Back to Journals » Orthopedic Research and Reviews » Volume 13
Monophasic Synovial Sarcoma in the Elbow Misclassified but Successfully Treated as Ewing’s Sarcoma with Chemotherapy
Authors Madariaga MC, Duke A, Hoda ST, Khan F
Received 6 August 2021
Accepted for publication 16 November 2021
Published 27 November 2021 Volume 2021:13 Pages 241—245
DOI https://doi.org/10.2147/ORR.S332441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Clark Hung
Maria Cecilia Madariaga,1 Alexander Duke,1 Syed T Hoda,2 Fazel Khan1
1Department of Orthopaedic Surgery, Stony Brook University Medical Center, Health Sciences Tower – Level 18, Stony Brook, NY, 11794-8181, USA; 2Department of Surgical Pathology, NYU Langone Orthopedic Hospital, New York, NY, 10003, USA
Correspondence: Fazel Khan
Department of Orthopaedic Surgery, Stony Brook University Medical Center, Health Sciences Tower – Level 18, Stony Brook, NY, 11794-8181, USA
Tel +1 (631) 444-4233
Email [email protected]
Abstract: To the best of the authors’ knowledge, this is the first published case of monophasic synovial sarcoma (SS) initially diagnosed as Ewing’s sarcoma (ES), yet successfully treated with chemotherapy in a 24-year-old patient. The initial diagnosis showed a monotonous round cell tumor and positivity for CD99, characteristic of ES; however, the cytology was negative for the classic EWSR1 rearrangement of ES. The patient was treated with the standard chemotherapy protocol of ES – COG AEWS1031 Regimen A with vincristine, doxorubicin, cyclophosphamide, and mesna – as well as with wide resection. Post-resection tissue submission showed additional morphologic features which led to a re-evaluation of the classification of the tumor as well as additional molecular studies; these revealed positivity for translocations of SS18 (18q11.1) in 100% of the nuclei, which is most characteristic of SS, thus, reclassifying the neoplasm as a SS tumor. This case underscores the importance of considering several pathologic entities in the differential diagnosis of small, round blue cell tumors, including ES, SS, and lymphoma. It also demonstrates the importance of using chromosomal identification for a more definitive diagnosis, rather than relying on histological features and markers which are found in more than one tumor classification. There is conflicting evidence of the impact of chemotherapy on survival in SS, as it is primarily treated with radiation therapy. Since SS is rare, prospective studies on the effect of chemotherapy on survival are limited in number. However, our case study demonstrates that chemotherapy is another modality that can be used in the treatment of SS neoplasms.
Keywords: sarcoma, Ewing’s sarcoma, synovial sarcoma, Oncology, chemotherapy
Introduction
While malignant small, blue round cell tumors, Ewing’s sarcoma (ES), synovial sarcoma (SS), and lymphoma share histology, markers, and clinical presentation, the prognosis and treatment differ for each. In ES, the second most common primary malignant bone tumor which occurs most often in adolescents, 20152770 ES is characterized by markers including the sensitive but poorly specific CD99,1 EWSR1-FLI1 and EWSR1-ERG gene fusions.2 In 85% of ES cases, the translocation t(11;22)(q24;q12) is found.3 Because of micro-metastasis, the most successful management is polychemotherapy, such as COG AEWS1030, Regimen A, accompanied by adjuvant surgical treatment.4–6
Lymphoma is similarly characterized with the finding of mixed small round blue cells on histology; however, it is negative for the CD99 marker and lacks the translocation t(11;22). Its tumor marker profile includes CD20, CD45, and lymphocyte common antigen among others. The most successful treatments for lymphoma are multi-agent chemotherapy – cyclophosphamide, doxorubicin, prednisone, and vincristine – with radiation added for local control in persistent disease.
SS is a rare metastatic soft tissue tumor most commonly diagnosed in 15–30-year-olds.7 Ninety percent of SS cases are positive for the translocation t(X;18)(p11.2;q11.2),1 and 95% are positive for SS18/SSX1, SS18/SSX2, and SS18/SSX4 gene fusions.8 While surgery and radiation therapy have achieved excellent local control, distant metastasis limits overall survival.9 To date, we have found that no randomized trial has been established in order to study the role of chemotherapy as a treatment modality for SS. Our case study demonstrates the use of chemotherapy in augmenting positive results in the long-term treatment of SS tumors.10,11
Case Report
In January 2012, a 24-year-old male presented with a 5-month history of a growing soft tissue mass on the medial aspect of the right elbow. Magnetic resonance imaging (MRI) demonstrated a (6.5×4.9×8.5 cm) well-circumscribed, heterogeneously enhancing soft tissue mass within the medial aspect of the distal biceps with mass effect on the median nerve and brachial vasculature (Figure 1).
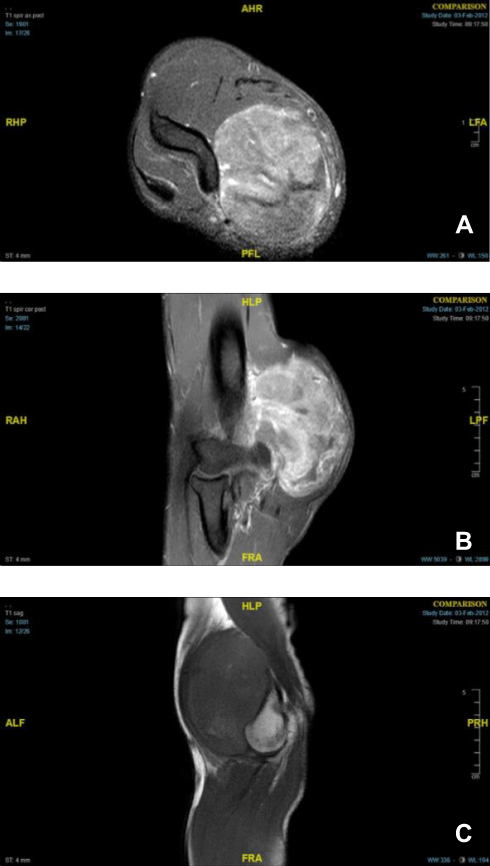 |
Figure 1 Right elbow MRI series showing a large heterogeneously enhancing and likely hemorrhagic mass with dilated vascular structures within the medial aspect of the distal biceps: axial view post-contrast (A), coronal view post-contrast (B), and sagittal view (C). |
A core needle biopsy revealed sheets of round to oval, medium-sized cells with atypical chromatin and nucleoli. Flow cytometry showed a population of cells consistent with ES, and the immunohistochemistry was positive for the CD99 marker characteristic of ES. The specimen did not demonstrate S100, CD45, myogenin, desmin, and panCK markers. Thus, the presumptive diagnosis was a soft tissue ES negative for classic EWSR1 rearrangement on chromosome 22, through both chromosomal and FISH analysis.
Subsequently, the patient was treated with chemotherapy COG AEWS 1031, Regimen A protocol with vincristine, doxorubicin, cyclophosphamide, and mesna. A post-chemo MRI demonstrated a successful response to the protocol with a documented reduction in tumor mass size of 4.8×3.6×5.5 cm. A follow-up surgical wide resection of the mass with medial epicondyle resection and medial and ulnar nerve neuroplasty was then performed. On pathology, the tumor size measured 4.7 cm in greatest diameter, with 85% of the mass demonstrating necrosis and myxoid degenerative changes. Examination of all biopsy margins showed no atypia or malignancy.
The resection yielded significantly more tumor to evaluate, and additional morphologic features such as overlapping hypercellularity as well as some focal spindling led to additional testing. Immunohistochemistry was then repeated on the surgical specimen and confirmed positivity for CD99 (most characteristic of ES) and for BCL-2 and EMA. EMA and BCL-2 staining are more routinely observed in synovial sarcoma rather than Ewing’s sarcoma. Bone marrow aspirates and core biopsies revealed erythropoiesis with normal tri-lineage hematopoiesis without evidence of metastatic ES. Following a re-evaluation of pathology, additional molecular FISH testing was ordered on the new resection specimen to rule out a synovial sarcoma.
Furthermore, the rearrangement of SS18 (18q11.1) was documented in 100% of the nuclei, a finding which is characteristic of SS tumors. An additional finding was the translocation involving the short arm of the X chromosome and the long arm of one chromosome 18 homolog [ie, t(X,18)(p11.2;q11.2)] or a three-way translocation involving the short arm of the X chromosome, the long arm of one chromosome 18 homolog, and the long arm of one chromosome 5 homolog [ie, t(X, 18;5)(p11.2;q11.2;11.2)]. With these additional identified translocations, the tumor was reclassified as a monophasic SS neoplasm.
A one-year post-excision MRI revealed enhancing soft tissue and intramuscular edema within the surgical bed, likely related to post-surgical changes, without evidence of solid nodular enhancing components to suggest recurrence (Figure 2). The overlying skin was resected and negative for malignancy.
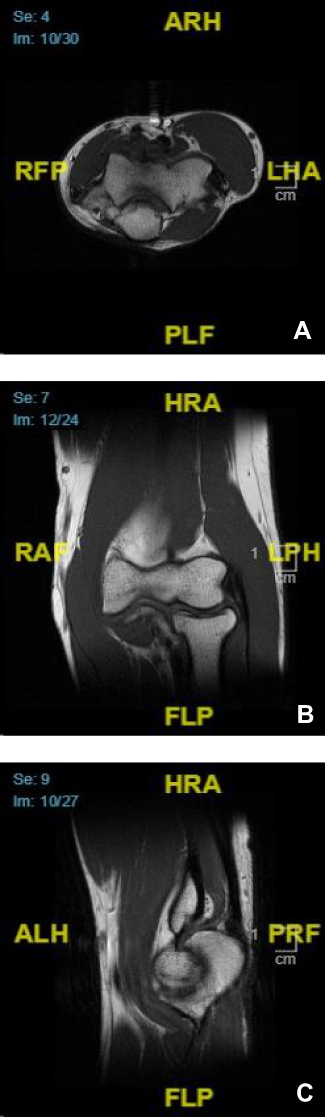 |
Figure 2 Right elbow MRI. No evidence of soft tissue mass or abnormal marrow signal: axial view (A), coronal view (B), and sagittal view (C). |
Discussion
ES, the second most common primary malignant bone tumor, occurs most often in adolescents.12 Its histologic profile is characterized by markers including CD99, which is sensitive but poorly specific for diagnosis.1 Other characteristics include EWSR1-FLI1 and EWSR1-ERG gene fusions2 and the translocation t(11;22)(q24;q12).3
Because the majority of ES patients have been found to have micro-metastasis, the most successful treatment of ES combines polychemotherapy with surgery and/or radiotherapy.6,13 Polychemotherapy for ES commonly consists of doxorubicin, vincristine, etoposide, and cyclophosphamide and/or ifosfamide.4 The Pediatric Oncology Group and the Children’s Cancer Group (COG) found that adding ifosfamide and etoposide to cyclophosphamide, doxorubicin, and vincristine for localized ES improves outcomes; this combination as well as the COG AEWS1030, Regimen A used in our study has become the basis on ES management in North America.4,5 Using surgery alone without chemotherapy results in higher relapse and mortality rates.6
The diagnostic value of immunohistochemistry in SS diagnosis is limited; however, a combination of EMA, BCL-2, and CD56 is commonly observed. CD99 staining may be positive in SS, and more recently TLE-1 immunohistochemistry has been used to help diagnose SS. Due to overlap with ES criteria, as evidenced by the overlap of CD99 and S100 positivity.1,14 Therefore, the utilization of gene arrangement analysis is vital for confirmation of definitive diagnosis between all small round blue cell tumors. The findings of t(X;18)(p11.2;q11.2) between SYT and SSX translocations are present in 90% of SS, and the fusion genes of SS18/SSX1, SS18/SSX2, and SS18/SSX4 are in 95% of SS, which would reclassify these soft tissue tumors, and therefore distinguish them from the others for perhaps a different treatment regimen.
While surgery and/or radiation have been most commonly used for treatment of SS, no consensus has been reached regarding the role of chemotherapy.9,11,15 Ifosfamide-based chemotherapy has been associated with survival in patients with high-risk extremity SS.16 An ifosfamide, vincristine, and doxorubicin regimen was found to have moderate activity against pediatric SS. Furthermore, 15767644 chemotherapy has also proved effective in adults, and in one study, the 4-year overall survival of adult patients treated with chemotherapy was 88% versus 67% for those not enrolled in a chemotherapeutic regimen.16,17 We can only speculate about the role of chemotherapy as a treatment for SS, as there is a lack of randomized trials and a definitive body of evidence in support of this modality.10,11
In our case study, the presumptive diagnosis of ES was based on an initial biopsy consistent with small round cell tumor appearance and positivity for CD99, two findings which support diagnosis of both ES and SS.1,14 The initial biopsy lacked the characteristic ES translocation, but the post-excision analysis did identify the SS18 rearrangement (1811.1) which is characteristic for SS. This resulted in the reclassification of the mass to a monophasic SS tumor. As such, the molecular analysis finding of SYT gene rearrangements supports the use of this technique for precise definitive tissue diagnosis of SS.18 It should be noted that an alternative explanation to our findings is that this was a biphasic tumor which had features of ES and SS. The chemotherapy successfully treated the ES element, and the SS element was cured by surgical resection.
In the past, more focused FISH testing had been used to specifically rule out certain translocations; however, with the advent of more widespread molecular testing techniques, multiple fusions can now be detected with modalities such as NGS fusion testing. These multi-fusion tests were not available at the time of this case’s presentation or diagnosis. These multi-gene fusion testing panels will play a major role to help further classify a tumor and test it for multiple different fusions in the list of differential diagnoses.19
To the best of the authors’ knowledge, this is the first reported case in which SS was successfully treated with standard ES chemotherapy regimen COG AEWS1030, Regimen A. This case supports the effectiveness of chemotherapy as part of the treatment regimen for SS in adult patients, as evidenced by the marked decrease in tumor size post-chemo and post-surgical excision, the negative remaining soft tissue bed, and the negative bony margins. Our patient presented one year later without recurrence. With the one year follow-up and documented lack of recurrence, the successful response to Regimen A as a part of treatment for this patient warrants further investigation into the routine incorporation of chemotherapy as an adjuvant arm to treatment in adult SS tumors.
Ethics Statement
Institutional approval was not required to publish the case details.
Patient Consent Statement
Written informed consent for publication or their details was obtained from the patient.
Disclosure
Dr Fazel Khan has patent pending to Navisect but no relationship or relevance to current manuscript at all. The authors report no other conflicts of interest in this work.
References
1. Sbaraglia M, Righi A, Gambarotti M, Dei Tos AP. Ewing sarcoma and Ewing-like tumors. Virchows Arch. 2020;476:109–119.
2. Wang WL, Patel NR, Caragea M, et al. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol. 2012;25(10):1378–1383. doi:10.1038/modpathol.2012.97
3. Park YK, Chi SG, Park HR, Yang MH, Unni KK. Detection of t(11;22)(q24;q12) translocation of Ewing’s sarcoma in paraffin embedded tissue by nested reverse transcription-polymerase chain reaction. J Korean Med Sci. 1998;13(4):395–399. doi:10.3346/jkms.1998.13.4.395
4. Meyers PA. Systemic therapy for osteosarcoma and Ewing sarcoma. Am Soc Clin Oncol Educ Book. 2015;e644–e647. doi:10.14694/EdBook_AM.2015.35.e644
5. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. doi:10.1056/NEJMoa020890
6. Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011;204(7):351–365. doi:10.1016/j.cancergen.2011.07.008
7. Stacchiotti S, Van Tine BA. Synovial sarcoma: current concepts and future perspectives. J Clin Oncol. 2018;36(2):180–187. doi:10.1200/JCO.2017.75.1941
8. Storlazzi CT, Mertens F, Mandahl N, et al. A novel fusion gene, SS18L1/SSX1, in synovial sarcoma. Genes Chromosomes Cancer. 2003;37(2):195–200. doi:10.1002/gcc.10210
9. Eilber FC, Dry SM. Diagnosis and management of synovial sarcoma. J Surg Oncol. 2008;97(4):314–320. doi:10.1002/jso.20974
10. Palmerini E, Staals EL, Alberghini M, et al. Synovial sarcoma: retrospective analysis of 250 patients treated at a single institution. Cancer. 2009;115(13):2988–2998. doi:10.1002/cncr.24370
11. Al-Hussaini H, Hogg D, Blackstein ME, et al. Clinical features, treatment, and outcome in 102 adult and pediatric patients with localized high-grade synovial sarcoma. Sarcoma. 2011;2011:231789. doi:10.1155/2011/231789
12. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184–192. doi:10.1016/S1470-2045(09)70286-4
13. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: current scenario and unmet need. World J Orthop. 2016;7(9):527–538. doi:10.5312/wjo.v7.i9.527
14. Xiao GY, Pan BC, Tian XY, Li Y, Li B, Li Z. Synovial sarcoma in cerebellum: a case report and literature review. Brain Tumor Pathol. 2014;31(1):68–75. doi:10.1007/s10014-012-0126-9
15. Wasif N, Smith CA, Tamurian RM, et al. Influence of physician specialty on treatment recommendations in the multidisciplinary management of soft tissue sarcoma of the extremities. JAMA Surg. 2013;148(7):632–639. doi:10.1001/jamasurg.2013.113
16. Eilber FC, Brennan MF, Eilber FR, et al. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann Surg. 2007;246(1):105–113. doi:10.1097/01.sla.0000262787.88639.2b
17. Wu Y, Bi W, Han G, Jia J, Xu M. Influence of neoadjuvant chemotherapy on prognosis of patients with synovial sarcoma. World J Surg Oncol. 2017;15(1):101. doi:10.1186/s12957-017-1165-9
18. Clark J, Rocques PJ, Crew AJ, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7(4):502–508. doi:10.1038/ng0894-502
19. Racanelli D, Brenca M, Baldazzi D, et al. Next-generation sequencing approaches for the identification of pathognomonic fusion transcripts in sarcomas: the experience of the Italian ACC sarcoma working group. Front Oncol. 2020;10:944. doi:10.3389/fonc.2020.00489
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.