischemia. Vasoconstriction in response to carbon dioxide decrease related to hyperventilation results in reduced cerebral perfusion.
While more than 20% of adult patients with MMD present with intracranial hemorrhage, intracranial bleeding is rare in MMD children.25,26 Cerebral hemorrhage is mainly due to rupture of fragile deep collateral vessels with deep intraparenchymal (basal ganglia region or periventricular deep white matter) or intraventricular hemorrhage. Subarachnoid hemorrhage is rare and, sometimes due to the rupture of saccular aneurysms located in the circle of Willis – most often in the vertebrobasilar system – developed in response to shifting circulatory patterns.
Intellectual disability is often reported in children with MMD. This cognitive disability was reported even among patients without history of stroke, suggesting the deleterious effect of cerebral chronic hypoxemia on cognitive functions, but tended to be greater in children who have had a stroke.27,28 Bilateral disease and duration of symptoms are the main factors associated with cognitive deficits.
Migraine-like headache is also a common symptom in MMD, probably due to stimulation of dural nociceptors by dilated transdural collaterals, or can be a symptom of chronic hypoxemia. It is sometimes difficult to establish if headache is related to the disease or not. Epilepsy is frequent in pediatric patients, related to cortical ischemia. More rarely, choreic movement disorders occur in pediatric or adult patients, secondary to basal ganglia damage.
Imaging data
Diagnosis of moyamoya angiopathy relies on both the visualization of distal stenosis of one or both internal carotid arteries and/or their branches (ACA and MCA), and on neovascularization reflecting the progressive nature of the stenoting process. Accurate radiological assessment is crucial for diagnosis and treatment planning.
Magnetic resonance imaging and magnetic resonance angiography
Magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) provides information on cerebral arteries, brain parenchyma, and cerebral perfusion, making it a key of the diagnostic assessment and follow-up of patients. Easy to perform, in a non-invasive way and without any radiation, MRI-MRA may be sufficient to diagnose MMD when the following criteria (which were first established on conventional angiography) are met (Figure 1):11
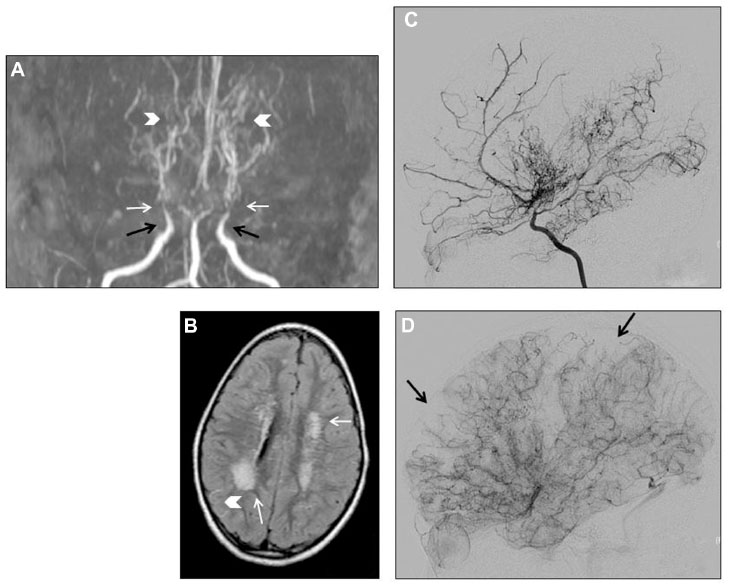 | Figure 1 Moyamoya angiopathy imaging in a 4-year-old girl. |
- stenosis or occlusion of the terminal part of the ICA and/or the proximal portion of the ACA and/or MCA;
- visualization of an abnormal arterial vascular network seen in the vicinity of the steno-occlusive lesions, ie, in the basal ganglia and thalamus, and with the appearance of a “puff of smoke”. These deep perforating dilated moyamoya vessels are usually seen on MRA, and they can also be identified on T1 and T2-weighted MRI when more than two apparent flow voids are seen in basal ganglia and thalamus;
- the diagnosis is “definite MMD” if these findings are bilateral and is “probable MMD” if they are unilateral; and
- the posterior circulation is spared.
MRI shows recent and old ischemic and hemorrhagic lesions (T1-, T2-, T2*-weighted sequences) in the brain parenchyma. White matter hyperintensities located in the distal vascular bed supplied by penetrating branches of MCA/ACA and related to border-zone infarcts are frequently seen.
Asymptomatic microbleeds are seen in a large proportion of adult patients, and may be predictive of the risks of developing a hemorrhagic stroke.29,30 Microbleeds are much more frequent in adult patients (from 28% to 46% according to MRI modalities) than in pediatric patients.30–32
Slowed cortical blood flow can be inferred from FLAIR sequences, showing linear hypersignals that follow a sulcal pattern and which are called the “ivy sign”.33
MRI perfusion sequences called dynamic susceptibility contrast and more recently arterial spin labeling allow the estimation of the degree of cerebral hypoperfusion (see the “Cerebral hemodynamics measurement” section).34
Conventional angiography
Cerebral conventional angiography (Figure 1) – with ICA, external carotid arteries (ECA) and vertebral arteries injection – is the gold standard for diagnosis (see criteria in the “MRI and MRA” section), and classification of MMD according to Suzuki grades.2,24
However, because of the invasivity of the technique, conventional angiography is realized in limited circumstances: uncertain diagnosis or progression of the angiopathy during follow-up with non-invasive techniques, pre-surgical management, and sometimes evaluation after surgical revascularization.
Disease severity is classified into six progressive stages according to Suzuki’s classification (Table 1).2 It is important to note that MMD is a progressive condition and that some imaging criteria can appear or disappear during the course of the disease. For example, deep collateral vessels can be missing in the early and end stages. Moreover, contralateral lesions can develop in patients initially presenting with unilateral findings. This progressive bilateralization is more frequent in pediatric patients.35–37
 | Table 1 Angiographic grades according to Suzuki classification |
Conventional angiography is the most accurate procedure to obtain a detailed mapping of collateral networks, which is essential for planning surgical treatment and avoiding disrupting existing collaterals during revascularization procedure. Indeed, several types of collateral networks typically develop in response to progressive occlusion of distal ICA: collateral deep vessel developed from thalamoperforating and lenticulostriate perforating arteries, that have the appearance of a puff of smoke on angiography imaging and are called “moyamoya vessels”, but also pial collateral arteries developed from the posterior circulation and transdural collateral arteries most often developed from ECA branches.
At late stages of the disease, ICAs are totally occluded and anterior parenchymal vascularization relies essentially on these collateral networks.
Cerebral hemodynamics measurement
Assessment of cerebral perfusion is useful for diagnosis, treatment decision, and follow-up of patients with moyamoya angiopathy.34
Cerebral hemodynamic (single photon emission computed tomography [SPECT], perfusion computed tomography [CT], xenon-enhanced CT and MRI-based methods: dynamic susceptibility contrast and arterial spin labeling) and metabolic (positron emission tomography [PET]-scan) patterns observed in pediatric patients with MMD are:
- decrease of global cerebral blood flow compared with controls, with a predominant posterior cerebral blood flow distribution;
- impaired cerebrovascular reactivity to acetazolamide or carbon dioxide in the ICA territory, suggesting reduced cerebrovascular reserve; and
- compensatory increase of cerebral blood volume by cerebral vessels’ vasodilation, and increase of oxygen fraction extraction.
These modifications are usually more prominent in pediatric than adult patients.
Transcranial Doppler ultrasonography
Transcranial Doppler (TCD) may be helpful in the monitoring of intracerebral hemodynamics in moyamoya angiopathy.38,39 However, data are scarce in literature and parameters assessed in ultrasonography differed across studies, although the main TCD parameters are the mean flow velocities and the pulsatility index. Moreover, TCD is a very operator-dependent tool. Therefore, TCD cannot be a sufficient tool for evaluation and follow-up in moyamoya angiopathy. In the same way, additional studies are needed to validate TCD in postoperative follow-up.40
Electroencephalography
Electroencephalography can show a specific pattern in pediatric patients with moyamoya, called “rebuild-up” phenomenon. This pattern consists of the reappearance of high amplitude slow waves after hyperventilation, indicating a cerebral hypoxemia induced by hyperventilation, and corresponds to a reduced cerebral perfusion reserve.41 However, because of the risk of hyperventilation-induced cerebral infarction, many physicians consider that electroencephalography hyperventilation should not be performed in these patients.
Investigations for usual causes of acquired or inherited moyamoya syndrome
A crucial step in the examination of patients who have moyamoya angiopathy is the research of underlying condition or neurological and extra-neurological signs that would suggest MMS. Acquired causes of MMS, including cranial irradiation which is the most frequently associated feature, should be screened by physicians (Table 2).110–114 Numerous other acquired conditions have been associated with moyamoya angiopathy but it is often difficult to determine whether the association is causal or incidental, as the number of cases is most often very limited. There are various clinical features associated with inherited MMS. (See “MMS” section and Table 3, where the main clinical symptoms found in inherited MMS are detailed).115–152
 | Table 2 Main acquired and inherited MMS and minimal etiologic investigations when discovering a moyamoya angiopathy |
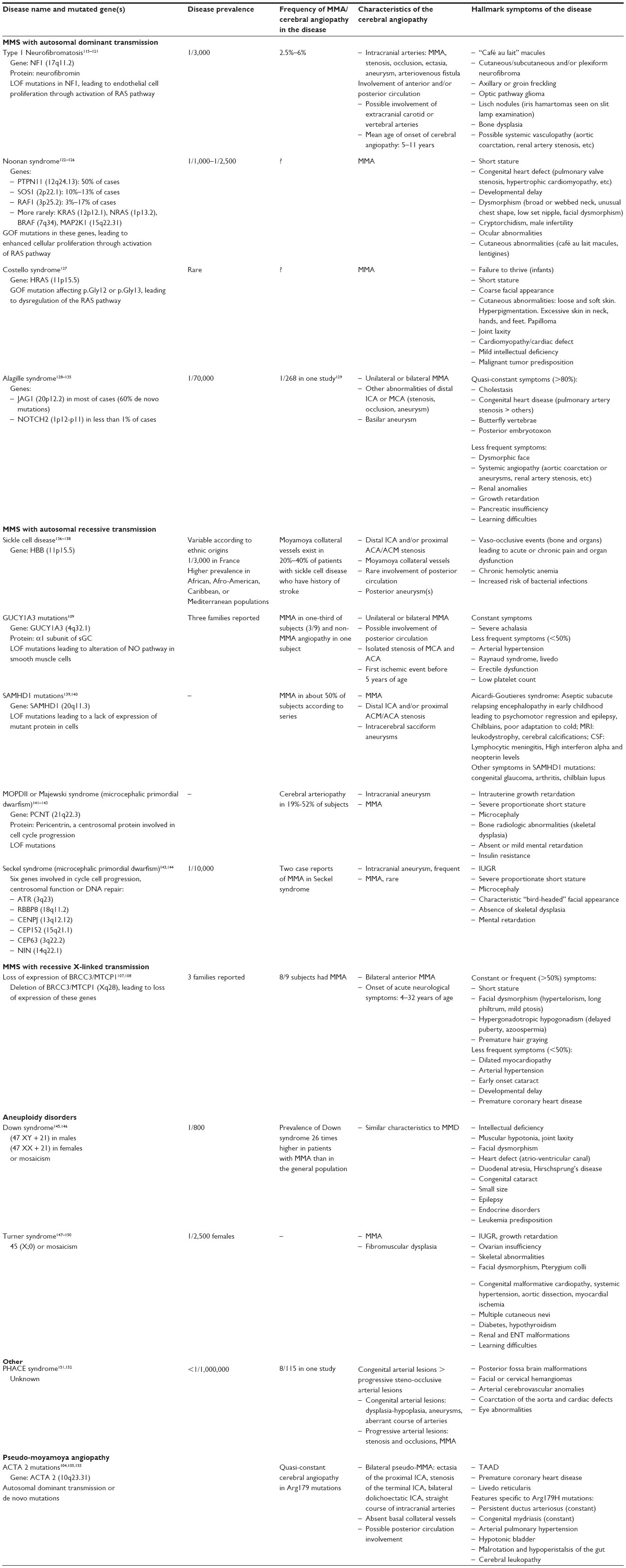 | Table 3 Characteristics of genetic moyamoya syndromes (and pseudo-moyamoya syndromes) |
Practically, the etiological investigation should include a rigorous interview of the patient, detailed physical examination, and a minimal set of complementary exams in order to rule out the main causes of MMS (Table 2).
Clinical course of MMD
The natural history of MMD in non-operated cases is not well described. Studies addressing natural history had limited patient samples and no systematic brain imaging follow-up.42 The course of the disease is variable and could go from a rapid progression leading to severe disabilities to clinical silence during several years. In most pediatric patients, there is a progressive worsening of the occlusive arterial lesions, even among asymptomatic patients or among patients with unilateral lesions.43,44 Studies addressing the prevalence of stroke in children with moyamoya are scarce but autonomy loss was observed in 60% of patients after a 5-year follow-up. In adults, the disease’s natural course seems to be less severe, as autonomy loss was observed in 20% of patients. However, the overall stroke rate in adults appears to be high, an estimated 10%–15% per year in symptomatic cases and 3% per year in asymptomatic cases.
The risk of developing an ischemic or hemorrhagic stroke increases in line with the progression of vascular lesions. In conservatively treated children, neurologic deterioration is most often due to repeated ischemic strokes that occur with progression of arterial occlusion. Non-operated MMD adult patients present a high risk of developing both ischemic and hemorrhagic stroke, and the latter is an important cause of functional deterioration or mortality in these patients. A regular follow-up of conservatively treated patients is crucial, even if they are asymptomatic or have unilateral lesions.26,45
Treatments in MMD
No curative treatment allowing regression of the occlusive arterial lesions has been proved in moyamoya vasculopathy. Treatments in the acute phase of ischemic or hemorrhagic stroke are symptomatic, but the use of intravenous tissue plasminogen activator is contraindicated for MMD manifesting as cerebral ischemia, because of the high risk of intracranial bleeding.46 After managing acute complications, patients should be referred to a reference center.
Common general measures are recommended in order to prevent deterioration of cerebral hemodynamics, such as avoidance of hypotension, dehydration, and hyperventilation, because of these patients’ deficit in cerebrovascular reserve. A very important point is specific care during the perioperative period, especially concerning the management of anesthetic risk to minimize the risk of perioperative stroke.47,48
Non-ischemic symptoms such as headache and seizure should be treated symptomatically with antiepileptic drugs and analgesics.
Medical therapy is usually started in every patient with ischemic moyamoya angiopathy. Long-term oral antiplatelet therapy is prescribed in children and adults to prevent thrombosis and thromboembolism at sites of arterial stenosis.26,46,48 Aspirin is most often used at the usual dose of 50–100 mg daily in children.24 Antiplatelet therapy may be the sole treatment when surgery revascularization is postponed because of recent infarction, or when surgical bypass is not indicated because the patient has relatively mild disease or good spontaneous revascularization. Data addressing aspirin’s short-term or long-term efficacy are scarce. Antiplatelet bi-therapy is not recommended because of the increased risk of intracerebral bleeding.46
The mainstay of treatment is based on surgical revascularization. The revascularization goal is to improve cerebral hemodynamics and to reduce moyamoya collateral network, thus reducing the risk of new ischemic or hemorrhagic events.26,45,49,50
The efficacy of surgical revascularization in moyamoya patients with ischemic stroke has not been evaluated in a randomized trial. In several non-comparative studies, the occurrence of ischemic events decreases after revascularization surgery, suggesting possible efficacy. In children, most symptomatic cases are operated on, because deteriorating natural course is frequent. In adults, surgery indications are variable between centers and considered on a case-by-case basis.
According to Japanese and US guidelines, indications for revascularization surgery include clinical ischemic symptoms or a decreased regional cerebral blood flow, vascular response and perfusion reserve, retrieved from cerebral circulation and metabolism studies.24,46,48 Moreover, the occurrence of transient ischemic attacks usually requires a short-term revascularization. However, the guidelines are unclear regarding the timing of surgery, “although the general principle of minimizing the time between diagnosis and revascularization is supported”, except in cases where there are immediate contraindications such as recent infarction or hemorrhage.24
After surgical revascularization, antiplatelet medication is usually sustained, in order to prevent cerebral embolization due to thrombi formation at sites of stenosis.26 Treatment in hemorrhagic moyamoya disease is more controversial.
There is no strong recommendation for the selection of surgical revascularization procedure. According to experts’ consensus, indirect revascularization is preferred in children because direct bypass is technically difficult on small-diameter vessels, whereas direct bypass is preferred in adult patients.48
Indirect revascularization is based on moyamoya physiopathology, which spares ECA and its branches. The development of cerebral neovascularization from ECA branches allows brain revascularization.51 Several techniques of indirect revascularization have been developed, all based on a synangiosis procedure. During synangiosis, tissues containing ECA branches (dura mater, temporal muscle, galeal tissue, or superficial temporal artery) are directly put in contact with the surface of the ischemic brain. Development of neovascularization from these tissues is supposed to be enhanced by angiogenic factors’ secretion from ischemic brain parenchyma. This procedure does not provide immediate benefit, since efficient neoangiogenesis requires at least 3 months to develop, and thus there is a risk of stroke during the perioperative period. On the contrary, in the direct revascularization technique, the superficial temporal artery – a branch of the ECA – is directly anastomosed with MCA or ACA.51 This allows an immediate improvement of cerebral hemodynamics and minimizes the risk of perioperative ischemic stroke when compared with indirect techniques. Conversely, direct bypass induces a higher risk of brain hemorrhage secondary to hyperperfusion syndrome, especially in patients with severe cerebral ischemia before surgery. Pre-surgery SPECT evaluation of brain perfusion allows risk estimation. In some cases, direct and indirect approaches can be combined.
Before surgical revascularization, an accurate assessment of vascular status by conventional angiography is required, to avoid disruption of pre-existing collateral vessels naturally developed from ECA.
Because of the possibility of angiopathy progression and persistent stroke risk, a clinical and imaging follow-up should be continued after surgical treatment, especially for patients with unilateral angiopathy who present a risk of bilateralization.
Of course, as mentioned before, as part of the general measures applying to MMD patients, revascularization surgery must be performed in a center experienced in the management of moyamoya, especially for the anesthetic procedures and the perioperative management.
Finally, to date, it is not recommended to use endovascular treatment such as angioplasty, intra-arterial vasodilators or thrombolytic agents in moyamoya vasculopathy.24
Pathophysiological features and genetics of MMD
Pathological studies showed a concentric and eccentric fibrocellular thickening of the intima in the terminal portion of ICA, leading to lumen stenosis, and fragmentation of internal elastic lamina.52 Immunohistochemical studies have shown that this intimal fibrocellular thickening contains alpha-actin-positive smooth muscle cells (SMCs) with a proliferative phenotype, while the media appears thinner.53 Inflammatory or atherosclerotic changes are absent.
Typical deep moyamoya collateral vessels result from the combination of a dilation of pre-existing penetrating vessels (thalamoperforating and lenticulostriate perforating arteries) and development of new vessels. They form a fragile vascular network with a thin media, a fragmented elastic lamina and microaneurysms, with an increased risk of rupture.54 Development of collateral vessels is thought to be the result of compensatory mechanisms in reaction to chronic brain ischemia, facilitated by secretion of pro-angiogenic factors by ischemic parenchyma.55 For others, these collateral vessels result from an abnormal neoangiogenesis in MMD.55,56 Patients also develop compensatory transdural and leptomeningeal collateral vessels. Stimulation of dural nociceptor by dilated transdural collaterals have been implicated in the genesis of headaches in moyamoya.
Pathophysiological mechanisms of MMD are unknown, and only hypotheses can be raised so far.57 A genetic contribution is strongly suspected, based on the existence of familial cases and the observation of a strong ethnicity effect. This genetic susceptibility has been confirmed by recent molecular data obtained in MMD (see Table 4). However, MMD appears to have a complex determinism, with both genetic predisposition and environmental triggers, as suggested by the observation of discordant monozygotic twins.
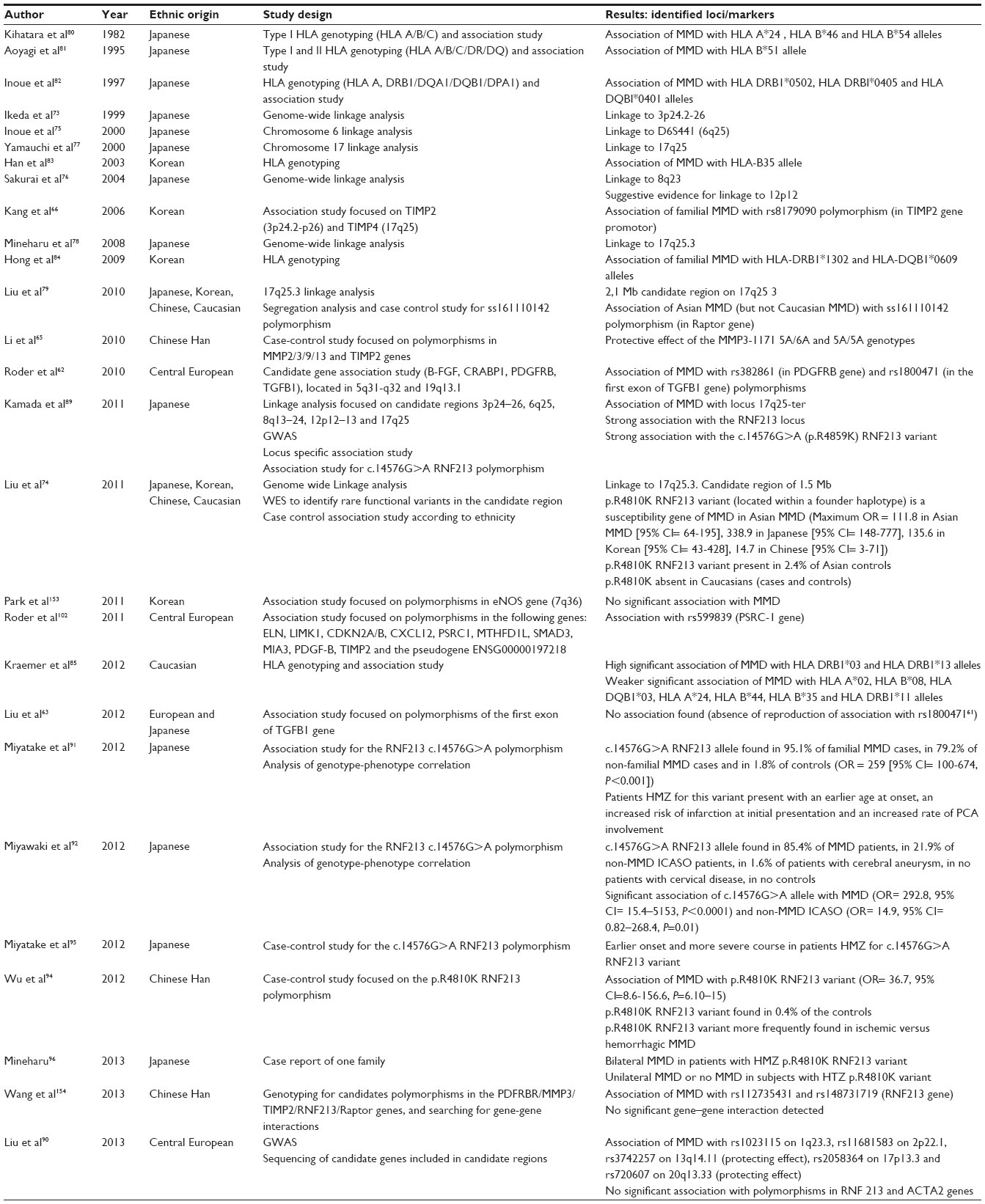 | Table 4 Main studies on MMD genetics |
Many studies highlighted an overexpression of proangiogenic factors in patients’ cerebrospinal fluid, blood, or tissues, but did not permit to determine whether this overexpression was primary or secondary to chronic cerebral ischemia.58,59
Two studies have shown that the expression of two angiogenic factors, fibroblast growth factor and hepatocyte growth factor, was significantly higher in cerebrospinal fluid of MMD patients compared to patients with atherosclerotic occlusion of ICA, suggesting that this overexpression was not a simple consequence of chronic brain ischemia in MMD, but a potential etiopathogenical factor.60,61
Based on these observations, it has been hypothesized that polymorphisms in genes coding for these factors would increase their expression or stability, leading to development of vascular abnormalities. However, association studies failed to show any correlation between the occurrence of MMD and polymorphisms in fibroblast growth factor and hepatocyte growth factor genes. One study showed an association between an increased risk of MMD and polymorphisms located in the promoter region of the platelet-derived growth factor (PDGF) receptor B gene and in the transforming growth factor beta 1 gene, two genes broadly implicated in cell growth and differentiation and thus potentially implicated in vascular growth.62 However, these results were not confirmed in other studies (Table 4).63 Increased plasmatic levels of PDGF were also hypothesized to be a compensatory mechanism to a decreased number of PDGF B receptors on vascular SMCs of MMD patients, and that over-exposition to this cytokine would lead to intimal thickening in MMD.59
Metalloproteinases and tissue inhibitor of metalloproteinases, extracellular matrix remodeling enzymes involved in angiogenesis and vasculopathies such as atherosclerosis, were also studied in MMD. A differential expression of metalloproteinases and tissue inhibitor of metalloproteinases between patients and controls and an association between polymorphisms in genes coding for these enzymes and the occurrence of MMD was shown in some studies, but not further confirmed (Table 4).58,64–66
Some studies showed intimal accumulation of cells expressing vascular smooth muscle actin, an antigen usually expressed by SMCs in the media. The exact nature of these intimal cells is not clear – SMCs or myofibroblasts – and they may be part of the pathophysiological process of moyamoya angiopathy. These cells could originate from circulating progenitors, as demonstrated in other stenotic vasculopathies (post-angioplasty restenosis, atherosclerosis).55,67 They could also originate from the underlying media through an endothelial-to-mesenchymal transition, in which SMCs would transit from media to intima while losing their contractile phenotype. Milewicz et al raised the hypothesis that moyamoya should be considered as a hyperplastic vasculomyopathy and that moyamoya physiopathology would be based on genetic predisposition that promotes migration and proliferation of SMCs and matrix production, in addition to an environmental trigger factor, such as cervical irradiation.68
Genetic factors are strongly suspected, based on a strong ethnicity-related effect, a high concordance rate of the disease in monozygotic twins, and the 10% proportion of familial cases. A polygenic inheritance combined with environmental factors may be involved in most cases. However, various Mendelian patterns of transmission have also been suggested in familial MMD. Transmission of the disease in multiple generations is the most frequently reported pattern of inheritance.69–72 Mineharu et al reported 15 large multigenerational pedigrees consistent with an autosomal dominant disorder with an incomplete penetrance.69 Mother-to-child transmission was the most common mode of inheritance in this paper but a few cases of father-to-child transmission were also observed, including father-to-son. A number of Japanese families suggesting a possible autosomal recessive inheritance were also reported.72,73 Whatever the mode of transmission, a larger preponderance of female patients was observed in both familial and sporadic cases.69–72
Since the 1990s, several genome-wide linkage analyses and Genome wide associations studies (GWAS) have been performed, mostly in East Asian populations in order to map and identify MMD genes.59,73–79,89,90 Five main loci were reported: 3p24.2–p26, 6q25, 8q23, 12p12, and 17q25.73,75–79 So far, the sole locus which has been confirmed is located at 17q25 (Table 4).
With the discovery of linkage with the 6q25 loci, several studies conducted in Asian and Caucasian European populations showed an association between various HLA alleles and MMD.80–85 However, these results were conflicting, except for HLA-DRB1*13, that was found to be associated with MMD with a high level of significance in a Korean and a European study (Table 4).84,85 Interestingly, HLA expression is involved in autoimmunity and several studies reported an increased prevalence of autoimmune diseases – such as diabetes mellitus and thyroid disease – in patients with MMD.86–88 These observations support the hypothesis of a role for autoimmunity in moyamoya pathophysiology.
In 2011, two research teams using different approaches identified ring finger protein 213 (RNF213), located in chromosome 17q25.3, as the first MMD susceptibility gene in Japanese moyamoya patients.74,89 A GWAS study detected a strong association between MMD and a single base substitution leading to an amino acid change p.R4810K (rs112735431 or ss179362673 corresponding to c.14429G>A on the basis of the National Center for Biotechnology Information NCBI Reference sequence NM_001256071.1, also corresponding to p.R4859K, c.14576G>A on the basis of NCBI reference sequence NM_020914.4) in both sporadic and familial cases.89 This variant conferred an odds ratio (OR) >190 and was located in a founder haplotype shared by patients from Japan. Genome-wide linkage combined with whole exome sequencing conducted in eight multigenerational families identified this variant in all probands.74 This variant was then shown to be strongly associated with MMD in both Japanese (maximum OR =339) and Korean patients and, to a lesser degree, Chinese patients. It was absent in Caucasians. The presence of this variant in 1.4%–2.4% of Asian controls strongly suggested that it is a susceptibility factor to MMD, with a probable involvement of other genes or environmental factors in the moyamoya phenotype. Neither this variant nor the founder haplotype was present in Caucasian patients.90 Additional RNF213 variants have also been detected at a lower frequency.74,89 These data have been confirmed in additional studies.91–94 In addition, patients homozygous for the p.R4810K variant have been shown to have an earlier age at onset and a more severe evolution (Table 4).91,95,96
The mechanisms leading from RNF213 to moyamoya vascular lesions are unknown so far. RNF213 gene encodes for a 591 kDa cytosolic protein that possesses a ring finger domain (suggesting an ubiquitine-ligase activity) and a pair of 2 AAA+ ATPase modules.97 To date, we do not know if p.R4810K polymorphism leads to gain or loss of function of RNF213. An in vitro functional study revealed that this mutation affected neither the transcription level nor the ubiquitin ligase activity of the protein.74 Zebrafish in which RNF213 has been knocked down had abnormalities in vascular development with irregular wall formation in trunk arteries and abnormal sprouting vessels.74 However, anatomical and histopathological studies of RNF213-deficient mice revealed no alteration in brain vasculature under physiological conditions.98,99 The development of mice harboring the R4810K mutations would be an important tool to investigate the role of this mutation in the cerebral vessel homeostasis. Recent experimental in vitro studies suggest that endothelial cells derived from induced pluripotent stem cells of R4810K mutated patients have a reduced angiogenic activity.100 In addition, genomic instability in fibroblasts and induced pluripotent stem cells derived from R4810K mutated patients was also observed.101 Interestingly, several genes involved in genome stability maintenance are associated with different MMS (see the “Moyamoya syndromes” section). How this genomic instability might relate to moyamoya mechanisms will benefit from further investigations.
In a Caucasian population, no locus or gene has been formally identified as being associated with MMD to date (Table 4). Studies conducted in MMD patients originated from central Europe found significant associations with polymorphisms located in PDGF receptor B and transforming growth factor beta 1 genes62 and with a polymorphism located in PSRC-1, a gene known to be associated with coronary heart disease.102 As previously mentioned, a study showed an association between several HLA markers and MMD in Caucasians, but these results have not been replicated.85 A recent GWAS conducted among 38 European patients failed to identify any major founder variant associated with MMD.90 In Caucasian MMD patients, no association was found between MMD and the p.R4810K variant in RNF213.74,90 However, few data suggest a potential role of RNF213 in susceptibility to develop MMD in a Caucasian population. First, sequencing of RNF213 among 50 Caucasian MMD patients revealed four additional variants in four distinct patients which were absent among controls.74 Second, clusters of variants located in the RNF213 region were weakly associated with Caucasian MMD in GWAS.90 Additional work on a larger number of patients and controls would be needed to clarify this point.
Moyamoya syndromes
MMS refers to a moyamoya angiopathy associated with other neurological and/or extra-neurological symptoms, or due to a well identified acquired (see Table 2) or inherited cause (see Table 3).
Cerebral angiopathy in MMS affects the anterior circulation, unilaterally or bilaterally, and can also involve the posterior circulation.
MMS have been described in several chromosomal disorders and Mendelian diseases some of whose causative genes have already been identified. Thus, MMS constitute a highly heterogeneous group regarding clinical presentation and patterns of inheritance. The main inherited MMS known to date are described in Table 3. Penetrance of moyamoya angiopathy among the different inherited MMS is highly variable, from 2.5%–6% in NF1 to quasi-constant in BRCC3 - MTCP1 deletions, a newly discovered Mendelian MMS.
The proportion of MMS seems to be higher in Western countries than in East Asian countries.27,103 A nationwide survey conducted in Japan in 2006 showed that the prevalence and annual incidence of MMS were 0.34/100,000 and 0.11/100,000 respectively, more than ten times lower than those of MMD but close to those observed in Western countries.10 The difference may also relate to the epidemiology of underlying conditions. For example, sickle cell disease is a frequent cause of moyamoya in people from African origin, which population is more frequent in the US and Europe than in Asia. Interestingly, moyamoya prevalence in Afro-American and Caucasian people was similar when patients with sickle cell disease were excluded.5
In the following section, we will focus on three newly discovered inherited MMS in which the cerebrovascular phenotype is highly penetrant.
Heterozygous missense mutations in the ACTA2 gene coding for the smooth muscle alpha actin, one of the major contractile proteins of SMCs, are involved in a pseudo-moyamoya angiopathy.104–106 The vasculopathy includes stenosis of the terminal part of ICA that can extend to the proximal segments of the MCA and ACA, dilation of the proximal ICA, an abnormal straight course of intracranial arteries and absence of basal collaterals. The posterior cerebral circulation is often impaired. However, the lack of collateral deep vessels and the coexistence of arterial ectasia and stenosis are features which differ from those observed in classical moyamoya angiopathy.104
A new recessive X-linked syndrome due to loss of expression of BRCC3/MTCP1 was recently described.107,108 In the three reported families, all patients were males and pedigrees showed a maternal inheritance. Bilateral moyamoya angiopathy was quasi-constant with an onset of acute neurological symptoms ranging from 4 to 32 years of age. Other frequent symptoms were short stature, hypergonadotropic hypogonadism, stereotyped facial dysmorphism, and heart involvement (isolated left ventricular enlargement or symptomatic dilated cardiomyopathy). Inconstantly, patients presented with arterial hypertension, premature coronary heart disease, and early onset bilateral cataracts. Developmental delay was reported in one family. The disease is caused by an Xq28 deletion that leads to a complete loss of expression of BRCC3 and MTCP1 genes. BRCC3 encodes for a member of two protein complexes: a nuclear DNA repair complex and a cytoplasmic complex that might have a role in cardiomyocyte protection. Knockdown studies in Zebrafish suggest that BRCC3 plays an important role in angiogenesis. The function of MTCP1 is unknown to date.108
Another MMS associated with moyamoya and achalasia was identified recently.109 It is caused by mutations in GUCY1A3, which encodes the α1 subunit of soluble guanylate cyclase (sGC), the major receptor for nitric oxide (NO). Three consanguineous families including a total of nine affected children (six males and three females) were reported. Early onset moyamoya angiopathy was present in one-third of mutated subjects and was revealed by ischemic events occurring before 5 years of age. Moyamoya was unilateral or bilateral and the posterior circulation could be affected. One child presented a unilateral long arterial stenosis of MCA and ACA without moyamoya neovessels network. Affected children also had constant and severe achalasia, leading to severe regurgitations beginning in the first few months of life. Other hallmark features were arterial hypertension, present in 50% of mutated subjects, and more rarely, vasomotor dysfunctions such as Raynaud phenomenon or livedo, erectile dysfunction, and low platelet count without history of abnormal bleeding. In each family, a homozygous loss-of-function mutation was identified in affected subjects that led to absence of expression of the sGC protein (one nonsense mutation, one splice-site mutation, and one frameshift deletion).
GUCY1A3 encodes for one subunit of the most abundant isoform of the sGC, an NO-dependent enzyme that regulates the SMCs’ relaxation in vascular and extravascular systems, which probably explains why vascular and digestive smooth muscles are both affected in this syndrome.
Translating genetic research into the clinics: future perspectives, potential treatments
Moyamoya angiopathy constitutes a “patchwork” including two main nosological entities: on the one hand, MMD, probably multifactorial and polygenic in most patients and on the other hand, MMS, a very heterogeneous group with various clinical presentations, various modes of inheritance, and a variable penetrance of the cerebrovascular phenotype.
Identification of the genes involved in MMD and several monogenic MMS unraveled different pathways involved in the development of this angiopathy, for example the NO-sGC pathway (with GUCY1A3 mutations), the RAS pathway (with NF1, Noonan, and Costello syndromes) or the NOTCH pathway (with Alagille syndrome), as well as pathways involved in genomic stability (with RNF213, Pericentrin and other genes involved in dwarfism, or BRCC3). Genes encoding additional members of these pathways may themselves be involved in this angiopathy genesis.
Identification of MMS causative mutations has several applications in clinical practice including the development of diagnostic tests and genetic counseling for the patients and their relatives.
Knowledge of the pathways involved in either MMD or MMS also provides valuable clues to investigate the pathophysiology of this disorder, and to identify pharmacological targets that could block the development of arterial lesions. Identification of the genes involved in MMD and highly penetrant MMS would also allow the development of moyamoya animal models, which are essential for pathophysiology investigations and for preclinical trials.
In addition, the use of next-generation sequencing tools will undoubtedly speed up the identification of novel familial MMD/MMS remaining to be discovered.
Disclosure
The authors declare that they have no conflict of interest.
References
Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerve. 1957;9:37–43. | |
Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20(3):288–299. | |
Kuriyama S, Kusaka Y, Fujimura M, et al. Prevalence and clinicoepidemiological features of moyamoya disease in Japan: findings from a nationwide epidemiological survey. Stroke. 2008;39(1):42–47. | |
Goto Y, Yonekawa Y. Worldwide Distribution of moyamoya disease. Neurol Med Chir (Tokyo). 1992;32(12):883–886. | |
Uchino K, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington State and California. Neurology. 2005;65(6):956–958. | |
Yonekawa Y, Ogata N, Kaku Y, Taub E, Imhof HG. Moyamoya disease in Europe, past and present status. Clin Neurol Neurosurg. 1997;99(Suppl 2):S58–S60. | |
Wakai K, Tamakoshi A, Ikezaki K, et al. Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey. Clin Neurol Neurosurg. 1997;99(Suppl 2):S1–S5. | |
Baba T, Houkin K, Kuroda S. Novel epidemiological features of moyamoya disease. J Neurol Neurosurg Psychiatr. 2008;79(8):900–904. | |
Hoshino H, Izawa Y, Suzuki N, Research Committee on Moyamoya Disease. Epidemiological features of moyamoya disease in Japan. Neurol Med Chir (Tokyo). 2012;52(5):295–298. | |
Hayashi K, Horie N, Suyama K, Nagata I. An epidemiological survey of moyamoya disease, unilateral moyamoya disease and quasi-moyamoya disease in Japan. Clin Neurol Neurosurg. 2013;115(7):930–933. | |
Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg. 1997;99(Suppl 2):S238–S240. | |
Im SH, Yim SH, Cho CB, et al. Prevalence and epidemiological features of moyamoya disease in Korea. J Cerebrovasc Endovasc Neurosurg. 2012;14(2):75–78. | |
Ahn IM, Park DH, Hann HJ, Kim KH, Kim HJ, Ahn HS. Incidence, prevalence, and survival of moyamoya disease in Korea: a nationwide, population-based study. Stroke. 2014;45(4):1090–1095. | |
Kleinloog R, Regli L, Rinkel GJ, Klijn CJ. Regional differences in incidence and patient characteristics of moyamoya disease: a systematic review. J Neurol Neurosurg Psychiatry. 2012;83(5):531–536. | |
Chen PC, Yang SH, Chien KL, Tsai IJ, Kuo MF. Epidemiology of moyamoya disease in Taiwan: a nationwide population-based study. Stroke. 2014;45(5):1258–1263. | |
Graham JF, Matoba A. A survey of moyamoya disease in Hawaii. Clin Neurol Neurosurg. 1997;99(Suppl 2):S31–S35. | |
Kossorotoff M, Hervé D, Toulgoat F, et al. Paediatric moyamoya in mainland France: a comprehensive survey of academic neuropaediatric centres. Cerebrovasc Dis. 2012;33(1):76–79. | |
Han DH, Nam DH, Oh CW. Moyamoya disease in adults: characteristics of clinical presentation and outcome after encephalo-duro-arterio-synangiosis. Clin Neurol Neurosurg. 1997;99(Suppl 2):S151–S155. | |
Fukui M, Kono S, Sueishi K, Ikezaki K. Moyamoya disease. Neuropathology. 2000;20(Suppl):S61–S64. | |
Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. 2004;100(2 Suppl Pediatrics):142–149. | |
Chiu D, Shedden P, Bratina P, Grotta JC. Clinical features of moyamoya disease in the United States. Stroke. 1998;29(7):1347–1351. | |
Hallemeier CL, Rich KM, Grubb RL Jr, et al. Clinical features and outcome in North American adults with moyamoya phenomenon. Stroke. 2006;37(6):1490–1496. | |
Kraemer M, Heienbrok W, Berlit P. Moyamoya disease in Europeans. Stroke. 2008;39(12):3193–3200. | |
Smith ER, Scott RM. Spontaneous occlusion of the circle of Willis in children: pediatric moyamoya summary with proposed evidence-based practice guidelines. A review. J Neurosurg Pediatr. 2012;9(4):353–360. | |
Hallemeier CL, Rich KM, Grubb RL Jr, et al. Clinical features and outcome in North American adults with moyamoya phenomenon. Stroke. 2006;37(6):1490–1496. | |
Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. 2004;100(2 Suppl Pediatrics):142–149. | |
Williams TS, Westmacott R, Dlamini N, et al. Intellectual ability and executive function in pediatric moyamoya vasculopathy. Dev Med Child Neurol. 2012;54(1):30–37. | |
Kossorotoff M. Cognitive decline in moyamoya: influence of chronic cerebral hypoxia, history of stroke, or comorbid conditions? Dev Med Child Neurol. 2012;54(1):5–6. | |
Kikuta K, Takagi Y, Nozaki K, Sawamoto N, Fukuyama H, Hashimoto N. The presence of multiple microbleeds as a predictor of subsequent cerebral hemorrhage in patients with moyamoya disease. Neurosurgery. 2008;62(1):104–112. | |
Kuroda S, Kashiwazaki D, Ishikawa T, Nakayama N, Houkin K. Incidence, locations, and longitudinal course of silent microbleeds in moyamoya disease: a prospective T2*-weighted MRI study. Stroke. 2013;44(2):516–518. | |
Kikuta KI, Takagi Y, Nozaki K, et al. Asymptomatic microbleeds in moyamoya disease: T2*-weighted gradient-echo magnetic resonance imaging study. J Neurosurg. 2005;102(3):470–475. | |
Qin Y, Ogawa T, Fujii S, et al. High incidence of asymptomatic cerebral microbleeds in patients with hemorrhagic onset-type moyamoya disease: a phase-sensitive MRI study and meta-analysis. Acta Radiol. Epub February 20, 2014. | |
Chabbert V, Ranjeva JP, Sevely A, Boetto S, Berry I, Manelfe C. Diffusion- and magnetisation transfer-weighted MRI in childhood moya-moya. Neuroradiology. 1998;40(4):267–271. | |
Lee M, Zaharchuk G, Guzman R, Achrol A, Bell-Stephens T, Steinberg GK. Quantitative hemodynamic studies in moyamoya disease: a review. Neurosurg Focus. 2009;26(4):E5. | |
Hirotsune N, Meguro T, Kawada S, Nakashima H, Ohmoto T. Long-term follow-up study of patients with unilateral moyamoya disease. Clin Neurol Neurosurg. 1997;99(Suppl 2):S178–S181. | |
Park EK, Lee YH, Shim KW, Choi JU, Kim DS. Natural history and progression factors of unilateral moyamoya disease in pediatric patients. Childs Nerv Syst. 2011;27(8):1281–1287. | |
Lee SC, Jeon JS, Kim JE, et al. Contralateral progression and its risk factor in surgically treated unilateral adult moyamoya disease with a review of pertinent literature. Acta Neurochir (Wien). 2014;156(1):103–111. | |
Lee YS, Jung KH, Roh JK. Diagnosis of moyamoya disease with transcranial Doppler sonography: correlation study with magnetic resonance angiography. J Neuroimaging. 2004;14(4):319–323. | |
Takase K, Kashihara M, Hashimoto T. Transcranial Doppler ultrasonography in patients with moyamoya disease. Clin Neurol Neurosurg. 1997;99(Suppl 2):S101–S105. | |
Wu M, Huang Z, Zhang D, et al. Color doppler hemodynamic study of the superficial temporal arteries in superficial temporal artery-middle cerebral artery (STA-MCA) bypass surgery for Moyamoya disease. World Neurosurg. 2011;75(2):258–263. | |
Kodama N, Aoki Y, Hiraga H, Wada T, Suzuki J. Electroencephalographic findings in children with moyamoya disease. Arch Neur. 1979;36(1);16–19. | |
Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol. 2008;7(11):1056–1066. | |
Ezura M, Yoshimoto T, Fujiwara S, Takahashi A, Shirane R, Mizoi K. Clinical and angiographic follow-up of childhood-onset moyamoya disease. Childs Nerv Syst. 1995;11(10):591–594. | |
Kuroda S, Ishikawa T, Houkin K, Nanba R, Hokari M, Iwasaki Y. Incidence and clinical features of disease progression in adult moyamoya disease. Stroke. 2005;36(10):2148–2153. | |
Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst. 2005;21(5):358–364. | |
Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis, Health Labour Sciences Research Grant for Research on Measures for Infractable Diseases. Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir (Tokyo). 2012;52(5):245–266. | |
Nomura S, Kashiwagi S, Uetsuka S, Uchida T, Kubota H, Ito H. Perioperative management protocols for children with moyamoya disease. Childs Nerv Syst. 2001;17(4–5):270–274. | |
Roach ES, Golomb MR, Adams R, et al. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39(9):2644–2691. | |
Ng J, Thompson D, Lumley JP, Saunders DE, Ganesan V. Surgical revascularisation for childhood moyamoya. Childs Nerv Syst. 2012;28(7):1041–1048. | |
Guzman R, Lee M, Achrol A, et al. Clinical outcome after 450 revascularization procedures for moyamoya disease. Clinical article. J Neurosurg. 2009;111(5):927–935. | |
Kuroda S, Houkin K. Bypass surgery for moyamoya disease: concept and essence of sugical techniques. Neurol Med Chir. 2012;52(5):287–294. | |
Haltia M, Iivanainen M, Majuri H, Puranen M. Spontaneous occlusion of the circle of Willis (moyamoya syndrome). Clin Neuropathol. 1982;1(1):11–22. | |
Masuda J, Ogata J, Yutani C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in moyamoya disease. Stroke. 1993;24(12):1960–1967. | |
Yamashita M, Oka K, Tanaka K. Histopathology of the brain vascular network in moyamoya disease. Stroke. 1983;14(1):50–58. | |
Achrol AS, Guzman R, Lee M, Steinberg GK. Pathophysiology and genetic factors in moyamoya disease. Neurosurg Focus. 2009;26(4):E4. | |
Rafat N, Beck GC, Pefia-Tapia PG, Schmiedek P, Vajkoczy P. Increased levels of circulating endothelial progenitor cells in patients with Moyamoya disease. Stroke. 2009;40:432–438. | |
Houkin K, Ito M, Sugiyama T, etal. Review of past research and current concepts on the etiology of moyamoya disease. Neurol Med Chir (Tokyo). 2012;52(5):267–277. | |
Kang HS, Kim JH, Phi JH, et al. Plasma matrix metalloproteinases, cytokines and angiogenic factors in moyamoya disease. J Neurol Neurosurg Psychiatr. 2010;81(6):673–678. | |
Roder C, Nayak NR, Khan N, Tatagiba M, Inoue I, Krischek B. Genetics of Moyamoya disease. J Hum Genet. 2010;55(11):711–716. | |
Yoshimoto T, Houkin K, Takahashi A, Abe H. Angiogenic factors in moyamoya disease. Stroke. 1996;27(12):2160–2165. | |
Nanba R, Kuroda S, Ishikawa T, Houkin K, Iwasaki Y. Increased expression of hepatocyte growth factor in cerebrospinal fluid and intracranial artery in moyamoya disease. Stroke. 2004;35(12):2837–2842. | |
Roder C, Peters V, Kasuya H, et al. Polymorphisms in TGFB1 and PDGFRB are associated with Moyamoya disease in European patients. Acta Neurochir (Wien). 2010;152(12):2153–2160. | |
Liu C, Roder C, Schulte C, et al. Analysis of TGFB1 in European and Japanese Moyamoya disease patients. Eur J Med Genet. 2012;55(10):531–534. | |
Fujimura M, Watanabe M, Narisawa A, Shimizu H, Tominaga T. Increased expression of serum Matrix Metalloproteinase-9 in patients with moyamoya disease. Surg Neurol. 2009;72(5):476–480. | |
Li H, Zhang ZS, Liu W, et al. Association of a functional polymorphism in the MMP-3 gene with Moyamoya Disease in the Chinese Han population. Cerebrovasc Dis. 2010;30(6):618–625. | |
Kang HS, Kim SK, Cho BK, Kim YY, Hwang YS, Wang KC. Single nucleotide polymorphisms of tissue inhibitor of metalloproteinase genes in familial moyamoya disease. Neurosurgery. 2006;58(6):1074–1080. | |
Sata M, Saiura A, Kunisato A, et al. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8(4):403–409. | |
Milewicz DM, Kwartler CS, Papke CL, Regalado ES, Cao J, Reid AJ. Genetic variants promoting smooth muscle cell proliferation can result in diffuse and diverse vascular diseases: evidence for a hyperplastic vasculomyopathy. Genet Med.2010;12(4):196–203. | |
Mineharu Y, Takenaka K, Yamakawa H, et al. Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting. J Neurol Neurosurg Psychiatr. 2006;77(9):1025–1029. | |
Yamauchi T, Houkin K, Tada M, Abe H. Familial occurrence of moyamoya disease. Clin Neurol Neurosurg. 1997;99(Suppl 2):S162–S167. | |
Kaneko Y, Imamoto N, Mannoji H, Fukui M. Familial occurrence of moyamoya disease in the mother and four daughters including identical twins. Neurol Med Chir (Tokyo). 1998;38(6):349–354. | |
Nanba R, Kuroda S, Tada M, Ishikawa T, Houkin K, Iwasaki Y. Clinical features of familial moyamoya disease. Childs Nerv Syst. 2006;22(3):258–262. | |
Ikeda H, Sasaki T, Yoshimoto T, Fukui M, Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am J Hum Genet. 1999;64(2):533–537. | |
Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE. 2011;6(7):e22542. | |
Inoue TK, Ikezaki K, Sasazuki T, Matsushima T, Fukui M. Linkage analysis of moyamoya disease on chromosome 6. J Child Neurol. 2000;15(3):179–182. | |
Sakurai K, Horiuchi Y, Ikeda H, et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004;49(5):278–281. | |
Yamauchi T, Tada M, Houkin K, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. 2000;31(4):930–935. | |
Mineharu Y, Liu W, Inoue K, et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 2008;70(24 Pt 2):2357–2363. | |
Liu W, Hashikata H, Inoue K, et al. A rare Asian founder polymorphism of Raptor may explain the high prevalence of Moyamoya disease among East Asians and its low prevalence among Caucasians. Environ Health Prev Med. 2010;15(2):94–104. | |
Kitahara T, Okumura K, Semba A, Yamaura A, Makino H. Genetic and immunologic analysis on moya-moya. J Neurol Neurosurg Psychiatry. 1982;45(11):1048–1052. | |
Aoyagi M, Ogami K, Matsushima Y, Shikata M, Yamamoto M, Yamamoto K. Human leukocyte antigen in patients with moyamoya disease. Stroke. 1995;26(3):415–417. | |
Inoue TK, Ikezaki K, Sasazuki T, et al. DNA typing of HLA in the patients with moyamoya disease. Jpn J Hum Genet. 1997;42(4):507–515. | |
Han H, Pyo CW, Yoo DS, Huh PW, Cho KS, Kim DS. Associations of Moyamoya patients with HLA class I and class II alleles in the Korean population. J Korean Med Sci. 2003;18(6):876–880. | |
Hong SH, Wang KC, Kim SK, Cho BK, Park MH. Association of HLA-DR and -DQ Genes with Familial Moyamoya Disease in Koreans. J Korean Neurosurg Soc. 2009;46(6):558–563. | |
Kraemer M, Horn PA, Roder C, et al. Analysis of human leucocyte antigen genes in Caucasian patients with idiopathic moyamoya angiopathy. Acta Neurochir (Wien). 2012;154(3):445–454. | |
Bower RS, Mallory GW, Nwojo M, Kudva YC, Flemming KD, Meyer FB. Moyamoya disease in a primarily white, midwestern US population: increased prevalence of autoimmune disease. Stroke. 2013;44(7):1997–1999. | |
Kim SJ, Heo KG, Shin HY, et al. Association of thyroid autoantibodies with moyamoya-type cerebrovascular disease: a prospective study. Stroke. 2010;41(1):173–176. | |
El Ramahi KM, Al Rayes HM. Systemic lupus erythematosus associated with moyamoya syndrome. Lupus. 2000;9:632–636. | |
Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56(1):34–40. | |
Liu W, Senevirathna STMLD, Hitomi T, et al. Genomewide association study identifies no major founder variant in Caucasian moyamoya disease. J Genet. 2013;92(3):605–609. | |
Miyatake S, Miyake N, Touho H, et al. Homozygous c.14576G.A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology. 2012;78(11):803–810. | |
Miyawaki S, Imai H, Takayanagi S, Mukasa A, Nakatomi H, Saito N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke. 2012;43(12):3371–3374. | |
Miyawaki S, Imai H, Shimizu M, et al. Genetic variant RNF213 c.14576G.A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke. 2013;44(10):2894–2897. | |
Wu Z, Jiang H, Zhang L, et al. Molecular analysis of RNF213 gene for moyamoya disease in the Chinese Han population. PLoS ONE. 2012;7(10):e48179. | |
Miyatake S, Touho H, Miyake N, et al. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G.A variant in RNF213 showed varying clinical course and severity. J Hum Genet. 2012;57(12):804–806. | |
Mineharu Y, Takagi Y, Takahashi JC, et al. Rapid progression of unilateral moyamoya disease in a patient with a family history and an RNF213 risk variant. Cerebrovasc Dis. 2013;36(2):155–157. | |
Morito D, Nishikawa K, Hoseki J, et al. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci Rep. 2014;4:4442. | |
Kobayashi H, Yamazaki S, Takashima S, et al. Ablation of Rnf213 retards progression of diabetes in the Akita mouse. Biochem Biophys Res Commun. 2013;432(3):519–525. | |
Sonobe S, Fujimura M, Niizuma K, et al. Temporal profile of the vascular anatomy evaluated by 9.4-T magnetic resonance angiography and histopathological analysis in mice lacking RNF213: a susceptibility gene for moyamoya disease. Brain Res. 2014;1552:64–71. | |
Hitomi T, Habu T, Kobayashi H, et al. Downregulation of Securin by the variant RNF213 R4810K (rs112735431, G.A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem Biophys Res Commun. 2013;438(1):13–19. | |
Hitomi T, Habu T, Kobayashi H, et al. The moyamoya disease susceptibility variant RNF213 R4810K (rs112735431) induces genomic instability by mitotic abnormality. Biochem Biophys Res Commun. 2013;439(4):419–426. | |
Roder C, Peters V, Kasuya H, et al. Common genetic polymorphisms in moyamoya and atherosclerotic disease in Europeans. Childs Nerv Syst. 2011;27(2):245–252. | |
Khan N, Schuknecht B, Boltshauser E, et al. Moyamoya disease and Moyamoya syndrome: experience in Europe; choice of revascularisation procedures. Acta Neurochir (Wien). 2003;145(12):1061–1071. | |
Milewicz DM, Østergaard JR, Ala-Kokko LM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A(10):2437–2443. | |
Munot P, Saunders DE, Milewicz DM, et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain. 2012;135(Pt 8):2506–2514. | |
Guo DC, Papke CL, Tran-Fadulu V, etal. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–627. | |
Hervé D, Touraine P, Verloes A, et al. A hereditary moyamoya syndrome with multisystemic manifestations. Neurology. 2010;75(3):259–264. | |
Miskinyte S, Butler MG, Hervé D, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. 2011;88(6):718–728. | |
Hervé D, Philippi A, Belbouab R, et al. Loss of α1β1 Soluble Guanylate Cyclase, the Major Nitric Oxide Receptor, Leads to Moyamoya and Achalasia. Am J Hum Genet. 2014;94(3):385–394. | |
Mathew NT, Abraham J, Chandy J. Cerebral angiographic features in tuberculous meningitis. Neurology. 1970;20(10):1015–1023. | |
Mori K, Takeuchi J, Ishikawa M, Handa H, Toyama M, Yamaki T. Occlusive arteriopathy and brain tumor. J Neurosurg. 1978;49(1):22–35. | |
Bitzer M, Topka H. Progressive cerebral occlusive disease after radiation therapy. Stroke. 1995;26(1):131–136. | |
Matsuki Y, Kawakami M, Ishizuka T, et al. SLE and Sjögren’s syndrome associated with unilateral moyamoya vessels in cerebral arteries. Scand J Rheumatol. 1997;26(5):392–394. | |
Bonduel M, Hepner M, Sciuccati G, Torres AF, Tenembaum S. Prothrombotic disorders in children with moyamoya syndrome. Stroke. 2001;32(8):1786–1792. | |
National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Neurofibromatosis. 1988;1(3):172–178. | |
Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64(3):553–555. | |
Cairns AG, North KN. Cerebrovascular dysplasia in neurofibromatosis type 1. J Neurol Neurosurg Psychiatr. 2008;79(10):1165–1170. | |
Rea D, Brandsema JF, Armstrong D, et al. Cerebral arteriopathy in children with neurofibromatosis type 1. Pediatrics. 2009;124(3):e476–e483. | |
Ghosh PS, Rothner AD, Emch TM, Friedman NR, Moodley M. Cerebral vasculopathy in children with neurofibromatosis type 1. J Child Neurol. 2013;28(1):95–101. | |
Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58(5):341–344. | |
Bajaj A, Li Q, Zheng Q, Pumiglia K. Loss of NF1 expression in human endothelial cells promotes autonomous proliferation and altered vascular morphogenesis. PLoS ONE. 2012;7(11):e49222. | |
Ganesan V, Kirkham FJ. Noonan syndrome and moyamoya. Pediatr Neurol. 1997;16(3):256–258. | |
Schuster JM, Roberts TS. Symptomatic moyamoya disease and aortic coarctation in a patient with Noonan’s syndrome: strategies for management. Pediatr Neurosurg. 1999;30(4):206–210. | |
Tang KT, Yang W, Wong J, Lee KY. Noonan syndrome associated with moyamoya disease: report of one case. Acta Paediatr Taiwan. 1999;40(4):274–276. | |
Yamashita Y, Kusaga A, Koga Y, Nagamitsu S, Matsuishi T. Noonan syndrome, moyamoya-like vascular changes, and antiphospholipid syndrome. Pediatr Neurol. 2004;31(5):364–366. | |
Hung PC, Wang HS, Wong AM. Moyamoya syndrome in a child with Noonan syndrome. Pediatr Neurol. 2011;45(2):129–131. | |
Shiihara T, Kato M, Mitsuhashi Y, Hayasaka K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr Neurol. 2005;32(5):361–363. | |
Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257. | |
Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354–1358. | |
Rachmel A, Zeharia A, Neuman-Levin M, Weitz R, Shamir R, Dinari G. Alagille syndrome associated with moyamoya disease. Am J Med Genet. 1989;33(1):89–91. | |
Woolfenden AR, Albers GW, Steinberg GK, Hahn JS, Johnston DC, Farrell K. Moyamoya syndrome in children with Alagille syndrome: additional evidence of a vasculopathy. Pediatrics. 1999;103(2):505–508. | |
Connor SE, Hewes D, Ball C, Jarosz JM. Alagille syndrome associated with angiographic moyamoya. Childs Nerv Syst. 2002;18(3–4):186–190. | |
Emerick KM, Krantz ID, Kamath B, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41(1):99–107. | |
Gaba RC, Shah RP, Muskovitz AA, Guzman G, Michals EA. Synchronous moyamoya syndrome and ruptured cerebral aneurysm in Alagille syndrome. J Clin Neurosci. 2008;15(12):1395–1398. | |
Rocha R, Soro I, Leitão A, Silva ML, Leão M. Moyamoya vascular pattern in Alagille syndrome. Pediatr Neurol. 2012;47(2):125–128. | |
Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. 2006;5(6):501–512. | |
Moritani T, Numaguchi Y, Lemer NB, et al. Sickle cell cerebrovascular disease: usual and unusual findings on MR imaging and MR angiography. Clin Imaging. 2004;28(3):173–186. | |
Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99(9):3144–3150. | |
Ramesh V, Bernardi B, Stafa A, et al. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol. 2010;52(8):725–732. | |
Xin B, Jones S, Puffenberger EG, et al. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci U S A. 2011;108(13):5372–5377. | |
Bober MB, Khan N, Kaplan J, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II):expanding the vascular phenotype. Am J Med Genet A. 2010;152A(4):960–965. | |
Perry LD, Robertson F, Ganesan V. Screening for cerebrovascular disease in microcephalic Osteodysplastic primordial dwarfism type II (MOPD II):an evidence-based proposal. Pediatr Neurol. 2013;48(4):294–298. | |
Moftakhar P, Smith ER, Choulakian A, Scott RM, Danielpour M. Moyamoya disease in children with congenital dwarfing conditions. Pediatr Neurosurg. 2010;46(5):373–380. | |
Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya. J Neurosurg Pediatr. 2009;3(4):320–324. | |
Kainth DS, Chaudhry SA, Kainth HS, Suri FK, Qureshi AI. Prevalence and characteristics of concurrent down syndrome in patients with moyamoya disease. Neurosurgery. 2013;72(2):210–215. | |
Jea A, Smith ER, Robertson R, Scott RM. Moyamoya syndrome associated with Down syndrome: outcome after surgical revascularization. Pediatrics. 2005;116(5):e694–e701. | |
Ajimi Y, Uchida K, Kawase T, Toya S. [A case of Turner’s syndrome associated with moyamoya disease]. No Shinkei Geka. 1992;20(9):1021–1024. Japanese. | |
Spengos K, Kosmaidou-Aravidou Z, Tsivgoulis G, Vassilopoulou S, Grigori-Kostaraki P, Zis V. Moyamoya syndrome in a Caucasian woman with Turner’s syndrome. Eur J Neurol. 2006;13(10):e7–e8. | |
Jagannath AD, Rastogi U, Spooner AE, Lin AE, Agnihotri AK. Aortic dissection and moyamoya disease in Turner syndrome. Am J Med Genet A. 2010;152A(8):2085–2089. | |
Manjila S, Miller BR, Rao-Frisch A, et al. Moyamoya Disease Associated with Asymptomatic Mosaic Turner Syndrome: A Rare Cause of Hemorrhagic Stroke. J Stroke Cerebrovasc Dis. 2014;23(5):1242–1244. | |
Siegel DH, Tefft KA, Kelly T, et al. Stroke in children with posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities (PHACE) syndrome: a systematic review of the literature. Stroke. 2012;43(6):1672–1674. | |
Heyer GL, Dowling MM, Licht DJ, et al. The cerebral vasculopathy of PHACES syndrome. Stroke. 2008;39(2):308–316. | |
Park YS, Min KT, Kim TG, et al. Age-specific eNOS polymorphisms in moyamoya disease. Childs Nerv Syst. 2011;27(11):1919–1926. | |
Wang X, Zhang Z, Liu W, et al. Impacts and interactions of PDGFRB, MMP-3, TIMP-2, and RNF213 polymorphisms on the risk of Moyamoya disease in Han Chinese human subjects. Gene. 2013;526(2):437–442. | |
Guo DC, Papke CL, Tran-Fadulu V, etal. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–627. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.