")
Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Monitoring and Management of Bardet-Biedl Syndrome: What the Multi-Disciplinary Team Can Do
Authors Caba L, Florea L , Braha EE, Lupu VV , Gorduza EV
Received 9 July 2022
Accepted for publication 16 September 2022
Published 27 September 2022 Volume 2022:15 Pages 2153—2167
DOI https://doi.org/10.2147/JMDH.S274739
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Lavinia Caba,1,* Laura Florea,2,* Elena Emanuela Braha,3,* Valeriu Vasile Lupu,4,* Eusebiu Vlad Gorduza1,*
1Department of Mother and Child Medicine – Medical Genetics, “Grigore T. Popa” University of Medicine and Pharmacy, Iaşi, Romania; 2Department of Nephrology - Internal Medicine, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania; 3“C. I. Parhon” National Institute of Endocrinology, Bucharest, Romania; 4Department of Mother and Child Medicine – Pediatrics, “Grigore T. Popa” University of Medicine and Pharmacy, Iaşi, Romania
*These authors contributed equally to this work
Correspondence: Lavinia Caba, Department of Medical Genetics, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, 16 University Street, Iasi, 700115, Romania, Email [email protected]
Abstract: Bardet – Biedl syndrome is a rare autosomal recessive multisystem non-motile ciliopathy. It has heterogeneous clinical manifestations. It is caused by mutations in 26 genes encoding BBSome proteins, chaperonines, and IFT complex. The main clinical features are: retinal cone-rod dystrophy, central obesity, postaxial polydactyly, cognitive impairment, hypogonadism and genitourinary anomalies, and kidney disease. The onset of clinical manifestations is variable which makes the diagnosis difficult in some patients. Because of the multiple system involvement, a multidisciplinary approach is necessary. The purpose of this review is to provide monitoring and management directions for a better approach to these patients.
Keywords: Bardet-Biedl syndrome, genetic heterogeneity, multidisciplinary, management
Introduction
Bardet-Biedl syndrome is a rare, autosomal recessive inherited, genetic syndrome caused by defects in the genes encoding proteins of the cilium / basal body complex. This non-motile ciliopathy is characterized by rods and cones cell dystrophy, learning difficulties, polydactyly, obesity, genital malformations and renal malformations.1,2 The phenotype of Bardet-Biedl syndrome develops progressively in the first and the second decade of life, although there are large differences from patient to patient. Thus, most people are diagnosed in late childhood or even when they are young adults.3
The clinical diagnosis of Bardet-Biedl syndrome is made by the presence of 4 major features or 3 major and 2 minor features. The major and the minor features are listed in Table 1.3,4
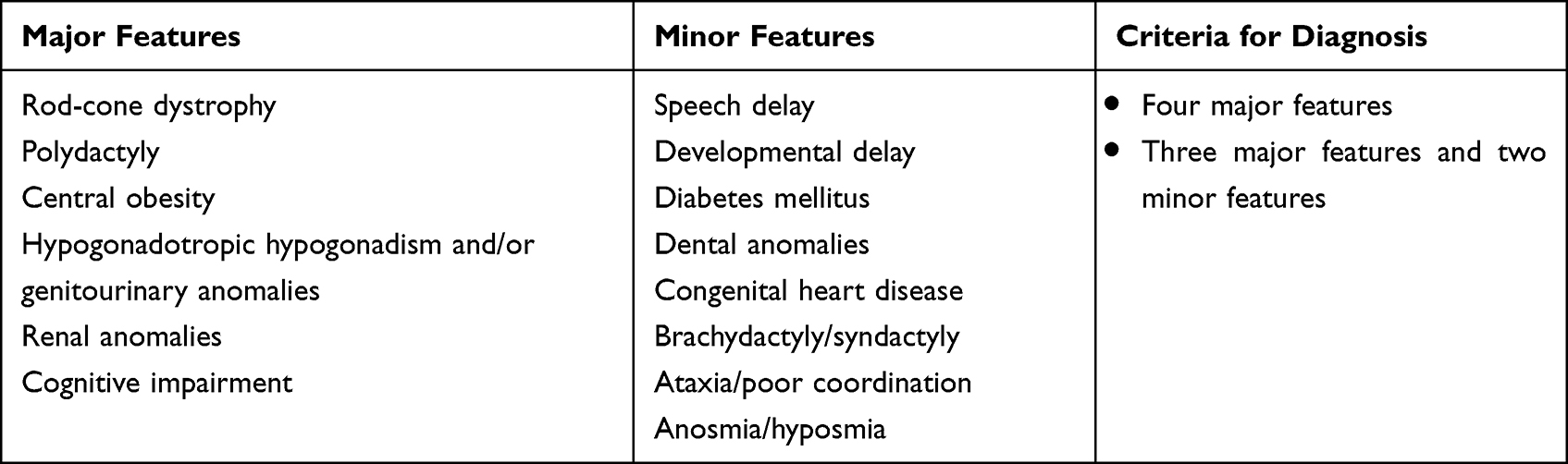 |
Table 1 Criteria for Diagnosis of Bardet-Biedl Syndrome |
BBS belongs to a group of ciliopathies and there is a clinical overlap between the different ciliopathies, which makes the diagnosis sometimes difficult. The involvement of eyes (retinal dystrophy), nose (anosmia), ears (hearing loss), brain (ataxia, epilepsy, mental disability, brain malformations), facial anomalies, energy homeostasis (central obesity), skeleton (skeletal anomalies), reproductive system (hypogonadism, genital anomalies), kidney (renal anomalies), liver (hepatic disease – liver fibrosis) are present primarily in non-motile cilia disorders. Involvement of the reproductive system with infertility, the brain with hydrocephaly and the respiratory system with chronic respiratory problems are especially associated with motile cilia disorders. The organ placement (organ laterality defects) and the involvement of heart with the congenital heart defects can be present in both motile and non-motile ciliopathies.5
Genetics
Bardet-Biedl syndrome (BBS) is a rare condition with autosomal recessive inheritance. BBS is characterized by genetic heterogeneity (locus, allelic, mutational and clinical).6 Table 2 summarizes the genes whose mutations have been associated with Bardet-Biedl syndrome.
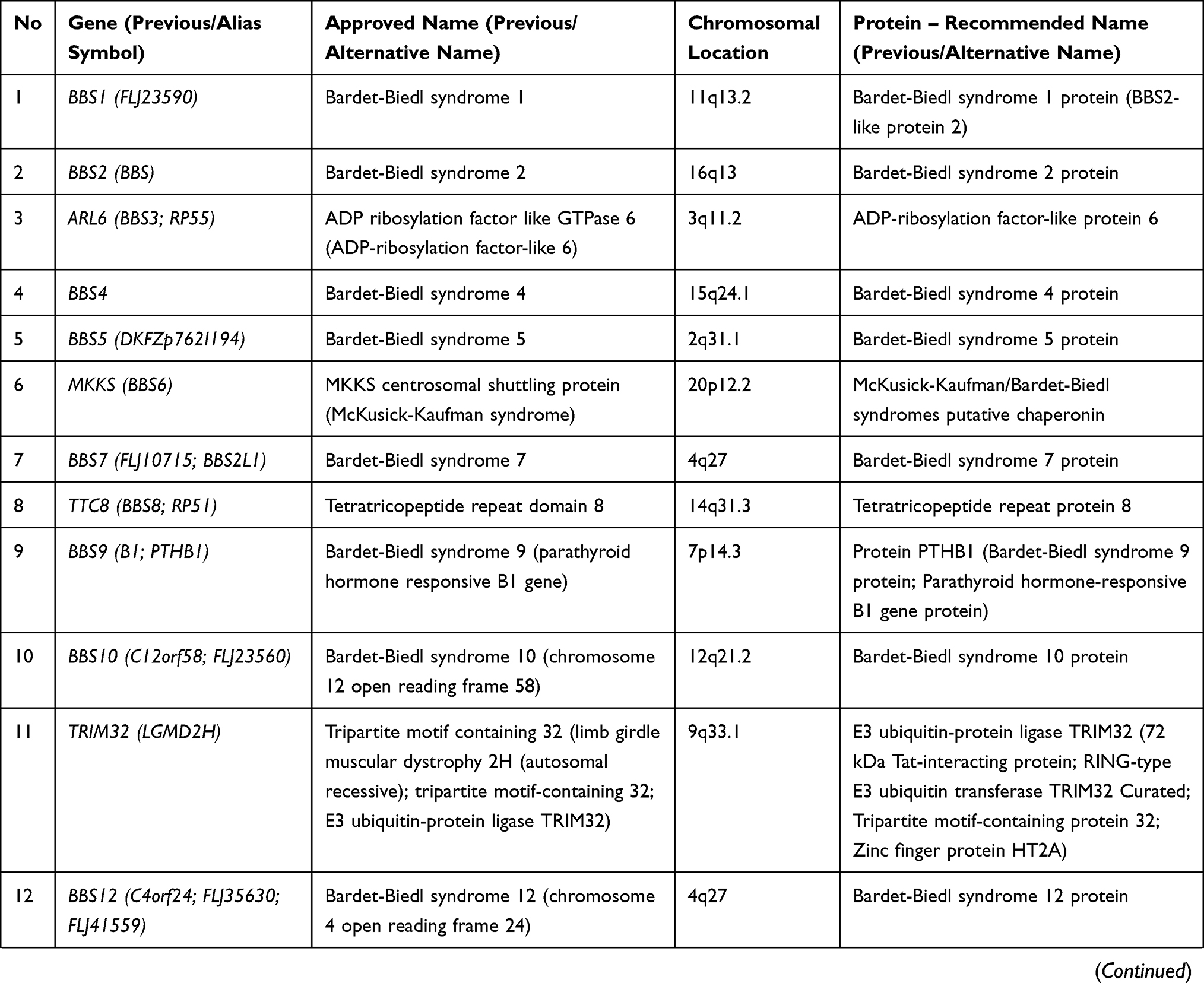 | 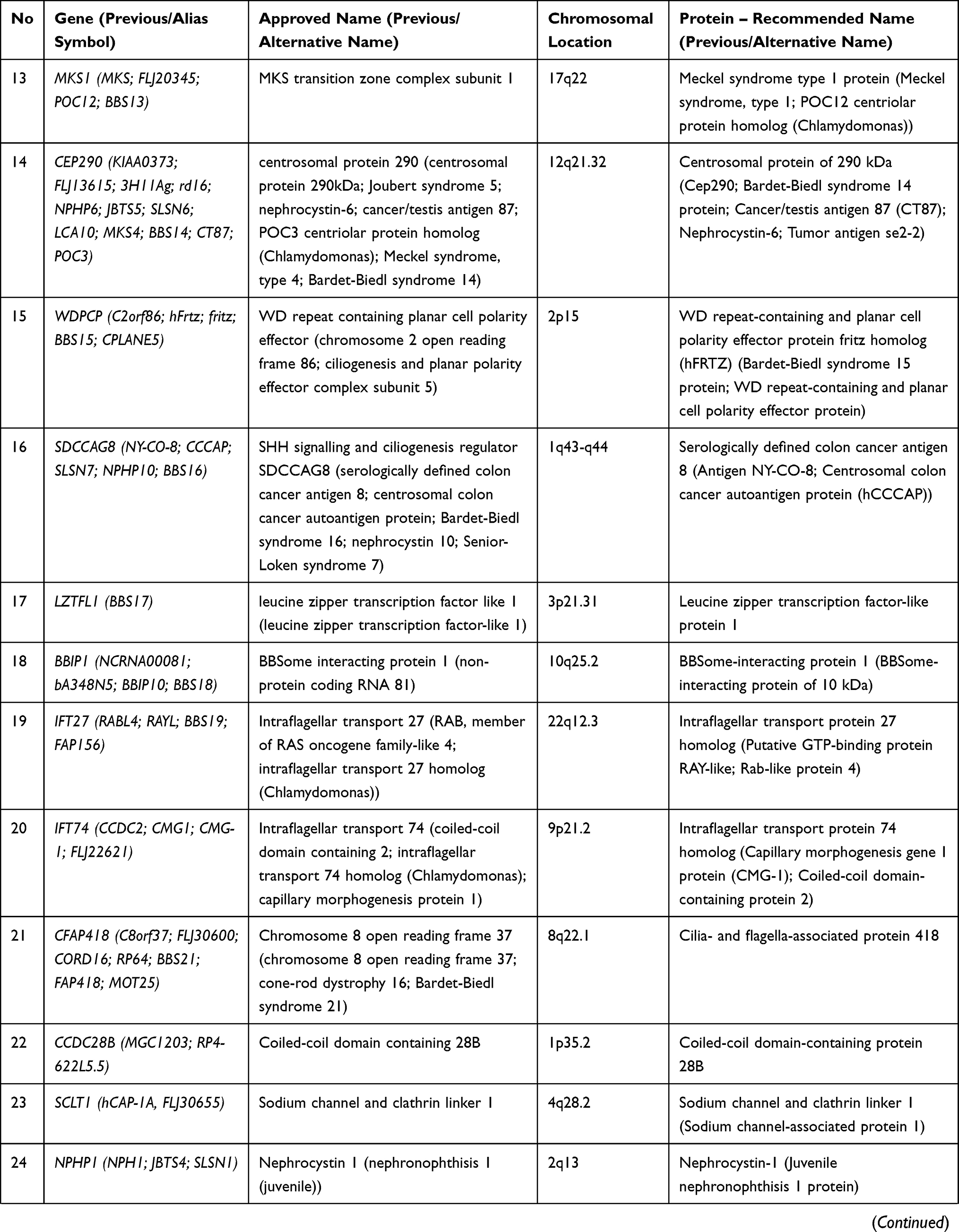 | 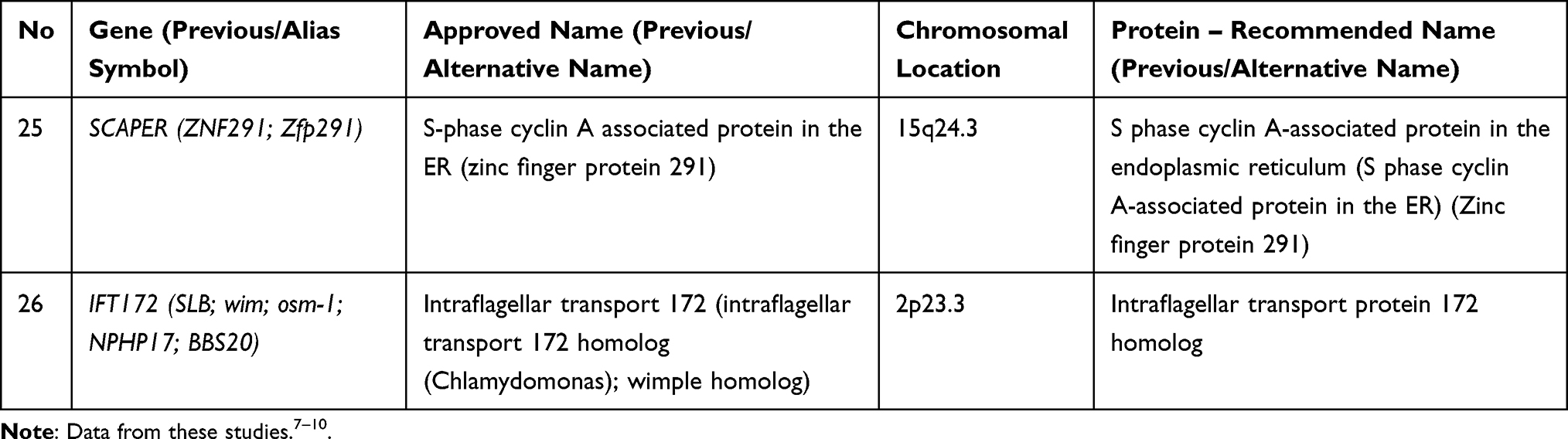 |
Table 2 Genes and Proteins Involved in Bardet-Biedl Syndrome |
The genes involved in BBS encode components of the BBSome (Bardet-Biedl syndrome 1 protein, Bardet-Biedl syndrome 2 protein, Bardet-Biedl syndrome 4 protein, Bardet-Biedl syndrome 5 protein, Bardet-Biedl syndrome 7 protein, Tetratricopeptide repeat protein 8, Protein PTHB1, BBSome-interacting protein 1) and also chaperonins (McKusick-Kaufman/Bardet-Biedl syndromes putative chaperonin, Bardet-Biedl syndrome 10 protein, Bardet-Biedl syndrome 12 protein).7 The BBS proteins are involved in several signalling pathways: GPCR (G protein-coupled receptors), Wnt (wingless-related integration site), mTOR (mammalian target of rapamycin). GPCR are important proteins for the structure and the function of the cilia both in the prenatal and postnatal development.11 Some GPCRs appears during postnatal developmental according to the neuronal type and function (somatostatin receptor 3 SSTR3, melanin-concentrating hormone receptor 1 - MCHR1, serotonin receptor 6–5HTR6, kisspeptin 1 receptor - KISS1R, dopamine receptors 1, 2, and 5, neuropeptide Y receptors - NPY2R and NPY5R).11
Niederlova et al suggested some genotype-phenotype correlations: the phenotype of the patients with mutations in ARL6 is typically a milder one compared to the patients with mutations in genes that encodes BBSome or chaperonines; mutations in BBS4 gene are not associated with a more severe phenotype than other BBS patients; mutations in BBS2 or BBS10 genes associates more often polydactyly and renal anomalies than patients with mutations in BBS1 gene; the penetrance of kidney anomalies is low in patients with mutations in BBS1, BBS4 or TTC8 genes and is high in patients with mutations in BBS2, BBS7, BBS9 genes.10
Multidisciplinary Approach
Both characteristics of BBS – the pleiotropy and the variable onset of the signs and symptoms - request a multidisciplinary approach (Figure 1). The multidisciplinary team has a dynamic component correlated with various stages of life and imply different medical specialties: paediatrics, neurology, dentistry, ophthalmology, endocrinology, ENT, genetics, cardiology, psychology, surgery, gastroenterology, nephrology.
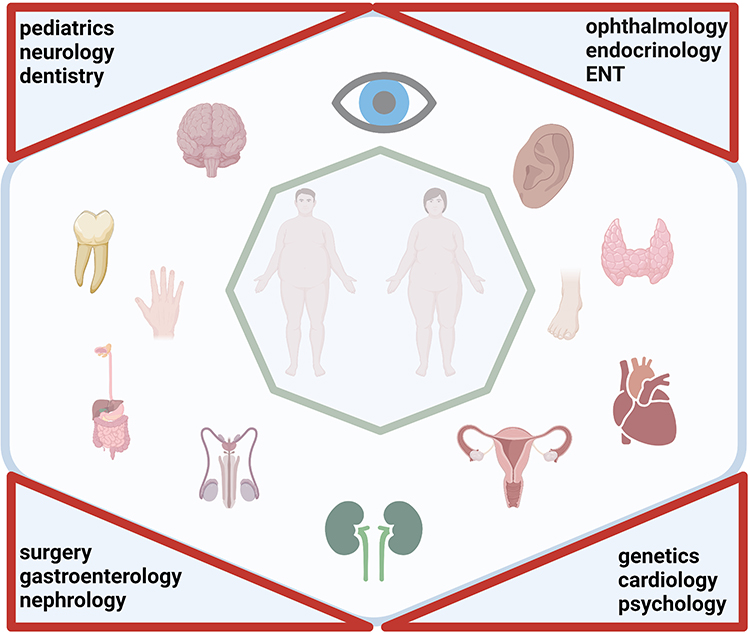 |
Figure 1 Pleiotropy and multidisciplinary management in Bardet-Biedl syndrome. Created with BioRender.com. |
Ocular Involvement - Rod-Cone Dystrophy
The main ophthalmic manifestation is the rod-cone dystrophy (RCD), found in 94% of the patients and considered to be the highest penetrance sign (100% penetrance in some studies).12–14 The rod-cone dystrophy is characterized by primary loss of rod photoreceptors and subsequent loss of cone photoreceptors, with a pigmentary retinopathy pattern.3,13 RCD is the second major cause of syndromic retinal degeneration.14,15 Other ophthalmic phenotypes described in the literature are: central cone-rod dystrophy, global severe retinal dystrophy, choroidal dystrophy.13,16 The symptoms appear in the first decade of life with nyctalopia, which is usually evident by the age of 7–8 years old.13,17 Progressive peripheral vision loss, decreased colour discrimination, impairment and loss of visual acuity can be observed throughout the evolution.12,14 Most patients are legally blind in the second or third decade of life.18 Other ocular anomalies are posterior subcapsular cataract and refractive errors, strabismus, nystagmus.3,6,17
Electroretinography (ERG) is the test of choice.3 Thus, early changes can be highlighted in the first two years of life, even before the development of fundus abnormalities.19
The baseline evaluation at the time of diagnosis includes: ophthalmic examination, electroretinogram, visual field testing, fundus examination, ERG and optical coherence tomography (OCT).12,20 Fundoscopic photographs are recommended for later reference.6,20 Infants and young children must be evaluated for strabismus and nystagmus, while older children and adults must be evaluated also for cataract.12
The surveillance must be done every year or as directed by ophthalmologist. The follow-up must include: visual acuity, visual field testing, fundus examination, ERG and screen for cataract, glaucoma and diabetic retinopathy.20
The management aims the correction of refractive errors. The tinted glasses are recommended if photophobia is present.20 Educational programs must be adapted to progressive visual impairment. The needs of a blind adult in educational programming must be anticipated.3,6,20,21
Polydactyly
Digital anomalies are represented by postaxial polydactyly (PAP), a major feature found in 79% of the patients. It is characterized by additional digits on the ulnar side of the hand and/or on the fibular side of the foot.12,20 It can affect both hands and feet, only the feet and, less often, only the hands.5,6 It should be noted that brachydactyly and/or syndactyly, with or without polydactyly, are considered minor features of BBS.12 It has also been described mesoaxial polydactyly in patients with pathogenic variants of the LZTFL1 gene.22,23 Surgical removal of accessory digits is recommended. It is recommended an early intervention, especially for feet polydactyly.
Obesity
Obesity is a feature of the Bardet-Biedl syndrome that is found in 89% of the diagnosed patients.12 Weight at birth is often within normal limits, although it is often towards the upper limit of normal. More than a third of the patients with Bardet-Biedl syndrome and normal birth weight develop overweight or obesity by the age of 1 year old. In adults, obesity is especially truncal, but in children it is evenly distributed throughout the body.3 The average value of BMI is 35.7 ± 7.8 kg/m2.24
A recent study of people with Bardet-Biedl syndrome found the prevalence of overweight and obesity according to WHO standards for raising children as follows in Table 3.25
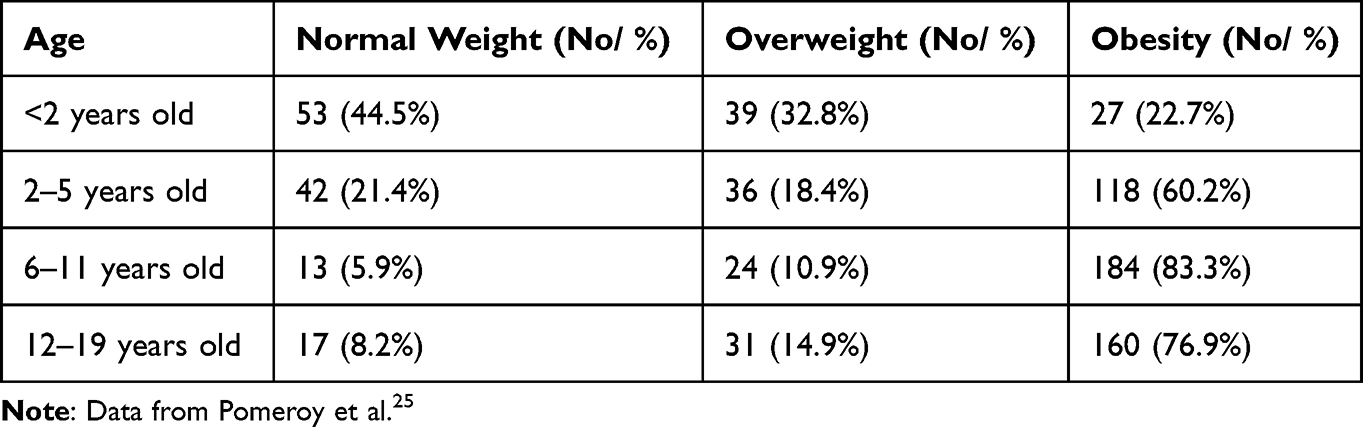 |
Table 3 Prevalence of Overweight and Obesity |
Hunger, satiety and energy consumption are deeply controlled processes in which neuroendocrine factors play a significant role. Some authors report polymorphisms in BBS4 (rs7178130), BBS6 (rs6108572, rs221667) and BBS2 (rs4784675) genes in early onset childhood and common adult severe obesity, which suggests their involvement in its development.26
Most BBS proteins are expressed in the primary cilia, which has so far failed to explain the occurrence of obesity in BBS. There is a hypothesis that BBS proteins are involved in loss of appetite regulation at the central level due to a defect in the ciliary.27 The BMP-8B protein (bone morphogenetic protein 8B) has been reported to play an important role in the metabolism through endocrine and paracrine actions, modulating the thermogenesis of the brown adipose tissue.28,29 It seems that BBSome is a critical determinant of the neuronal BMB-8B sensitivity.30 The potential mechanism involving BBS1 gene in inducing obesity is supported by the loss of central BMP-8B response in mice carrying a single missense mutation in Bbs1 gene.30 Other hypothesis of the development of obesity in BBS is that of impaired membrane expression of the leptin receptor (LepRB) in the hypothalamic cell population. Leptin, a polypeptide hormone, binding to its receptor in the brain, decreases food intake and increases energy expenditure. Genetic alterations of LepRB will promote signaling pathway associated with the development of obesity.31,32
Baseline evaluation includes: measurements of height, weight, head and waist circumference, calculation of BMI and waist-hip ratio, dietary evaluation with caloric intake and dietary components and the assessment of daily physical activity level.6,12,20 All these parameters will be evaluated at each medical visit.12
Obesity treatment is difficult for BBS patients and includes also, as for other obese patients, diet and lifestyle changes. Regular exercise program for weight control and behavioral and family therapy are recommended.6 For those who had a body mass index (BMI)> 30 kg/m² pharmacotherapy is recommended (eg, orlistat, lorcaserin, phentermine-topiramate, naltrexone-bupropion). In patients with a BMI >40 kg/m2 or those with BMI> 35 kg/m2 and comorbidities bariatric surgery can be discussed.33–36
Obesity complications are recommended to be treated as in the general population.3 To anticipate these complications, there should be made annual measurements of the blood pressure, blood glucose and haemoglobin A1C levels and the serum cholesterol and lipid levels.
A novelty in the therapy of the Bardet-Biedl syndrome is the administration of targeted therapies. An example is the use of melanocortin receptor agonists as a potential treatment for obesity in the syndrome.37 A research of the mechanisms leading to aberrant signalling by mutant proteins is ongoing, but there is evidence that there are defects in the leptin-melanocortin hypothalamic axis occurring in the Bardet-Biedl syndrome.38 This leads to resistance to leptin, which generate obesity.37
There are promising results in studies of subcutaneous injection treatment with setmelanotide, a melanocortin 4 receptor agonist, studied now in Phase 3 clinical trials.39 The clinical group evaluated by Haws RM et al included individuals aged >6 years old, with a diagnosis of BBS or Alström syndrome, treated with randomized, double-blind, placebo-controlled therapy. They selected individuals with body mass index ≥30 kg/m2 (aged ≥16 years old) or weight >97th percentile (aged 6–15 years old). The therapeutic regimen was different depending on the age of the patients. For patients aged >16 years old, the dose of setmelanotide was initially 2 mg/day in the first 2 weeks and 3 mg per day from the 3rd week. For patients aged between 6 and 16 years old, the following regimen was used: first week - 1 mg/day; second week 2 mg/day; from the 3rd week on 3 mg per day. Weight changes were noted after 52 weeks of treatment. These results include, in BBS patients, weight loss and feelings of early satiety.39
Hypogonadotropic Hypogonadism and/or Genitourinary Anomalies
Hypogonadism and genitourinary anomalies have been reported in 59% of the cases.10 The hypogonadism may be clinically manifest only at puberty by delaying the onset of secondary sexual characteristics. Anomalies described in males include cryptorchidism, short scrotum, micropenis, and low testicular volume, suggesting the possibility of infertility.3
In women, anatomical anomalies described are hypoplastic or duplex uterus, hypoplastic fallopian tubes and/or ovaries, septate vagina, partial or complete vaginal atresia, absent vaginal and/or urethral orifices, hydrocolpos/hydrometrocolpos, persistent urogenital sinus, and vesicovaginal fistula.40 Polycystic ovary syndrome has been rarely reported.24
Hormone testing revealed hypogonadotropic hypogonadism in some patients, while hypogonadism due to testicular origin was reported in others.41–44
Some studies describe a 63% frequency of pituitary abnormalities visible by MRI (pituitary hypoplasia, empty sella, Rathke’s cyst, enlarged pituitary gland) accompanied by hormonal imbalances in 45% of the cases (hyperprolactinemia, low FSH/LH and growth hormone deficiency).41 The treatment of the growth hormone deficiency and the hyperprolactinemia is being administered accordingly to the current guidelines.
The control of the GnRH release by kisspeptin signalling is reported to be involved in the gonadotropic axis activity during foetal life period, the pubertal onset and in maintaining fertility in adults.45,46 Congenital ciliary cell dysfunction in BBS may explain the decreased hypothalamic kisspeptin receptor KISS1R with secondary hypogonadism.45,47 This appears to be reversible in many cases, supporting the hypothesis that, at puberty, GnRH neurons increase the number of expressed cilia and, thus, could lead to normal development of the gonadotrophic axis in adults.48,49 It should not be overlooked that cryptorchidism and obesity can both impact on gonadic function with the decrease of the ratio testosterone/LH.49
Safa Mujahid et al, 2018, reported low SHBG in most of the patients with hypogonadism.24 Decreased SHBG levels are reported in obesity, glucocorticoid use, insulin resistance, type II diabetes mellitus and hypothyroidism and, thus, we consider that low SHBG in BBS patient could be interpreted also because of the obesity status.50–53
Other mechanism of infertility in men with BBS appears to be due to inappropriate conditions for spermatogenesis (short scrotum and obesity which disturb scrotal thermoregulation), unilateral agenesis of a seminal vesicle and partial obstruction of the genital duct by cysts.49,54 However, in adult men, sperm structure does not seem to be affected by primary cilia disfunction in BBS.49
Some authors recommend annual laboratory assessment for hypogonadism starting at the age of 13 years old. Other authors propose to refrain from initiating testosterone treatment in asymptomatic BBS men but, also, to continue studying this type of treatment that would improve metabolic parameters, including body composition and would increase muscle strength.24 The androgen replacement therapy regimen and doses used are those recommended for the paediatric and adult population with hypogonadism.55 General recommendations for hypogonadotropic hypogonadism treatment, both in women and men, are based on experience and current guidelines.56–58
Fertility is reported to be diminished for both sexes but they may have offsprings.24 There are few reports of the association of BBS and endometrial carcinoma. It should be considered in women with BBS and risk factors for hyperoestrogenism (obesity, hyperinsulinemia, ovulatory dysfunction).59–61
Renal Anomalies
Renal abnormalities in BBS can be a major cause of morbidity and mortality.62 The prevalence of renal disease has been estimated at 53–82%.63–65
Polycystic kidney disease, a typical feature of ciliopathies with renal manifestations can be associated with BBS.62–70
In a foetus/infant with a structural kidney disease or genitourinary malformations and/or other features (postaxial polydactyly) BBS must be suspected. Prenatal ultrasonography is useful in detection of renal cysts, but in 39% of the patients the results of the foetal ultrasonography are normal and the renal cysts are detected after birth.71
Renal anomalies are both structural and functional.
Renal tract malformations which can be discovered by ultrasonography are: renal dysplasia, unilateral renal agenesis, horseshoe kidneys, ectopic, duplex kidneys, foetal lobulation and diffuse cortical scarring, calyceal or parenchymal cysts, cystic tubular disease.
Other reported anomalies are glomerulosclerosis and tubulo-interstitial fibrosis, glomerular basement membrane disease, mesangial proliferation and sclerosis.
Urinary concentration defects (polyuria and polydipsia) caused by reduced concentrating ability due to reduced responsiveness to vasopressin with diabetes-like syndrome, kidney failure, renal anemia, hypertension can be detected.
In 31% of the children and 42% of the adults with BBS chronic kidney disease is present. End stage renal disease can be detected in early childhood (before the age of 5) or in adulthood related to BBS or to complications as hypertension or type 2 diabetes. 6% of the children and 8% of the adults developed end stage renal disease requiring dialysis or transplantation.63
Meyer et al reported some genotype-phenotype correlations in BBS associated with kidney disease. The severe kidney disease is more often associated with pathogenis variants in BBS10 gene (predominantly truncating variants) and BBS1 gene (predominantly missense variants). The patients with BBS determined by truncating variants in other genes (not only BBS10) have an increased risk for kidney failure. The author showed that truncating variants in SDCCAG8 gene has a highly predictive risk for early onset of kidney failure. Other genes associated with early onset of kidney failure were IFT172, SDCCAG8.72
However, some patients with structural renal anomalies do not develop functional renal disease.73
Urological anomalies included urinary incontinence, vesicoureteral reflux, neuropathic bladder and bladder outflow obstructions.
Investigation of clinical symptoms as anemia, polyuria, and polydipsia, hypertension, urinary tract infection, renal colic, symptoms of neurogenic bladder or bladder outflow obstruction is important at each evaluation visit.
Baseline investigations and yearly follow-up include basal blood pressure assessment, 24-hour blood pressure monitoring, complete blood cell count (CBC), serum creatinine, urea, cystatin C, electrolytes, glomerular filtration rate (GFR), urinary analysis for glucose, urinary protein (early morning urine analysis for albumin/creatinine ratio), hematuria and osmolarity, bladder and renal ultrasound examination.3,6,12
For detecting the calyceal anomalies abdominal magnetic resonance imaging can be done.
If structural or functional renal anomalies are present the patient must be referral to a nephrologist and should be monitored as directed by the nephrologist.
Another pathology rarely reported is nephrogenic diabetes insipidus (NDI), explained by the lack of renal response to desmopressin, probable due to vasopressin V2 receptor (V2R) damage.24 V2R, normally activated by antidiuretic hormone (ADH), is localized in the basolateral membrane of the principal cells alongside the collecting duct (CD). Marion et al describe that silencing BBS10 gene in human CD cell line leads primary to the loss of the cilium, and secondary to diminishing functions of V2R.69 Due to renal resistance to ADH, patients do not have a good response to desmopressin treatment. In patients with NDI, treatment is intended to decrease the polyuria and avoiding hypernatremia and volume depletion (low-sodium diet, drink adequate amounts of water).74
Metabolic Syndrome
Metabolic syndrome has an incidence of 54.3% in patients with BBS.24 Early detection of metabolic syndrome followed by appropriate treatment is necessary in order to prevent complications such as cardiovascular disease and diabetes later in life. The International Diabetes Federation (IDF) consensus definition of metabolic syndrome in children and adolescents (IDF) is being used. In the age group 6–10 years, the metabolic syndrome cannot be diagnosed, but weight reduction is recommended for children with abdominal obesity. In patients aged 10 years or older, metabolic syndrome is defined in the presence of abdominal obesity associated with two or more of the following features (elevated triglyceride, low HDL-cholesterol, high blood pressure, increased plasma glucose). The values used for diagnosis can be found in Table 4.75
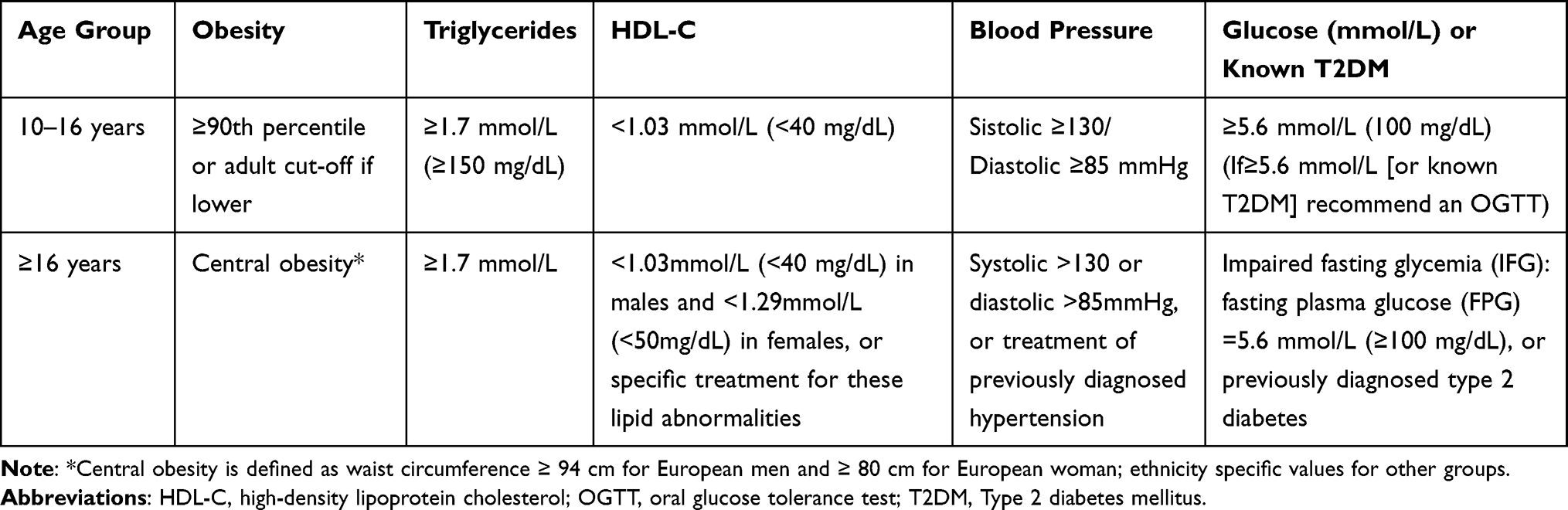 |
Table 4 The International Diabetes Federation Consensus Definition of Metabolic Syndrome in Children, Adolescents and Adults |
Baseline evaluation consists of a lipid panel (triglycerides, HDL-C, LDL-C, total cholesterol) and fasting blood glucose and HgbA1c. These will be repeated annually from the age of 4, if they are normal. In people with metabolic syndrome, the monitoring should be more frequent.12
Diabetes
Type 2 Diabetes Mellitus has an incidence of 15.8% in patients with BBS.24 The diagnostic criteria established by The American Diabetes Association (ADA) are used for diagnosis.76 One of the following tests can be used:
- Fasting plasma glucose (FPG) ≥126 mg/dL (7.0 mmol/L) OR
- 2-h plasma glucose (PG) ≥200 mg/dL (11.1 mmol/L) during OGTT (after age of 12 years old) OR
- A1C ≥6.5% (48 mmol/mol) OR
- In a patient with classic symptoms of hyperglycaemia or hyperglycaemic crisis, a random plasma glucose ≥200 mg/dL (11.1 mmol/L).
Baseline investigations for insulin resistance/diabetes mellitus include: fasting plasma glucose, OGTT after age of 6 years old. Fasting plasma insulin concentration and hyperinsulinemia may be present from childhood.20
The follow-up of the adolescent patients involves the promotion of healthy eating, physical activity according to visual impairment.20 Acanthosis nigricans (35.4% of patients with BBS) is a sign of insulin resistance / diabetes mellitus.39
Cognitive Impairment
Cognitive impairment is present in 66% of the patients with BBS.12 Kerr et al showed that most of the patients have difficulties in perceptual intellectual abilities, auditory attentional capacities, adaptive independence, behaviour. Thus, impairments in verbal fluency is present in 22–40% of the patients, abnormal attention capacity in 69% and severe impairment in functional independence in 74% of the patients.77
The baseline assessment should include a full developmental and/or neurocognitive assessment. The specialists involved are: clinical psychologist, developmental paediatrician/specialist in child development, behavioural psychologist and speech therapist.6 The methods used in the evaluation will be adapted to the visual status. Language assessment should be postponed after the age of 2 years old. In children with BBS, the intelligible speech and sentence formation can be delayed up to 4 years. Other speech anomalies are: articulation defects, nasal and/or breathy speech quality. Depending on the clinical situation, the existence of pharyngeal and/or laryngeal muscle incoordination must be taken into account. In this case, videofluoroscopy and palatal articulation studies must be performed.20 Early speech therapy should be initiated at the first signs of speech impairment.
A mental health assessment is also necessary because patients may have some psychiatric and behavioural conditions, such as: anxiety, mood disorders, depression, psychosomatic manifestations, bipolar disorder and emotional outbursts, hyperactivity, frustration, inflexibility, obsessive/compulsive tendencies, preference for fixed routines, inability to recognize social cues, inappropriate and disinhibited behaviour, shallow affect.6,20,78,79
Subclinical Hypothyroidism
Subclinical hypothyroidism has been reported in BBS patients more often than expected, with an incidence of 19.4%.24 The patient’s hypothyroidism was treated with levothyroxine hormone replacement in order to prevent any possible risk of negative effect on growth and development.24
The management of hypothyroidism in children is similar to adults, with the particularity that in children the doses are adjusted according to the child’s weight (commonly for newborns 10 μg/Kg/day, 1-year-old children 4–6 μg/Kg/day, adolescents 2–4 μg/Kg/day, with conversion to the adult dose of 1.6 μg/Kg/day once endocrine maturation is completely reached.80,81
According to the guidelines, the target of treatment is to keep the FT4 level in the mid to upper half of the reference range and the TSH level in the mid to lower half of the reference range.82,83
Tsyklauri et al (2021) reported autoimmune Hashimoto’s thyroiditis as having a higher prevalence in BBS patients than in the overall population, probably due to impaired immunity in patients with ciliopathies.84
At baseline evaluation, the investigation of the thyroid function (the FT4 and TSH levels corroborated with the ultrasonography of the thyroid) is recommended, and then an yearly evaluation of the thyroid gland function.12
When pregnancy is possible, preconception level of FT4 and TSH should be evaluated in order to modulate levothyroxine hormone replacement during pregnancy and prevent impairment of foetal brain development.85
ENT
Evaluation of the upper airways is important because laryngeal webs and bifid epiglottis can lead to life-threatening complications.86,87 Another rare abnormality that may be present is the choanal stenosis.6
The literature is poor in assessing anaesthetic risks. In a case study, Smith et al showed that there were differences between anaesthetic management in paediatric patients (<18 years old) and adults in terms of intubation. Thus, in paediatric patients, intubation was successfully performed by direct laryngoscopy. In adult patients, however, indirect laryngoscopy techniques were required (40% awake fiberoptic technique, 27% video laryngoscopy). The authors estimate that this was due to airway difficulty that worsen with age due to oral and facial anomalies in correlation with progressive morbid obesity.88 Clinical features that are associated with airway difficulty include: dental anomalies, high arched palate, facial dysmorphism and morbid obesity, bifid epiglottis, laryngeal web.86–88
There is a special management in terms of both anaesthetic and perioperative risk in patients with BBS. The anaesthesiologist and the multidisciplinary team must follow some steps: evaluation for difficult airways, cardiac anomalies, renal impairment, morbid obesity and intellectual disability.6,88
Neurology
Neurological development delay occurs in 81% of the patients.10 Ataxia or poor coordination can be found in 85% of the patients with BBS and may contribute to gross motor and fine motor delays.6,12 The baseline assessment includes careful neurological examination of the coordination and gait. An electroencephalogram should be performed for seizures (found in 9.6% of the patients).6,12,20 Olfactory dysfunction is present with a high incidence and requires a smell identification test. Usually, the olfactory bulb is abnormal on the brain MRI.12,89 MRI is also requested for evaluation of other neurological conditions (ataxia, hypotonia, seizures).
Dentistry
Oral or dental abnormalities are present in 50% of the patients and include the following: hypodontia or microdontia, high arched palate, crowding, shortened roots and taurodontism, enamel hypoplasia, posterior crossbite.12,90 There are primary craniofacial abnormalities and oral manifestations and secondary anomalies determined by other comorbidities.91
Recommended baseline evaluation includes routine dental care with the assessment for hygiene, dental crowding and hypodontia. In case of malocclusion, extractions or dental crowding, standard interventions are recommended. Antibiotic prophylaxis will be considered in case of dental interventions in individuals with heart disease.20 The follow-up is done every 6 months starting from the age of 1 year old.6,12
Gastroenterology
Liver disease (any abnormality on liver imaging and/or abnormal alanine aminotransferase - ALT - level) is present in 30% of the patients. The following have been described: bile duct abnormalities with cystic dilatation, perilobular and periportal fibrosis, increased length of the bile ducts and biliary cirrhosis.12,92,93 The following conditions have a lower incidence: Hirschsprung disease (2.8%), inflammatory bowel disease (1.1%), and celiac disease (1.5%).12 If gastrointestinal symptoms are present, investigations should be initiated for possible structural and functional abnormalities. For hepatic disease the baseline investigations include: liver ultrasonography to evaluate a possible liver fibrosis and steatosis, measurements of plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT) level and tests for synthetic function (prothrombin time - PT, partial thromboplastin time - PTT).6,12,20 If these parameters are normal, they are reassessed annually. People with liver disease should be monitored as directed by the hepatologist.12,20
Cardiology
Cardiovascular defects are described in the medical literature as having a frequency of 1.6% −29% and include: bicuspid aortic valve, pulmonary valve stenosis, tricuspid incompetence, atrioventricular septal defects, patent ductus arteriosus, cardiomyopathies, bilateral persistent superior vena cava, interrupted inferior vena cava, hemiazygos vein continuation, tetralogy of Fallot, single ventricle with transposition of the great vessels, hypoplasia of the aorta.3,6,12 Laterality defects have a prevalence 170 times higher than in general population, but the prevalence of dextrocardia and situs inversus totalis in patients with BBS is low (1.6%).6,12,94
The baseline evaluation involves auscultation of the heart, electrocardiogram and echocardiogram to assess for congenital heart defects and/or cardiomyopathy. If these conditions are present, the follow-up is recommended at the cardiologist’s indication. If the baseline evaluation is normal, re-evaluation is recommended only if symptoms appear.6,12,20 If there are structural cardiac defects, antibiotic prophylaxis is recommended in case of surgical and dental procedures.6
Hypertension
Hypertension is present in 30% of the patients with BBS.93 Guo et al showed that the neuronal BBSome plays an important role in hemodynamic, sympathetic and vascular regulation. Another cause of hypertension in BBS is the obesity and kidney disease.31,64
Dermatology
Cutaneous signs are common in BBS, although they are not diagnostic criteria. The mechanisms by which they occur could be: disturbance of keratinisation and keratinocyte function. Encountered dermatosis are: keratosis pilaris, alopecia, nevi, cherry angiomas, acanthosis nigricans, onychodystrophy, striae, ichthyosis vulgaris, pigmentation anomalies, psoriasis, acne vulgaris, confluent and reticulated papillomatosis, seborrheic dermatitis, intertrigo 1, acrochordons, lichen simplex, eczema, asteatotic dermatitis, hidradenitis.95 In the presence of one of these features, a dermatologic examination is requested and the specialist will decide which follow-up path must be applied.
Genetic Testing
Genetic testing is necessary to both certify the diagnosis in those cases in which the clinical diagnostic criteria are not present, and to provide correct genetic counselling to the family and perform a prenatal diagnosis. Testing a single gene is not recommended as there are no genotype-phenotype correlations to justify this approach.12 Sequencing of the BBS-causing genes by NGS (Next Generation Sequencing) confirms the diagnosis in 80% of the patients.3
The percentage of gene mutations in BBS are different. The most involved genes are BBS1 (23.4%), BBS10 (14.5%) followed by BBS2 (9.6%), BBS12 (6.4%), MKKS (6.3%), CEP290 (6.3%), BBS4 (5.3%), ARL6 (5.1%), SDCCAG8 (4.3%), BBS7 (4.2%), BBS5 (3.7%), BBS9 (3.4%), TTC8 (2.0%), CFAP418 (1.6%), MKS1 and IFT172 (1%), BBIP1, TRIM32, WDPCP, LZTFL1, IFT27, IFT74 and SCLT1 (<1%).12
For a cost reduction approach, the genetic testing can start by testing recurrent mutations in BBS1 genes (exon 12, NM_024649: c.1169T> G, p.M390R) and in BBS10 genes (exon 2, NM_024685: c.271dupT, p.Cys91Leufs * 5). The two mutations are identified in 30% of the patients.6 Currently, testing specific BBS-gene panels or larger ciliopathy genes panels is recommended. This is justified because there is a significant clinical overlap between ciliopathies.96 If the result is negative, the sequencing of the rarely involved genes is continued.6 Comprehensive genomic testing is another test that can be used. The advantage of genomic testing is that it can identify mutations in genes that have not yet been included in gene panels or mutations in new genes not previously known to be associated with BBS.12 If genetic testing is not available, it is recommended to initiate the management procedures if the clinical diagnostic criteria are met.6
Conclusion
Bardet - Biedl syndrome is a rare, multisystem, non-motile ciliopathy characterized by a high degree of genetic heterogeneity. The diagnosis and the monitoring and management require a multidisciplinary team. The main features to be evaluated are: ocular involvement, polydactyly, obesity, genitourinary anomalies, renal anomalies, metabolic features, cognitive impairment, hypothyroidism, ENT changes, neurologic modifications, dental particularities, gastrointestinal, cardiologic, and dermatologic aspects. The genetic examination and tests are essential to confirm the clinic diagnosis. The best test to use is the gene panel containing the 26 genes involved in the BBS pathogenic mechanisms.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet–Biedl syndrome—now and in the future. Front Pediatr. 2018;6:23. doi:10.3389/fped.2018.00023
2. Zacchia M, Capolongo G, Trepiccione F, Marion V. Impact of local and systemic factors on kidney dysfunction in Bardet-Biedl syndrome. Kidney Blood Press Res. 2017;42(5):784–793. doi:10.1159/000484301
3. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. doi:10.1038/ejhg.2012.115
4. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter F. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–446. doi:10.1136/jmg.36.6.437
5. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–547. doi:10.1038/nrm.2017.60
6. Carey JC, Cassidy SB, Battaglia A, Viskochil D, Eds. Cassidy and Allanson’s Management of Genetic Syndromes. John Wiley & Sons; 2020.
7. Florea L, Caba L, Gorduza EV. Bardet–Biedl syndrome—Multiple kaleidoscope images: insight into mechanisms of genotype–phenotype correlations. Genes. 2021;12(9):1353. doi:10.3390/genes12091353
8. HGNC, HUGO Gene Nomenclature Committee Home Page. Available from: http://www.genenames.org/.
9. UniProt: Universal Protein Knowledgebase. Available from: https://www.uniprot.org/.
10. Niederlova V, Modrak M, Tsyklauri O, et al. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019;40(11):2068–2087. doi:10.1002/humu.23862
11. Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol. 2018;6:8. doi:10.3389/fcell.2018.00008
12. Forsyth RL, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2003. Available from https://www.ncbi.nlm.nih.gov/books/NBK1363/.
13. Denniston AK, Beales PL, Tomlins PJ, et al. Evaluation of visual function and needs in adult patients with Bardet–Biedl syndrome. Retina. 2014;34(11):2282–2289. doi:10.1097/IAE.0000000000000222
14. Weihbrecht K, Goar WA, Pak T, et al. Keeping an eye on Bardet-Biedl syndrome: a comprehensive review of the role of Bardet-Biedl syndrome genes in the eye. Med Res Arch. 2017;5:9. doi:10.18103/mra.v5i9.1526
15. Stone EM, Andorf JL, Whitmore SS, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314–1331. doi:10.1016/j.ophtha.2017.04.008
16. Mockel A, Perdomo Y, Stutzmann F, et al. Retinal dystrophy in Bardet–Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res. 2011;30(4):258–274. doi:10.1016/j.preteyeres.2011.03.001
17. Berezovsky A, Rocha DM, Sacai PY, et al. Visual acuity and retinal function in patients with Bardet-Biedl syndrome. Clinics. 2012;67(2):145–149. doi:10.6061/clinics/2012(02)09
18. Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28(3):113–125. doi:10.1080/13816810701537424
19. Scheidecker S, Hull S, Perdomo Y, et al. Predominantly cone-system dysfunction as rare form of retinal degeneration in patients with molecularly confirmed Bardet-Biedl syndrome. Am J Ophthalmol. 2015;160(2):364–372. doi:10.1016/j.ajo.2015.05.007
20. Bardet -Biedl syndrome guideline development group. Management of Bardet -Biedl syndrome A clinical guideline, Version 14; April 28, 2014. Available from: http://www.orpha.net/national/data/IE-EN/www/uploads/BardetBiedl2014.pdf.
21. Christian PH. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2(1):7. doi:10.1186/1750-1172-2-7
22. Schaefer E, Lauer J, Durand M, et al. Mesoaxial polydactyly is a major feature in Bardet–Biedl syndrome patients with LZTFL1 (BBS17) mutations. Clin Genet. 2014;85(5):476–481. doi:10.1111/cge.12198
23. Marion V, Stutzmann F, Gerard M, et al. Exome sequencing identifies ´ mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet–Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet. 2012;49(5):317–321. doi:10.1136/jmedgenet-2012-100737
24. Mujahid S, Hunt KF, Cheah YS, et al. The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. J Clin Endocrinol Metab. 2018;103(5):1834–1841. doi:10.1210/jc.2017-01459
25. Pomeroy J, Krentz AD, Richardson JG, Berg RL, VanWormer JJ, Haws RM. Bardet‐Biedl syndrome: weight patterns and genetics in a rare obesity syndrome. Pediatr Obes. 2021;16(2):e12703. doi:10.1111/ijpo.12703
26. Benzinou M, Walley A, Lobbens S, et al. Bardet-Biedl syndrome gene variants are associated with both childhood and adult common obesity in French Caucasians. Diabetes. 2006;55(10):2876–2882. doi:10.2337/db06-0337
27. Büscher AK, Cetiner M, Büscher R, et al. Obesity in patients with Bardet-Biedl syndrome: influence of appetite-regulating hormones. Pediatr Nephrol. 2012;27(11):2065–2071. doi:10.1007/s00467-012-2220-y
28. Whittle AJ, Carobbio S, Martins L, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell. 2012;149(4):871–885. doi:10.1016/j.cell.2012.02.066
29. Martins L, Seoane-Collazo P, Contreras C, et al. A functional link between AMPK and orexin mediates the effect of BMP8B on energy balance. Cell Rep. 2016;16(8):2231–2242. doi:10.1016/j.celrep.2016.07.045
30. Rial-Pensado E, Freire-Agulleiro O, Ríos M, et al. Obesity induces resistance to central action of BMP8B through a mechanism involving the BBSome. Mol Metab. 2022;59:101465. doi:10.1016/j.molmet.2022.101465
31. Guo DF, Cui H, Zhang Q, et al. The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet. 2016;12(2):e1005890. doi:10.1371/journal.pgen.1005890
32. Rouabhi M, Guo DF, Morgan DA, et al. BBSome ablation in SF1 neurons causes obesity without comorbidities. Mol Metab. 2021;48:101211. doi:10.1016/j.molmet.2021.101211
33. Hamlington B, Ivey LE, Brenna E, et al. Characterization of courtesy stigma perceived by parents of overweight children with Bardet-Biedl syndrome. PLoS One. 2015;10(10):e0140705. doi:10.1371/journal.pone.0140705
34. Daskalakis M, Till H, Kiess W, et al. Roux-en-Y gastric bypass in an adolescent patient with Bardet-Biedl syndrome, a monogenic obesity disorder. Obes Surg. 2010;20(1):121–125. doi:10.1007/s11695-009-9915-6
35. Mujahid S, Huda MS, Beales P, et al. Adjustable gastric banding and sleeve gastrectomy in Bardet-Biedl syndrome. Obes Surg. 2014;24(10):1746–1748. doi:10.1007/s11695-014-1379-7
36. Garvey WT, Mechanick JI, Brett EM, et al. Reviewers of the AACE/ACE Obesity Clinical Practice Guidelines. American association of clinical endocrinologists and American college of endocrinology comprehensive clinical practice guidelines for medical care of patients with obesity. Endocr Pract. 2016;22 Suppl 3:1–203. doi:10.4158/EP161365.GL
37. Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet. 2009;18(7):1323–1331. doi:10.1093/hmg/ddp031
38. Mason K, Page L, Balikcioglu PG. Screening for hormonal, monogenic, and syndromic disorders in obese infants and children. Pediatr Ann. 2014;43(9):e218–e224. doi:10.3928/00904481-20140825-08
39. Haws RM, Gordon G, Han JC, et al. The efficacy and safety of setmelanotide in individuals with Bardet-Biedl syndrome or Alström syndrome: phase 3 trial design. Contemp Clin Trials Commun. 2021;22:100780. doi:10.1016/j.conctc.2021.100780
40. Deveault C, Billingsley G, Duncan JL, et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat. 2011;32(6):610–619. doi:10.1002/humu.21480
41. Guran T, Ekinci G, Atay Z, et al. Radiologic and hormonal evaluation of pituitary abnormalities in patients with Bardet-Biedl syndrome. Clin Dysmorphol. 2011;20(1):26–31. doi:10.1097/MCD.0b013e32833fd528
42. Leroith D, Farkash Y, Bar-Ziev J, et al. Hypothalamic-pituitary function in the Bardet-Biedl syndrome. Isr J Med Sci. 1980;16(7):514–518.
43. Mozaffarian G, Nakhjavani MK, Farrahi A. The Laurence-Moon-Bardet-Biedl syndrome: unresponsiveness to the action of testosterone, a possible mechanism. Fertil Steril. 1979;31(4):417–422. doi:10.1016/s0015-0282(16)43940-3
44. Green JS, Parfrey PS, Harnett JD, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321(15):1002–1009. doi:10.1056/NEJM198910123211503
45. Uenoyama Y, Pheng V, Tsukamura H, et al. The roles of kisspeptin revisited: inside and outside the hypothalamus. J Reprod Dev. 2016;62(6):537–545. doi:10.1262/jrd.2016-083
46. Silveira LG, Tusset C, Latronico AC. Impact of mutations in kisspeptin and neurokinin B signaling pathways on human reproduction. Brain Res. 2010;1364:72–80. doi:10.1016/j.brainres.2010.08.087
47. Koemeter-Cox AI, Sherwood TW, Green JA, et al. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A. 2014;111(28):10335–10340. doi:10.1073/pnas.1403286111
48. Desai A, Jha O, Iyer V, et al. Reversible hypogonadism in Bardet-Biedl syndrome. Fertil Steril. 2009;92(1):391.e13–5. doi:10.1016/j.fertnstert.2009.02.023
49. Koscinski I, Mark M, Messaddeq N, et al. Reproduction function in male patients with Bardet Biedl syndrome. J Clin Endocrinol Metab. 2020;105(12):e4417–29. Erratum in: J Clin Endocrinol Metab. 2021;106(4):e1936. doi:10.1210/clinem/dgaa551
50. Le TN, Nestler JE, Strauss JF 3rd, et al. Sex hormone-binding globulin and type 2 diabetes mellitus. Trends Endocrinol Metab. 2012;23(1):32–40. doi:10.1016/j.tem.2011.09.005
51. Dumoulin SC, Perret BP, Bennet AP, et al. Opposite effects of thyroid hormones on binding proteins for steroid hormones (sex hormone-binding globulin and corticosteroid-binding globulin) in humans. Eur J Endocrinol. 1995;132(5):594–598. Erratum in: Eur J Endocrinol 1995 Sep;133(3):381. doi:10.1530/eje.0.1320594
52. Wallace IR, McKinley MC, Bell PM, et al. Sex hormone binding globulin and insulin resistance. Clin Endocrinol. 2013;78(3):321–329. doi:10.1111/cen.12086
53. Giagulli VA, Kaufman JM, Vermeulen A. Pathogenesis of the decreased androgen levels in obese men. J Clin Endocrinol Metab. 1994;79(4):997–1000. doi:10.1210/jcem.79.4.7962311
54. Mieusset R, Fouda PJ, Vaysse P, et al. Increase in testicular temperature in case of cryptorchidism in boys. Fertil Steril. 1993;59(6):1319–1321. doi:10.1016/s0015-0282(16)55999-8
55. Chioma L, Cappa M. Hypogonadism in male infants and adolescents: new androgen formulations. Horm Res Paediatr. 2021;1–9. doi:10.1159/000521455
56. Sanfilippo JS, Lara-Torre E, Edmonds DK, et al. Clinical Pediatric and Adolescent Gynecology. New York: Informa Healthcare; 2009:450.
57. Al-Sharefi A, Quinton R. Current national and international guidelines for the management of male hypogonadism: helping clinicians to navigate variation in diagnostic criteria and treatment recommendations. Endocrinol Metab. 2020;35(3):526–540. doi:10.3803/EnM.2020.760
58. Young J, Xu C, Papadakis GE, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. 2019;40(2):669–710. doi:10.1210/er.2018-00116
59. Grechukhina O, Gressel GM, Munday W, et al. Endometrial carcinoma in a 26-year-old patient with Bardet-Biedl syndrome. Case Rep Obstet Gynecol. 2018;2018:1952351. doi:10.1155/2018/1952351
60. Peiker G, Boehm W, Carol W. Laurence-Moon-Bardet-Biedl-Syndrom und Endometriumkarzinom [Laurence-Moon-Bardet-Biedl syndrome and endometrial carcinoma]. Zentralbl Gynakol. 1978;100(17):1126–1130.
61. Schwab G, Kriz K. Two sisters with Laurence Bardet-Biedl-Moon syndrome and concomitant carcinoma of the body of the uterus. Geburtshilfe Frauenheilkd. 1980;40(3):279–281. doi:10.1055/s-2008-1037011
62. O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996;27(6):776–783. doi:10.1016/S0272-6386(96)90513-2
63. Forsythe E, Sparks K, Best S, et al. Risk factors for severe renal disease in Bardet-Biedl syndrome. J Am Soc Nephrol. 2017;28(3):963–970. doi:10.1681/ASN.2015091029
64. Imhoff O, Marion V, Stoetzel C, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6(1):22–29. doi:10.2215/CJN.03320410
65. Harnett JD, Green JS, Cramer BC, et al. spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med. 1988;319(10):615–618. doi:10.1056/NEJM198809083191005
66. Tieder M, Levy M, Gubler MC, Gagnadoux MF, Broyer M. Renal abnormalities in the Bardet-Biedl syndrome. Int J Pediatr Nephrol. 1982;3(3):199–203.
67. Gourdol O, David L, Colon S, et al. Renal involvement in the Laurence-Moon-Bardet-Biedl syndrome. Apropos of 3 cases. Pediatrie. 1984;39(3):175–181.
68. Putoux A, Mougou-Zerelli S, Thomas S, et al. BBS10 mutations are common in ‘Meckel’-type cystic kidneys. J Med Genet. 2010;47(12):848–852. doi:10.1136/jmg.2010.079392
69. Marion V, Schlicht D, Mockel A, et al. Bardet-Biedl syndrome highlights the major role of the primary cilium in efficient water reabsorption. Kidney Int. 2011;79(9):1013–1025. doi:10.1038/ki.2010.538
70. Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol. 2012;27(1):7–15. doi:10.1007/s00467-010-1751-3
71. Mary L, Chennen K, Stoetzel C, et al. Bardet-Biedl syndrome: antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clin Genet. 2019;95(3):384–397. doi:10.1111/cge.13500
72. Meyer JR, Krentz AD, Berg RL, et al. Kidney failure in Bardet–Biedl syndrome. Clin Genet. 2022;101(4):429–441. doi:10.1111/cge.14119
73. Forsythe E, Sparks K, Hoskins BE, et al. Genetic predictors of cardiovascular morbidity in Bardet-Biedl syndrome. Clin Genet. 2015;87(4):343–349. doi:10.1111/cge.12373
74. Christ-Crain M, Winzeler B, Refardt J. Diagnosis and management of diabetes insipidus for the internist: an update. J Intern Med. 2021;290(1):73–87. doi:10.1111/joim.13261
75. International Diabetes federation: the IDF consensus worldwide definition on the metabolic syndrome. Available from:https://idf.org/our-activities/advocacy-awareness/resources-and-tools/60:idfconsensus-worldwide-definitionof-the-metabolic-syndrome.html.
76. American Diabetes Association. Diabetes Overview. Diagnosis. Available from: https://www.diabetes.org/diabetes/a1c/diagnosis.
77. Kerr EN, Bhan A, Heon E. Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet–Biedl syndrome. Clin Genet. 2016;89(4):426–433. doi:10.1111/cge.12614
78. Barnett S, Reilly S, Carr L, Ojo I, Beales PL, Charman T. Behavioural phenotype of Bardet-Biedl syndrome. J Med Genet. 2002;39(12):e76. doi:10.1136/jmg.39.12.e76
79. Zelihić D, Hjardemaal FR, von der Lippe C. Caring for a child with Bardet-Biedl syndrome: a qualitative study of the parental experiences of daily coping and support. Eur J Med Genet. 2020;63(4):103856. doi:10.1016/j.ejmg.2020.103856
80. Jonklaas J. Sex and age differences in levothyroxine dosage requirement. Endocr Pract. 2010;16(1):71–79. doi:10.4158/EP09257.OR
81. Lafranchi S. Thyroiditis and acquired hypothyroidism. Pediatr Ann. 1992;21(1):29,32–9. doi:10.3928/0090-4481-19920101-07
82. American Academy of Pediatrics. Committee on Genetics. American Academy of Pediatrics: health supervision for children with Down syndrome. Pediatrics. 2001;107(2):442–449. doi:10.1542/peds.107.2.442.
83. Jonklaas J, Bianco AC, Bauer AJ, et al. Guidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association task force on thyroid hormone replacement. Thyroid. 2014;24(12):1670–1751. doi:10.1089/thy.2014.0028
84. Tsyklauri O, Niederlova V, Forsythe E, et al. Bardet-Biedl syndrome ciliopathy is linked to altered hematopoiesis and dysregulated self-tolerance. EMBO Rep. 2021;22(2):e50785. doi:10.15252/embr.202050785
85. Krassas GE, Poppe K, Glinoer D. Thyroid function and human reproductive health. Endocr Rev. 2010;31(5):702–755. doi:10.1210/er.2009-0041
86. Kaur P, Chaudhry C, Neelam H, Panigrahi I. Bardet-Biedl syndrome presenting with laryngeal web and bifid epiglottis. BMJ Case Rep. 2021;14(1):e236325. doi:10.1136/bcr-2020-236325
87. Poulin MA, Laframboise R, Blouin MJ. Association of bifid epiglottis and laryngeal web with Bardet-Biedl syndrome: a case report. Int J Pediatr Otorhinolaryngol. 2019;122:138–140. doi:10.1016/j.ijporl.2019.04.019
88. Smith BB, Barbara DW, Hyder JA, Smith MM. Anesthetic considerations for patients with Bardet–Biedl syndrome: a case series and review of the literature. Paediatr Anaesth. 2016;26(4):429–437. doi:10.1111/pan.12848
89. Braun JJ, Noblet V, Kremer S, et al. Value of MRI olfactory bulb evaluation in the assessment of olfactory dysfunction in Bardet–Biedl syndrome. Clin Genet. 2016;90(1):79–83. doi:10.1111/cge.12697
90. Abbas B, Abbas S, Malik SM, Rahim M, Umair M, Khurshid Z. Consanguineous marriages and dental anomalies: a cross-sectional analytical study. Int J Dent. 2022;2022:9750460. doi:10.1155/2022/9750460
91. Panny A, Glurich I, Haws RM, Acharya A. Oral and craniofacial anomalies of Bardet-Biedl syndrome: dental management in the context of a rare disease. J Dent Res. 2017;96(12):1361–1369. doi:10.1177/0022034517716913
92. Branfield Day L, Quammie C, Heon E, et al. Liver anomalies as a phenotype parameter of Bardet–Biedl syndrome. Clin Genet. 2016;89(4):507–509. doi:10.1111/cge.12684
93. Khan SA, Muhammad N, Khan MA, Kamal A, Rehman ZU, Khan S. Genetics of human Bardet–Biedl syndrome, an updates. Clin Genet. 2016;90(1):3–15. doi:10.1111/cge.12737
94. Olson AJ, Krentz AD, Finta KM, Okorie UC, Haws RM. Thoraco-abdominal abnormalities in Bardet-Biedl syndrome: situs inversus and heterotaxy. J Pediatr. 2019;204:31–37. doi:10.1016/j.jpeds.2018.08.068
95. Haws RM, McIntee TJ, Green CB. Cutaneous findings in Bardet‐Biedl syndrome. Int J Dermatol. 2019;58(10):1160–1164. doi:10.1111/ijd.14412
96. Zaki MS, Sattar S, Massoudi RA, Gleeson JG. Co‐occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am J Med Genet A. 2011;155(12):3042–3049. doi:10.1002/ajmg.a.34173
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.