Back to Journals » Drug Design, Development and Therapy » Volume 13
Molecular Requirements for the Expression of Antiplatelet Effects by Synthetic Structural Optimized Analogues of the Anticancer Drugs Imatinib and Nilotinib
Authors Pantazi D ![]() , Ntemou N, Brentas A, Alivertis D, Skobridis K, Tselepis AD
, Ntemou N, Brentas A, Alivertis D, Skobridis K, Tselepis AD
Received 17 April 2019
Accepted for publication 12 August 2019
Published 12 December 2019 Volume 2019:13 Pages 4225—4238
DOI https://doi.org/10.2147/DDDT.S211907
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Despoina Pantazi,1 Nikoleta Ntemou,2 Alexios Brentas,2 Dimitrios Alivertis,3 Konstantinos Skobridis,2 Alexandros D Tselepis1
1Department of Chemistry, Atherothrombosis Research Centre, Laboratory of Biochemistry, University of Ioannina, Ioannina 45110, Greece; 2Department of Chemistry, Section of Organic Chemistry and Biochemistry, University of Ioannina, Ioannina 45110, Greece; 3Department of Biological Applications and Technology, University of Ioannina, Ioannina 45110, Greece
Correspondence: Konstantinos Skobridis
Department of Chemistry, Section of Organic Chemistry and Biochemistry, University of Ioannina, Ioannina 45110, Greece
Tel + 30 26510 08598
Fax + 30 26510 08682
Email [email protected]
Background: Platelets play important roles in cancer progression and metastasis, as well as in cancer-associated thrombosis (CAT). Tyrosine kinases are implicated in several intracellular signaling pathways involved in tumor biology, thus tyrosine kinase inhibitors (TKIs) represent an important class of anticancer drugs, based on the concept of targeted therapy.
Purpose: The objective of this study is the design and synthesis of analogues of the TKIs imatinib and nilotinib in order to develop tyrosine kinase inhibitors, by investigating their molecular requirements, which would express antiplatelet properties.
Methods: Based on a recently described by us improved approach in the preparation of imatinib and/or nilotinib analogues, we designed and synthesized in five-step reaction sequences, 8 analogues of imatinib (I–IV), nilotinib (V, VI) and imatinib/nilotinib (VII, VIII). Their inhibitory effects on platelet aggregation and P-selectin membrane expression induced by arachidonic acid (AA), adenosine diphosphate (ADP) and thrombin receptor activating peptide-6 (TRAP-6), in vitro, were studied. Molecular docking studies and calculations were also performed.
Results: The novel analogues V–VIII were well established with the aid of spectroscopic methods. Imatinib and nilotinib inhibited AA-induced platelet aggregation, exhibiting IC50 values of 13.30 μ&Mgr; and 3.91 μ&Mgr;, respectively. Analogues I and II exhibited an improved inhibitory activity compared with imatinib. Among the nilotinib analogues, V exhibited a 9-fold higher activity than nilotinib. All compounds were less efficient in inhibiting platelet aggregation towards ADP and TRAP-6. Similar results were obtained for the membrane expression of P-selectin. Molecular docking studies showed that the improved antiplatelet activity of nilotinib analogue V is primarily attributed to the number and the strength of hydrogen bonds.
Conclusion: Our results show that there is considerable potential to develop synthetic analogues of imatinib and nilotinib, as TKIs with antiplatelet properties and therefore being suitable to target cancer progression and metastasis, as well as CAT by inhibiting platelet activation.
Keywords: cancer, kinase inhibitors, platelets, synthesis, thrombosis
Introduction
Platelets play a crucial role in hemostasis having as a major function to prevent bleeding and reduce blood loss following vascular injury.1 Platelets are also key regulators of thrombotic complications in various disease states including cancer.2–5 Indeed, many studies have demonstrated that the development of malignancy is associated with a plethora of alterations in the coagulation pathways, characterized by activation of clotting mechanisms to a different extent, depending on the tumor type.3,6,7 The pathogenesis of thrombosis in cancer is multifactorial, including various platelet alterations, which may significantly contribute to cancer-associated thrombosis (CAT).6–8 In this regard, P-selectin and platelet count are among the CAT risk factors identified and may also represent predictive biomarkers of venous thromboembolism (VTE) in cancer patients.8,9
Besides their important role in coagulation, platelets may significantly contribute to inflammation, cancer progression, metastasis and angiogenesis.10–12 Activated platelets secrete an array of biologically active molecules by which they can modulate tumor growth and metastasis.10,11,13 Within the blood compartment, tumor cells bind to platelets, thus escaping natural killer cell-mediated cytotoxicity.11,14,15 Therefore, tumor cell adhesion to platelets is a crucial step for tumor cell survival within the blood circulation.11,16,17 An important role in platelet adhesion to cancer cells is played by their P-selectin which binds to primarily to the receptor PSGL-1 (P-selectin glycoprotein ligand 1).18 Platelets can also promote an epithelial–mesenchymal transition in cancer cells, an essential step in cancer invasion and metastasis.19
Given the crucial roles of platelets in cancer progression and metastasis, as well as in CAT, inhibiting platelet activation is considered as a promising strategy to reduce both cancer progression and CAT.20,21 Among the antiplatelet drugs, the effect of low aspirin dose (around 75 mg/day) on cancer progression has attracted considerable interest.22,23 In this regard, population and clinical studies have demonstrated that aspirin significantly reduces the risk of colon cancer development and inhibits cancer growth and invasiveness.22,23 A number of clinical trials are currently ongoing to evaluate the therapeutic effect of aspirin treatment in cancer patients.20,21
Therefore, to limit platelet activation in cancer patients is a relevant aspect to take into account when designing new therapeutic approaches to treat cancer. However, such approaches would have to be carefully designed to avoid risk for bleedings, considering the key role of platelets in hemostasis. Nevertheless, the available evidence suggests that targeting platelets in cancers known to have a high risk of thrombotic events is therapeutically beneficial.20–23
Tyrosine kinases play important roles in the intracellular signaling pathways. At present, around 40 drugs with TKI activity have been approved by the FDA or are under investigation in clinical trials for anti-cancer treatment.24,25 Because of the comparable structure of the catalytic pocket of tyrosine kinases, TKIs often target multiple kinases that play a role in several different signaling pathways.26,27 Although the approved kinase inhibitors are active against more than one type of cancer, however only a few of them have been used for the treatment of non-oncological diseases.
Platelets display high tyrosine kinase activities in comparison to other cell types and tyrosine phosphorylation is one of the key signal transduction mechanisms of platelet activation.28 Therefore, it is expected that several TKIs used to treat malignancies may interfere with platelet activation processes thus exhibiting off-target antiplatelet effects.26,29,30
The ATP antagonists/mimetics imatinib and nilotinib represent privileged scaffolds in drug discovery. The non-receptor tyrosine kinase c-Src is involved in the intracellular signaling pathways leading to platelet activation and therefore represents a potential target for developing new antiplatelet therapies.31 Upon platelet activation, multiple intracellular proteins become phosphorylated at tyrosine residues.32 This process is mediated by at least four families of tyrosine kinases, Src, Syk, FAK, and JAK.33 Src-family kinases (SFKs) play a central role in mediating the rapid response of platelets to vascular injury, transmitting activation signals from a diverse repertoire of platelet surface receptors.33
Moreover, the crystal structure of regulated Abl closely resembles that seen in structures of regulated Src-family kinases.34 Structural differences between the inactive conformations of tyrosine kinase domains of Abl, identical in sequence with the kinase domain of Bcr-Abl, and its close relative c-Src suggest why imatinib can only inhibit the kinase domain of Abl but not that of c-Src.35
During optimization of the imatinib structure, it has been established that insubstantial structural modifications of the molecule/compound resulted in increased inhibitory and selectivity properties. In particular, it was shown that the introduction of only a “flag-methyl” at the imatinib phenyl ring C (Figure 1) would completely abolish the inhibitory effect of imatinib against protein kinase C alpha (PKCα), while the inhibitory effect against Bcr-Abl was retained or even enhanced.31 The dramatic loss of activity against PKCα was explained by a forced change of the preferred conformation of imatinib upon the introduction of this “flag-methyl” group.36 Furthermore, in our previous study, we had established that the elimination of the N-methylpiperazine ring and the incorporation of different groups at the final imatinib phenyl ring D (Figure 1) had greater activity against other kinase family members and poorer activity against Abl.37
 |
Figure 1 Modifications of prepared imatinib and nilotinib derivatives. Upper panel left: Following removal of the piperazinyl ring of imatinib, modifications were made to the final phenyl moiety as shown (I–IV). Upper panel right: Removal of the imidazolyl ring, modifications were made to the final phenyl ring (V, VI). Lower panel: Structure of the imatinib or nilotinib analogues by replacement of the amide bond with the urea moiety (VII, VIII). |
In light of the above developments, we hypothesized that slight changes in the imatinib and nilotinib structure might exert an inhibitory activity on these enzymes and therefore screened the three-dimensional architecture of Src-family kinases (SFKs), which are involved in platelet activation, and are considered as potential targets for the development of new antiplatelet agents.38 The synthetic analogues were designed in order to increase spatial flexibility and to explore potentially favorable halogen bond interactions and strong interactions of the nitro groups, incorporating at the final phenyl ring, with residues typically positioned at the ATP-binding pocket of the c-Src, especially with positively charged residues. To further explore the Structure–Activity Relationship (SAR), the modifications involve, except the removal of the N-methylpiperazine group in ring D of imatinib and the 4-methylimidazolyl group on the final phenyl ring D of nilotinib and replacement of the amide function with the urea (Figure 1).
In an effort to investigate the molecular requirements for these drugs to influence the platelet functionality, we specifically focused on their effects on platelet activation induced by the platelet agonists, arachidonic acid (AA) and adenosine diphosphate (ADP), which activate platelets through the main pathways targeted by the established antiplatelet drugs (aspirin and ADP receptor P2Y12 antagonists)39,40 as well by thrombin receptor activating peptide-6 (TRAP-6), which activates platelets through protease-activated receptor-1 (PAR-1).41
Materials and Methods
General
Solvents used were puriss, with the exception of THF, which was purified by fresh distillation over Na/benzophenone. Commercially available reagents were used as received, without any further purification. Nilotinib was purchased from Selleck Chemicals, Munich, Germany. Analytical TLC was performed on commercial Merck silica gel 60 F254. Flash chromatography was carried out using silica gel 60 (230–400 mesh). HPLC experiments were performed using a Shimadzu system, consisting of a DGU-20A controller, an LC-20AD pump, an SPD-M20A photodiode-array detector and a CTO-10AS column oven. 1H and 13C NMR spectra were recorded on a Brucker AMX (400/100 MHz 1H/13C) spectrometer using tetramethyl silane (TMS) as an internal standard. Chemical shifts are reported in ppm (δ) referenced to TMS, coupling constants J in Hz. High-resolution ESI mass spectra were measured on a Thermo Fisher Scientific LTQ ORBITRAP/LC−MS system. Elemental analyses were performed on a Heraeus CHN-Rapid Analyser.
Chemistry
The synthesis of the intermediates 2, 4–6 and 8–10, as well as of the final compounds, imatinib analogues I–IV, were based on a recently described optimized approach in the synthesis of imatinib intermediates and analogues and its spectroscopic data are consistent with the reported ones.42 The experimental procedure for the final step of the target compounds, V–VIII, and specific details are given below.
4-Methyl-N-(4-chloro-3-nitrophenyl)-3-[(4-pyridin-3-ylpyrimidin-2-yl) amino]benzamide (V)
To an ice-cold solution of acid 10 (1.00 equiv, 0.326 mmol) in dry DMF (3 mL) was added DIPEA (0.6 mL) under argon, followed by the addition of HATU (1.20 equiv, 0.39 mmol). After 30 min, a solution of 4-chloro-3-nitroaniline (1.20 equiv, 0.39 mmol) in dry DMF (2 mL) was added. The mixture was stirred vigorously at room temperature for 24 hrs and at 90°C for 3 hrs. The reaction mixture was cooled to room temperature, concentrated in vacuo, and then portioned between water and CH2Cl2. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried (Na2SO4). The solvent was evaporated in vacuo and the residue was purified by flash chromatography on silica gel (dichloromethane/methanol 15:1) to give the product as a pale yellow solid (36% yield). 1H NMR (400 MHz, DMSO-d6): δ 2.35 (s, 3H, CH3), 7.38 (d, J = 8.00 Hz, 1H), 7.44 (d, J = 8.00 Hz, 1H), 7.47–7.55 (m, 4H), 7.65 (d, J = 7.20 Hz, 1H), 7.75 (m, 1H), 8.08 (d, J = 8.80 Hz, 1H), 8.31 (s, 1H), 8.41–8.45 (m, 2H), 8.55 (t, J = 5.60 Hz, 1H), 8.60 (s, 1H), 8.69 (m, 2H), 9.05 (s, 1H), 9.15 (s, 1H), 9.27 (s, 1H), 9.31 (s, 1H), 10.67 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 18.14, 107.87, 108.06, 116.49, 123.48, 123.72, 124.19, 124.40, 124.52, 124.99, 130.31, 130.95, 131.77, 134.16, 137.73, 138.45, 139.26, 148.03, 151.36, 159.48, 161.00, 161.55, 167.31; HRMS (ESI): Calcd for C23H17ClN6O3 [M+H]+ m/z 461.1129, found [M+H]+ m/z 461.1127; Anal. calcd for C23H17ClN6O3 (460.87): C 59.94, H 3.72, N 18.24, found: C 59.71, H 3.56, N 18.31.
4-Methyl-N-(4-amino-3-trifluoromethyl)-3-[(4-pyridin-3-ylpyrimidin-2-yl) amino]benzamide (VI)
To an ice-cold solution of acid 10 (1.00 equiv, 0.48 mmol) in dry THF (5 mL) was added Et3N (0.5 mL) under argon, followed by the addition of TBTU (1.10 equiv, 0.53 mmoL). After 30 min, a solution of 4-(trifluoromethyl)benzene-1,3-diamine (1.20 equiv, 0.57 mmoL) in dry THF (2 mL) was added. The reaction mixture was stirred at 0°C for 30 min and at room temperature for 24 h. After completion monitored by TLC (CH2Cl2/MeOH 15:1, v/v), the mixture was concentrated in vacuo, and then portioned between water and CH2Cl2. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried (Na2SO4). The solvent was evaporated in vacuo and the residue was purified by flash chromatography on silica gel (CH2Cl2/MeOH 15:1, v/v) to give the product as a pale yellow solid (93% yield). 1H NMR (400 MHz, DMSO-d6): δ 2.33 (s, 3H, CH3), 6.82 (d, J = 8.80 Hz, 1H), 7.38 (d, J = 8.00 Hz, 1H), 7.46 (d, J = 5.20 Hz, 1H), 7.50 (dd, J = 7.20, 4.80 Hz, 1H), 7.64 (d, J = 8.80 Hz, 1H), 7.69 (d, J = 7.60 Hz, 1H), 7.81 (s, 1H), 8.22 (s, 1H), 8.43 (d, J = 8.00 Hz, 1H), 8.53 (d, J = 5.20 Hz, 1H), 8.68 (d, J = 4.80 Hz, 1H), 9.10 (s, 1H), 9.26 (s, 1H), 10.01 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 18.06, 107.73, 116.99, 118.16, 118.21, 123.25, 123.69, 124.11, 126.21, 127.84, 130.09, 132.04, 132.47, 134.17, 135.96, 137.88, 142.45, 148.04, 151.34, 159.46, 160.99, 161.49, 164.54; HRMS (ESI): Calcd for C24H19F3N6O [M+H]+ m/z 465.1651, found [M+H]+ m/z 465.1647; Anal. calcd for C24H19F3N6O (464.4425): C 62.07, H 4.12, N 18.09, found: C 62.22, H 4.03, N 18.31.
1-(4-Methyl-3-[(4-pyridin-3-yl)pyrimidin-2-ylamino)phenyl]-3-(Phenyl)urea (VII)
In a solution of amine 6 (1.00 equiv, 0.25 mmol) and Et3N (0.5 mL) in dry CH2Cl2 (10 mL) solution of phenyl isocyanate (1.10 equiv, 0.28 mmol) in dry CH2Cl2 (5 mL) was added dropwise under stirring at 5°C. The resulting mixture was stirred at 5°C for 30 mins and at room temperature for 24 hrs. The mixture was concentrated in vacuo, and then portioned between water and CH2Cl2. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried (Na2SO4). The solvent was evaporated in vacuo and the residue was purified by flash chromatography on silica gel (CH2Cl2/MeOH 10:1) to give the product as a pale yellow solid (78% yield). 1H NMR (400 MHz, DMSO-d6): δ 2.20 (s, 3H, CH3), 6.96 (d, J = 7.60 Hz, 1H), 7.14 (s, 2H), 7.27 (t, J = 7.60 Hz, 2H), 7.44 (m, 3H), 7.52 (m, 1H), 7.80 (s, 1H), 8.49 (m, 1H), 8.52 (d, J = 5.20 Hz, 1H), 8.60 (s, 1H), 8.61 (s, 1H), 8.70 (dd, J = 4.80, 1.20 Hz, 1H), 8.88 (s, 1H), 9.29 (d, J = 5.20 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 25.04, 115.12, 122.06, 122.30, 125.67, 129.25, 131.33, 132.88, 136.30, 137.79, 139.79, 141.99, 145.17, 145.49, 147.34, 155.74, 158.93, 160.09, 166.99, 168.64, 169.15; HRMS (ESI): Calcd for C23H20N6O [M+H]+ m/z 397.1777, found [M+H]+ m/z 397.1757; Anal. calcd for C23H20N6O (396.4445): C 69.68, H 5.08, N 21.20, found: C 69.81, H 5.02, N 21.33.
1-(4-Methyl-3-[(4-pyridin-3-yl)pyrimidin-2-ylamino)phenyl]-3-(4-chloro-3-nitrophenyl)urea (VIII)
To a stirred solution of amine 6 (1.00 equiv, 0.25 mmol) and Et3N (0.7 mL) in dry THF (10 mL) was added a solution of 4-chloro-3-nitrophenyl isocyanate (1.10 equiv, 0.28 mmoL) in dry THF (5 mL) dropwise with stirring at 5°C. The resulting mixture was stirred at 5°C for 30 mins and at room temperature for 24 hrs. The mixture was concentrated in vacuo, and then portioned between water and AcOEt. The aqueous layer was extracted with AcOEt, and the combined organic layers were dried (Na2SO4). The solvent was evaporated in vacuo and the residue precipitated by the addition of 2–3 mL CH2Cl2. The precipitated product was filtered off and dried to yield a first crop of the product. A second crop of product was obtained from the filtrate, which was concentrated in vacuo and the residue purified by flash chromatography on silica gel (CH2Cl2/MeOH 20:1) to give the product as a pale yellow solid (71% total yield). 1H NMR (400 MHz, DMSO-d6): δ 2.21 (s, 3H, CH3), 7.15 (m, 2H), 7.44 (d, J = 5.20 Hz, 1H), 7.52 (m, 1H), 7.61–7.66 (m, 2H), 7.89 (d, J = 1.20 Hz, 1H), 8.35 (d, J = 2.4 Hz, 1H), 8.50 (dt, J = 8.20, 2.40 Hz,1H), 8.52 (d, J = 5.20 Hz, 1H), 8.68 (dd, J = 4.80, 1.60 Hz, 1H), 8.87 (s, 1H), 8.93 (s, 1H), 9.32 (t, J = 2.00, 1H); 13C NMR (100 MHz, DMSO-d6): δ 25.06, 115.22, 121.68, 122.36, 122.52, 123.92, 130.50, 131.30, 133.25, 137.82, 139.24, 139.72, 141.98, 144.57, 145.55, 147.59, 155.15, 155.81, 158.92, 159.85, 167.01, 168.55, 169.16; HRMS (ESI): Calcd for C23H18ClN7O3 [M+H]+ m/z 476.1238, found [M+H]+ m/z 476.1262; Anal. calcd for C23H18ClN7O3 (475.8871): C 58.05, H 3.81, N 20.60, found: C 58.27, H 3.66, N 20.52.
Biological Assays
Platelet Aggregation Studies
Platelet aggregation studies were performed in platelet-rich plasma (PRP) prepared from peripheral venous blood of apparently healthy normolipidemic volunteers, using as anticoagulant citric acid solution (ACD) as we have previously described.43 The study protocol was approved by the ethics committee of the University Hospital of Ioannina and adheres to the principles of the declaration of Helsinki. All participants gave their written informed consent before peripheral venous blood was drawn. Isolated platelets in PRP were enumerated and their number was adjusted to 250×106/mL with homologous platelet-poor plasma (PPP). Platelets were pre-incubated at 37°C with various concentrations of imatinib, nilotinib and their synthetic analogues solubilized in DMSO (or its vehicle; DMSO, considering as control) for 1 min or 5 min. The maximum concentration of each tested compound was 100 μΜ. Light transmittance aggregometry (LTA) was performed in 0.5 mL of PRP in the presence of the platelet agonists AA (500 μM), ADP (5 μΜ) and TRAP-6 (10 μΜ) in an aggregometer (Model 700-4DR, Chrono-Log, Havertown, PA, USA), using 0.5 mL of corresponding platelet-poor plasma (PPP) as a blank. In all studies, the concentration of DMSO added in PRP was 0.5% (v/v).44 Aggregation was monitored with the use of the software AggroLink (ver. 8.0). The inhibitory efficacy of imatinib, nilotinib and their synthetic analogues was expressed as IC50 values, in μΜ (the concentration that induces 50% inhibition of platelet aggregation). In parallel experiments, we incubated platelets with imatinib, nilotinib and related synthetic analogues at a concentration of 100 μΜ for up to 10 min at 37°C to investigate the possible existence of agonistic activity. All aggregation assays were conducted within 3 h after blood venipuncture.43,45
The surface expression of P-selectin was studied in a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) using the fluorescently labeled monoclonal antibody (anti-CD62P-PE) as we previously described with slight modifications.45 Platelets in PRP were incubated for 5 min at 37°C with imatinib, nilotinib or their synthetic analogues at various concentrations up to 100 μΜ. Platelets were then activated with AA (800 μΜ), ADP (50 μΜ), or TRAP-6 (50 μΜ) for 5 min at 37°C without stirring. After incubation, a small portion of the reaction mixture was incubated with the fluorescently labeled monoclonal antibody towards P-selectin, anti-CD62P-PE, for 20 min at room temperature in the dark, diluted 1:5 (v/v) with 10 mM PBS, pH=7.4 and immediately analyzed by flow cytometry, as previously described.45,46 Platelets were gated according to staining for the platelet antigen CD61, using the fluorescently labeled monoclonal antibody anti-CD61-PerCP. Analyses included the percentage of positive events facilitated by evaluating the mean fluorescence intensity (MFI) values.45,46
Molecular Docking
The X-ray structure of tyrosine kinase protein c-Src was obtained from the RSCB Protein Data Bank (PDB ID: 1Υ57). We used the known c-Src kinase domains to model the synthetic drug complexes through flexible docking calculations. Before docking, the inhibitors were optimized and docked onto the active site of enzyme used. The docking calculations were performed using discovery studio visualizer 2016 and Chimera 1.11c. with imatinib, nilotinib and their synthetic analogues in the c-Src kinase domain (5 dockings for each complex).
Statistics
All data are presented as mean ± SD for at least three independent experiments. Independent–samples Student’s t-test was performed using the Statistical Package for the Social Sciences (SPSS), version 25 (IBM). A p-value of <0.05 was considered significant.
Results and Discussion
Chemistry
The synthesis of imatinib and all imatinib (I-IV), nilotinib (V, VI) and imatinib/nilotinib (VII, VIII) analogues (Figure 1), as mentioned above, were based on a recently described by us improved and efficiently optimized approach in the preparation of imatinib and/or nilotinib analogues.42 The imatinib analogues, compounds I-IV, and the imatinib/nilotinib analogues, compounds VII and VIII, containing the urea moiety, were prepared as shown in Scheme 1. The nilotinib analogues, compounds V and VI, were prepared as shown in Scheme 2. Briefly, 3-acetyl pyridine 1 was converted into the corresponding enaminone 2 by the use of N,N-dimethylformamide-diethyl acetal which was subsequently reacted with N-(2-methyl-5-nitrophenyl)-guanidinium hydrochloride 4, prepared from aniline hydrochloride 3 with excess of molten cyanamide, to the corresponding phenylamino-pyrimidine 5. Catalytic hydrogenation of the nitro group of compound 5 in the presence of PtO2 (Adam’s catalyst) led to the corresponding aniline 6 quantitatively, which, subsequently, was coupled with the appropriate benzoyl chloride to afford the desired imatinib analogues I-IV (Scheme 1). The novel imatinib or nilotinib analogues VII and VIII, N,N’-diphenyl urea derivatives, were prepared in high yields and high purity from the pharmacophore 6 and the appropriate phenylisocyanate in THF or CH2Cl2 under basic conditions (Scheme 1).
 |
Scheme 1 Reactions and conditions for the synthesis of the imatinib I–IV and of the imatinib/nilotinib analogues VII and VIII. |
 |
Scheme 2 Reactions and conditions for the synthesis of the nilotinib analogues V and VI. |
Following the above-described route, we prepared the nilotinib analogues V and VI. The intermediate key compound phenylaminopyrimidine 9, analogue to the previous imatinib intermediate, bearing the ethyl ester group instead of the nitro group, was prepared by the reaction of the enaminone 2 and the corresponding guanidinium hydrochloride 8. Alkaline hydrolysis of the carboxyl ester 9 with our protocol in non-aqueous conditions, under very mild conditions, such as short time, room temperature and low concentration of alkali, by the use of dichloromethane/methanol (9:1, v/v) as solvent47 was then performed to produce the corresponding carboxylic acid 10 which subsequently coupled with the aniline fragment 4-chloro-3-nitroaniline, using 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU) as coupling agent and hydroxybenzotriazole (HOBt) as an additive in DMF in the presence of N,N-diisopropylethylamine (DIPEA), to give the nilotinib analogue V. The coupling of carboxylic acid 10 with 4-(trifluoromethyl)benzene-1,3-diamine, using 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) in THF in the presence of triethylamine (Et3N) proceeded readily and afforded the corresponding analogue VI in high yields (Scheme 2).
All the final compounds were purified by flash chromatography on a silica gel column. The purity of these synthetic analogues ranged from 99.0% to 99.5% as estimated by RP-HPLC and 1H NMR. The novel analogues V-VIII, reported herein, were well established by their 1H NMR, 13C NMR, as well as by high-resolution mass spectra and elemental analysis (Supplementary materials).
Biological Assays
Effect on Human Platelet Aggregation in PRP
All compounds were subsequently screened for their effect on human platelet aggregation induced by 3 different agonists, AA, ADP and TRAP-6. In preliminary experiments, we evaluated whether any of the studied compounds exhibited a platelet aggregating activity. None of these compounds at a concentration of 100 μΜ exhibited any agonistic effect (data not shown). The inhibitory effect of imatinib/nilotinib and their synthetic analogues were subsequently studied on platelet aggregation. Among the 3 agonists evaluated, the strongest dose-dependent inhibitory effect of imatinib and nilotinib (incubated for 5 min in the aggregometer at 37°C, under stirring), was observed towards AA-induced platelet aggregation, exhibiting IC50 values of 13.30 μΜ and 3.91 μΜ, respectively, indicating that nilotinib is about 4-fold more active than imatinib (Table 1). Among the imatinib synthetic analogues, compounds I and II exhibited an approximately 3- and 2-fold higher inhibitory activity, respectively, whereas compounds III and IV were significantly less active compared with imatinib (Table 1). Among the nilotinib analogues, compound V exhibited a 9-fold higher activity than nilotinib, being the most active among all compounds evaluated, whereas VI exhibited a slightly higher inhibitory activity compared with nilotinib (Table 1). Finally, the imatinib/nilotinib analogue VII was a very weak inhibitor, whereas VIII exhibited an inhibitory activity similar to that of nilotinib (Table 1). The inhibitory activity of imatinib, nilotinib and their analogues was significantly reduced when these compounds were incubated for 1 min with PRP instead of 5 min, before the initiation of the aggregation with AA, the compounds I to IV, VI and VII being inactive (Table 1). This suggests that the inhibitory effect of all compounds is time dependent. All compounds tested towards ADP- and TRAP-6 were much less efficient in inhibiting platelet aggregation compared with their effect towards AA (Figure 2A). Representative aggregation curves illustrating the dose-dependent inhibitory effect of the nilotinib analogue V on AA-, ADP- and TRAP-6-induced platelet aggregation are presented in Figure 2B. Finally, no inhibitory effect of all compounds tested was observed towards platelet aggregation induced by ADP or TRAP-6 when they were incubated for 1 min with PRP instead of 5 min prior to platelet activation (data not shown).
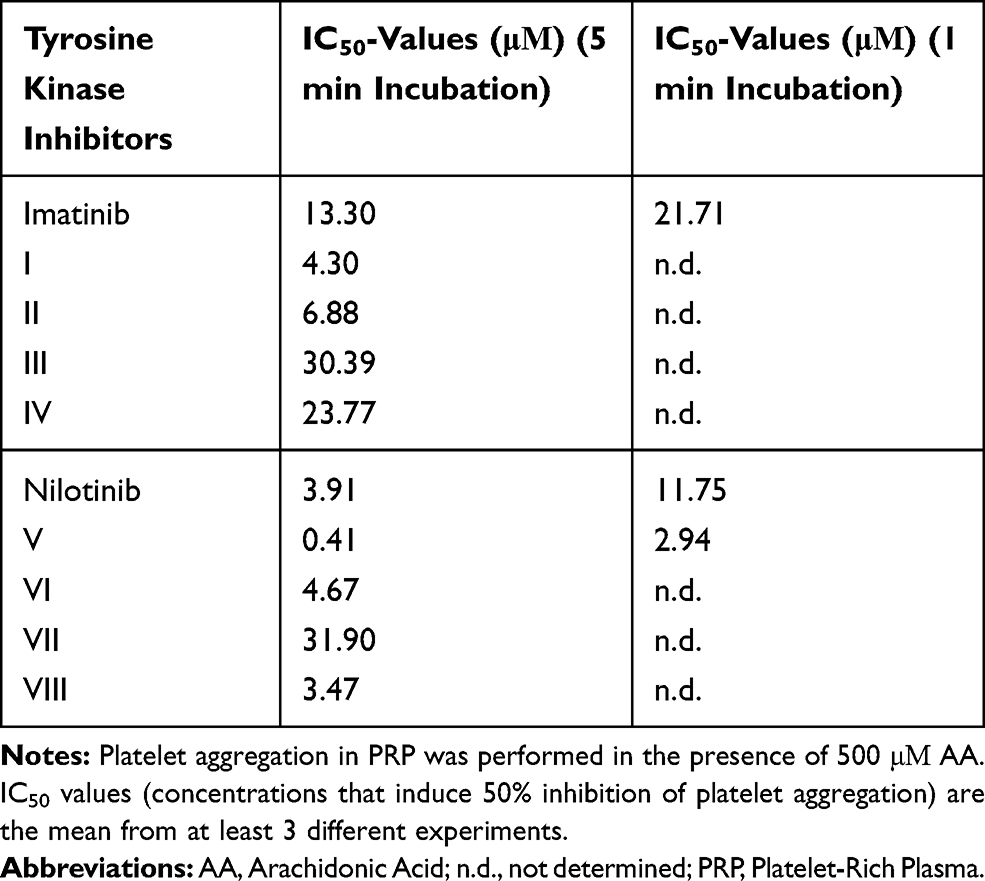 |
Table 1 The Inhibitory Effect of Imatinib, Nilotinib and Their Synthetic Analogues on AA-Induced Platelet Aggregation in PRP |
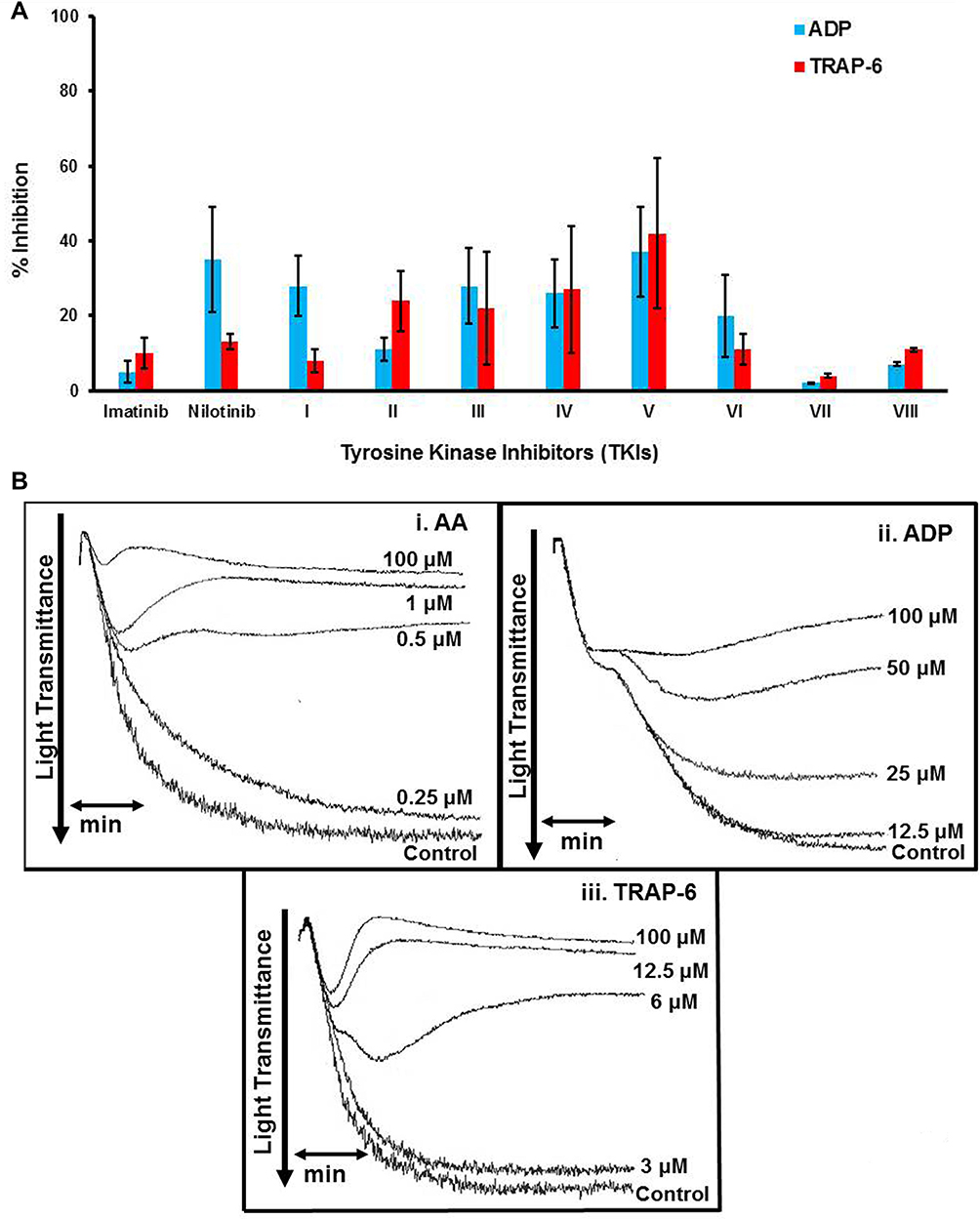 |
Figure 2 (A). Bar graph illustrating the % inhibition induced by imatinib (Im), nilotinib (N) and related synthetic analogues (at a concentration of 100 μΜ) on platelet aggregation induced by ADP or TRAP-6. Values represent the Mean±SD from three different platelet preparations. (B). Representative aggregation curves illustrating the inhibitory effect of compound V, at different concentrations, on platelet aggregation induced by i. AA ii. ADP, iii. TRAP-6. Abbreviations: ADP, Adenosine Diphosphate; SD, Standard Deviation; TRAP-6, Thrombin Receptor Activating Peptide-6. |
Ιnhibition of P-Selectin Exposure
The effect of imatinib, nilotinib and their synthetic analogues on platelet secretion, were also studied by determining the membrane expression of P-selectin on platelets activated by AA, ADP and TRAP-6. The results obtained were similar to those of the aggregation experiments, indicating that among the 3 agonists evaluated, the strongest inhibitory effect of imatinib, nilotinib and analogues, was observed towards AA-induced platelet aggregation. Furthermore, nilotinib was more active than imatinib, whereas among the synthetic compounds tested the most active was the nilotinib analogue V. Figure 3A illustrates the inhibitory effect of imatinib, nilotinib and the most active analogue V. Compounds tested towards ADP and TRAP-6 were much less efficient in inhibiting P-selectin membrane expression compared with their effect towards AA, with some compounds being inactive. Figure 3B and C illustrate the effects of imatinib, nilotinib and compound V.
 |
Figure 3 Bar graphs illustrating the effect of imatinib, nilotinib and synthetic compound V on P-selectin membrane expression induced by the platelet agonists (A) AA, (B) ADP and (C) TRAP-6. Platelets in PRP were labeled with anti-CD62P-PE monoclonal antibody and analyzed by flow cytometry to determine the membrane expression of P-selectin on activated platelets. The compounds imatinib, nilotinib and V were used at the concentration of 100 μΜ. Notes: Results are the mean±SD from at least three independent experiments. *p<0.01 compared with activated platelets, $p<0.05 compared with imatinib, #p<0.01 compared with nilotinib. **p<0.05 compared with respective activated platelets, &p<0.05 compared with nilotinib. Abbreviations: AA, Arachidonic Acid; ADP, Adenosine Diphosphate; MFI, Mean Fluorescence Intensity; PRP, Platelet-Rich Plasma; SD, Standard Deviation; TRAP-6, Thrombin Receptor Activating Peptide-6. |
Docking Calculations
The superposition of the model of the V-c-Src (in green) and the structures of nilotinib (in brown), imatinib (in pink) at the active site of the kinase c-Src are illustrated in Figure 4A. The differences are focused on the final phenyl moiety. Imatinib docked at the active site of the kinase c-Src (Figure 4B) develops hydrogen-bonding interactions with the residues Met341, via a pair of hydrogen bonds, as well as van de Waals, carbon-hydrogen bonds (C-H bonds), pi-alkyl and alkyl with Asp386, Asp404, Gly276, Gly274, Leu273, Val281, Lys295, Leu393 and Glu339. The docking score (maestro glide) is – 11.22 kcal/mol. As shown in Figure 4C, nilotinib docked at the active site of the c-Src, developing hydrogen-bonding interactions with the residues Met341, via a pair of hydrogen bonds, and with the residue Leu273 (one hydrogen bond), as well as van de Waals, carbon-hydrogen bonds (C-H bonds), pi-alkyl, pi-sigma and alkyl with Gly276, Gly274, Val281, Lys295, Tyr340, Leu293, Ala293 and Glu339 with a docking score of – 12.80 kcal/mol.
 |
Figure 4 Models of imatinib, nilotinib and of the most active analogue V bound to c-Src enzyme based on PDB (ID: 1Υ57): (A) Superposition of the model of the V-c-Src (in green) and the structures of nilotinib (in brown), imatinib (in pink) at the active site of the kinase c-Src. The differentiations are focused mainly on the final phenyl moiety. (B, C, D) Modeled binding modes presented as 2D ligand-interaction ligand (LID) of imatinib, nilotinib and compound V bound, respectively, positioned at the active site of the kinase c-Src, as derived from flexible docking calculations. Note: Protein residues forming hydrogen bonds (dotted green lines) are labelled. |
The docking pose of the more active nilotinib analogue V is shown in Figure 4D. Important hydrogen-bonding and hydrophobic interactions with the c-Src have been identified for the analogue V. The presence of the NO2 group at the final phenyl ring at the 3-position was a decisive part of the pharmacophore due to formation of strong interactions. On the contrary, it develops two pairs of hydrogen bonds with the residues Asp404 and Met341, two and one more hydrogen bonds than those of imatinib and nilotinib, respectively. Furthermore, compound V develops van de Waals, carbon-hydrogen bonds (C-H bonds), pi-alkyl and alkyl bonding interactions with Leu273, Tyr340, Leu293, Ala293, Glu339, Lys295 and Val281 with a docking score of – 14.30 kcal/mol, indicating high binding affinity (Figure 4D).
According to our results, the parent compounds imatinib and nilotinib and all their synthetic analogues inhibited platelet aggregation and secretion induced by AA, exhibiting a wide variability in their potency, the most active compound being V. Although clear structure relationships cannot conclusively be delineated, the improvements of 3.1 kcal/mol and 1.5 kcal/mol compared to imatinib and nilotinib, respectively, based on the docking score, suggest that the above variability could be at least partially attributed to the number and the strength of hydrogen bonds. The nitro oxygens of the nitro moiety of V, as strong proton acceptors, are involved in two additional hydrogen bonds with the Asp404 side chain residue of c-Src (Figure 4D), explaining probably its higher inhibitory activity.
In addition, we tested how the replacement of the amide bond with the urea bond (analogues VII and VIII) and the substituents at the final benzene ring affects platelet activation. Absence of the substituents at the final benzene ring (3-NO2, 4-Cl) resulted in a 9-fold weaker antiplatelet activity of VII compared with analogue VIII, making the impact of these substituents on antiplatelet potency apparent.
However, we should highlight that the nilotinib analogue V has the same molecular scaffold and the same substituents at the same positions at the final benzene ring, namely 3-NO2 and 4-Cl, with the much less (approximately 70-fold) active analogue III, the only difference between the 2 molecules being in the amide bond position (retro-amides) between the two phenyl rings C and D (Figure 1). Thus, it is confirmed that even insubstantial structural modifications, as hypothesized, on imatinib and nilotinib (i.e. amide bond like nilotinib in the analogue V) can have a strong affinity impact to the ATP-binding pocket and therefore a strong impact on their antiplatelet potency. Except the above, compound V fulfil Lipinski’s rule of five: molecular weight 460.87 (≤ 500Da), logPo/w (iLOGP) 2.77 (≤ 5), hydrogen bond donors 2 (≤ 5), hydrogen bond acceptors 6 (≤ 10), rotatable bonds 7 (≤ 10) and polar surface area (TPSA) 125.62 Å2 (≤ 140 Å2).
The results of the present study show that there is significant potential to develop tyrosine kinase inhibitors (synthetic imatinib and nilotinib analogues), which would express antiplatelet properties. Lack of the 4-methylimidazolyl group of nilotinib, with the amide bond between the two final phenyl rings, C and D, such as in nilotinib, as well as the incorporation of a strong electron-withdrawing substituent, specially NO2 group and of a halogen atom (F, Cl, Br) or CF3 substituent at the 2-, 3-, or 4-positions at the terminal phenyl ring, could be a promising method for the discovery of novel molecules with increased antiplatelet activity.
Our data further demonstrated that imatinib, nilotinib and their synthetic structural analogues were weak inhibitors of platelet activation induced by ADP and TRAP-6 compared with AA. It has been established that SFKs associate with G-protein-coupled receptors (GPCRs) and contribute to downstream signaling in platelets induced by the soluble ligands of these receptors.38 Consequently, SFKs are involved in the intracellular signalling resulting in platelet activation induced not only by Thromboxane A2 (TxA2), the metabolite of AA that mediates the AA-induced platelet activation, but also by ADP and TRAP-6 via their GPCRs, P2Y12 and PAR-1, respectively.38 Therefore, the higher inhibitory activity of our compounds towards AA-induced platelet activation compared with ADP and TRAP-6 may not be only due to the c-Src inhibition. Hence, the mechanisms underlying the antiplatelet effects of the compounds tested in the present study beyond the c-Src inhibition should be further investigated.
Conclusion
Platelets play important roles in cancer progression and metastasis, as well as in CAT, therefore inhibiting platelet activation would be a promising strategy to reduce both cancer progression and CAT. The present study indicates that based on the structure of the TKIs imatinib and nilotinib there is a significant potential to develop synthetic analogues, expressing improved antiplatelet effects, therefore being suitable to target cancer progression and metastasis, as well as CAT. Among all compounds tested, the synthetic analog of nilotinib V demonstrated the strongest antiplatelet action. Consequently, this analogue could be considered as a new lead compound for the development of inhibitors with improved antiplatelet properties and studies are underway to further optimize its activity.
Supporting Materials
1H NMR, 13C NMR and high-resolution ESI-MS spectra (Figures S1–12) of the nilotinib (V, VI) and imatinib/nilotinib (VII, VIII) analogues are provided as Supplementary materials.
Acknowledgments
The authors thank the Atherothrombosis Research Center of the University of Ioannina for providing access to the laboratory equipment and facilities. We appreciate the use of NMR Unit and the Unit of Environmental, Organic and Biochemical high-resolution analysis – ORBITRAP-LC-MS of the University of Ioannina for providing access to the facilities funded by the Network of Research Supporting Laboratories of the University of Ioannina. This research has been financially supported by General Secretariat for Research and Technology (GSRT) and the Hellenic Foundation for Research and Innovation (HFRI) (Scholarship Codes: 2000 and 82195).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93:327–358. doi:10.1152/physrev.00016.2011
2. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi:10.1056/NEJMra071014
3. Abdol Razak NB, Jones G, Bhandari M, et al. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers. 2018;10:1–21. doi:10.3390/cancers10100380
4. Connolly GC, Francis CW. Cancer-associated thrombosis. Hematology Am Soc Hematol Educ Program. 2013;2013:684–691. doi:10.1182/asheducation-2013.1.684
5. Ay C, Pabinger I, Cohen AT. Cancer-associated venous thromboembolism: burden, mechanisms, and management. Thromb Haemost. 2017;117:219–230. doi:10.1160/TH16-08-0615
6. Falanga A, Marchetti M, Russo L. The mechanisms of cancer-associated thrombosis. Thromb Res. 2015;135:S8–S11. doi:10.1016/S0049-3848(15)50432-5
7. Falanga A, Russo L, Milesi V. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol. 2017;118:79–83. doi:10.1016/j.critrevonc.2017.08.003
8. Connolly GC, Phipps RP, Francis CW. Platelets and cancer-associated thrombosis. Semin Oncol. 2014;41:302–310. doi:10.1053/j.seminoncol.2014.04.009
9. Falanga A, Marchetti M. Hemostatic biomarkers in cancer progression. Thromb Res. 2018;164:S54–S61. doi:10.1016/j.thromres.2018.01.017
10. Olsson AK, Cedervall J. The pro-inflammatory role of platelets in cancer. Platelets. 2018;29(6):569–573. doi:10.1080/09537104.2018.1453059
11. Schlesinger M. Role of platelets and platelet receptors in cancer metastasis. J Hematol Oncol. 2018;11:125. doi:10.1186/s13045-018-0669-2
12. Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017;36:249–262. doi:10.1007/s10555-017-9673-1
13. Gay LJ, Felding-Habermann B. Contribution of platelets to tumor metastasis. Nat Rev Cancer. 2011;11(2):123–134. doi:10.1038/nrc3004
14. Palumbo JS, Talmage KE, Massari JV, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–185. doi:10.1182/blood-2004-06-2272
15. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates nkg2d thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009;69:7775–7783. doi:10.1158/0008-5472.CAN-09-2123
16. Felding-Habermann B, Habermann R, Saldivar E, Ruggeri ZM. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J Biol Chem. 1996;271:5892–5900. doi:10.1074/jbc.271.10.5892
17. Egan K, Cooke N, Kenny D. Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis. 2014;31:697–704. doi:10.1007/s10585-014-9660-7
18. Gong L, Cai Y, Zhou XD, Yang HP. Activated platelets interact with lung cancer cells through p-selectin glycoprotein ligand-1. Pathol Oncol Res. 2012;18:989–996. doi:10.1007/s12253-012-9531-y
19. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. doi:10.1016/j.ccr.2011.09.009
20. Elaskalani O, Berndt MC, Falasca M, Metharom P. Targeting Platelets for the Treatment of Cancer. Cancers. 2017;9:94. doi:10.3390/cancers9070094
21. Gresele P, Momi S, Malvestiti M, Sebastiano M. Platelet-targeted pharmacologic treatments as anticancer therapy. Cancer Metastasis Rev. 2017;36:331–355. doi:10.1007/s10555-017-9679-8
22. Rothwell PM, Wilson M, Price JF, et al. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi:10.1016/S0140-6736(12)60209-8
23. Cao Y, Nishihara R, Wu K, et al. Population-wide impact of long-term use of aspirin and the risk for cancer. JAMA Oncol. 2016;2:762–769. doi:10.1001/jamaoncol.2015.6396
24. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi:10.1016/j.tips.2015.04.005
25. Tullemans BME, Heemskerk JWM, Kuijpers MJE. Acquired platelet antagonism: off-target antiplatelet effects of malignancy treatment with tyrosine kinase inhibitors. J Thromb Haemost. 2018;16:1686–1699. doi:10.1111/jth.2018.16.issue-9
26. Estevez B, Du X. New Concepts and mechanisms of platelet activation signaling. Physiology (Bethesda). 2017;32:162–177. doi:10.1152/physiol.00020.2016
27. Kanikarla-Marie P, Lam M, Sorokin AV, et al. Platelet metabolism and other targeted drugs; potential impact on immunotherapy. Front Oncol. 2018;8:107. doi:10.3389/fonc.2018.00107
28. Oda A, Ikeda Y, Ochs HD, et al. Rapid tyrosine phosphorylation and activation of Bruton’s tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross-linking. Blood. 2000;95:1663–1670.
29. Phillips DR, Nannizzi-Alaimo L, Prasad KS. Beta3 tyrosine phosphorylation in alphaIIbbeta3 (platelet membrane GP IIb-IIIa) outside-in integrin signaling. Thromb Haemost. 2001;86:246–258. doi:10.1055/s-0037-1616222
30. Sater HA, Gandhi AS, Dainer P, et al. Receptor tyrosine kinases in human platelets: a review of expression, function and inhibition in relation to the risk of bleeding or thrombocytopenia from phase I through phase III trials. J Cancer Prev Curr Res. 2017;8:1–13.
31. Wang WY, Hsieh PW, Wu YC, et al. Synthesis and pharmacological evaluation of novel beta-nitrostyrene derivatives as tyrosine kinase inhibitors with potent antiplatelet activity. Biochem Pharmacol. 2007;74:601–611. doi:10.1016/j.bcp.2007.06.001
32. Golden A, Brugge JS. Thrombin treatment induces rapid changes in tyrosine phosphorylation in platelets. Proc Natl Acad Sci U S A. 1989;86:901–905. doi:10.1073/pnas.86.3.901
33. Maguire PB, Wynne KJ, Harney DF, et al. Identification of the phosphotyrosine proteome from thrombin activated platelets. Proteomics. 2002;2:642–648. doi:10.1002/1615-9861(200206)2:6<642:AID-PROT642>3.0.CO;2-l
34. Seeliger MA, Nagar B, Frank F, et al. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure (London, England:1993). 2007;15:299–311. doi:10.1016/j.str.2007.01.015
35. Jackson SP, Schoenwaelder SM, Yuan Y, et al. Non-receptor protein tyrosine kinases and phosphatases in human platelets. Thromb Haemost. 1996;76:640–650. doi:10.1055/s-0038-1650637
36. Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi:10.1038/nrd839
37. Skobridis K, Kinigopoulou M, Theodorou V, et al. Novel imatinib derivatives with altered specificity between Bcr-Abl and FMS, KIT, and PDGF receptors. Chem Med Chem. 2010;5:130–139. doi:10.1002/cmdc.200900394
38. Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124:2013–2024. doi:10.1182/blood-2014-01-453134
39. Kalantzi KI, Tsoumani ME, Goudevenos IA, et al. Pharmacodynamic properties of antiplatelet agents: current knowledge and future perspectives. Expert Rev Clin Pharmacol. 2012;5:319–336. doi:10.1586/ecp.12.19
40. Tsoumani ME, Tselepis AD. Antiplatelet agents and anticoagulants: from pharmacology to clinical practice. Curr Pharm Des. 2017;23:1279–1293. doi:10.2174/1381612823666170124141806
41. Moschonas IC, Goudevenos JA, Tselepis AD. Protease-activated receptor-1 antagonists in long-term antiplatelet therapy. Current state of evidence and future perspectives. Int J Cardiol. 2015;185:9–18. doi:10.1016/j.ijcard.2015.03.049
42. Kinigopoulou M, Filippidou M, Gogou M, et al. An optimized approach in the synthesis of imatinib intermediates and analogues. RSC Adv. 2016;6:61458–61467. doi:10.1039/C6RA09812F
43. Mitsios JV, Tambaki AP, Abatzis M, et al. Effect of synthetic peptides corresponding to residues 313-332 of the alphaIIb subunit on platelet activation and fibrinogen binding to alphaIIbbeta3. Eur J Biochem. 2004;271:855–862. doi:10.1111/j.1432-1033.2004.03990.x
44. Yi X, Liu M, Luo Q, et al. Toxic effects of dimethyl sulfoxide on red blood cells, platelets, and vascular endothelial cells in vitro. FEBS Open Bio. 2017;7:485–494. doi:10.1002/2211-5463.12193
45. Kouki A, Mitsios JV, Sakarellos-Daitsiotis M, et al. Highly constrained cyclic (S,S) -CXaaC- peptides as inhibitors of fibrinogen binding to platelets. J Thromb Haemost. 2005;3:2324–2330. doi:10.1111/j.1538-7836.2005.01487.x
46. Dimitriou AA, Stathopoulos P, Mitsios JV, et al. Inhibition of platelet activation by peptide analogues of the beta(3)-intracellular domain of platelet integrin alpha(IIb)beta(3) conjugated to the cell-penetrating peptide Tat(48-60). Platelets. 2009;20:539–547. doi:10.3109/09537100903324219
47. Theodorou V, Skobridis K, Tzakos AG, et al. A simple method for the alkaline hydrolysis of esters. Tetrahedron Lett. 2007;48:8230–8233. doi:10.1016/j.tetlet.2007.09.074
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.