Back to Journals » Journal of Pain Research » Volume 19
Molecular Mechanisms and Therapeutic Targets for Pain Following Osteoporotic Vertebral Fractures
Received 14 September 2025
Accepted for publication 24 January 2026
Published 2 February 2026 Volume 2026:19 567567
DOI https://doi.org/10.2147/JPR.S567567
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alaa Abd-Elsayed
Huisheng Zhou, Kanglong Wu
Department of Orthopaedics, the First Hospital of Jiaxing, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China
Correspondence: Kanglong Wu, Department of Orthopaedics, the First Hospital of Jiaxing, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China, Email [email protected]
Abstract: Osteoporotic vertebral fractures (OVF) are among the most common fractures in older adults. They are strongly linked to severe pain, disability, and a reduced quality of life. Pain from OVFs often becomes chronic and differs from pain caused by fractures in normal bone. This review systematically summarizes the molecular mechanisms of OVF-related pain. It focuses on how changes in bone metabolism, inflammation, non-coding RNA regulation, and neural pathways affect each other. Major factors that drive the onset and persistence of OVF pain include increased osteoclast activity, abnormal Wnt/β-catenin signaling, inflammatory mediators, and neuropeptides. Recent studies also report new molecular targets that are closely related to OVF pain, such as TRPA1, WWP1, STK11, and specific microRNAs. Targeted treatments may improve pain control and function in patients with OVF. These include anti-neurosensitization drugs, anti-osteoporosis therapies, anti-inflammatory treatments, and neural modulators. More research is still needed to clarify these mechanisms and to develop safer, more effective, and more personalized treatments that improve outcomes and quality of life.
Keywords: osteoporosis, fracture, pain, mechanisms, therapy
Introduction
Osteoporosis (OP) is a prevalent metabolic bone disease characterized by increased bone fragility, reduced bone mass, and an elevated risk of fractures.1 According to data and estimates from the World Osteoporosis Foundation (WOF) for 2024, about one in three women and one in five men older than 50 will have an OP-related fracture during their lifetime.2 Among these, vertebral fractures are the most common type in individuals with OP and are associated with significant morbidity and increased mortality.3 Notably, vertebral fractures often occur independently of falls or major trauma, and due to the frequent absence of acute pain, they are frequently overlooked. This results in an underestimation of their true prevalence in the general population. Nevertheless, osteoporotic vertebral fracture (OVF) can lead to chronic back pain persisting for more than three months, disability, and a higher risk of subsequent fragility fractures at both vertebral and non-vertebral sites.4–6
Moreover, osteoporotic fractures are often more painful than fractures in otherwise healthy bone.7 Evidence from a single-center observational study suggests that, after excluding a history of osteoporotic fracture, OP itself may still be a source of pain.8 Compared with patients with OP but no vertebral fracture, those with vertebral fractures report higher back pain intensity and more frequent and longer pain episodes. The pain is usually located in the lumbar spine and can markedly limit daily activities.9 In addition to pain at the fracture site, patients often report pain in the ribs. Pain in areas such as the hip, groin and chest that is not directly related to the fracture, referred to as “non-midline pain”, is also common.10 A multivariate logistic regression analysis revealed that fractures in the middle thoracic vertebrae (odds ratio (OR) 5.520, P < 0.001), a higher ratio of injured vertebral width (OR 76.138, P = 0.025), and a greater reduction in foraminal area (OR 1.019, P = 0.027) were risk factors.10 These changes may increase instability in the vertebral body, intervertebral disc, and facet joints. This can further irritate the intercostal nerves and cause rib pain. Occult fractures, which can be assessed with computed tomography (CT) or magnetic resonance imaging (MRI), are often missed in routine care.11 They may also contribute to persistent pain in patients with OP.
Current treatment strategies for OVF focus on medications and surgery. Drug therapy includes calcium and vitamin D supplementation, antiresorptive agents, and bone forming agents.12 However, some patients cannot tolerate these drugs or respond poorly.12 Surgical treatment also carries risks related to the procedure itself, and some patients are not fit for surgery. In addition, because OVF pain has distinct features, current approaches often cannot fully control the pain. This highlights the need for better management and more targeted strategies than those used for fractures in general. However, the clinical features and mechanisms of OVF related pain are still not well understood. Based on the causes and biological processes of OVF related pain, we conducted a systematic review. We focused mainly on drug based strategies, as well as new evidence for existing therapies. Our goal was to provide an updated summary of mechanistic advances and clinical knowledge on pain after OVF.
Pain in OVF
The pain associated with OVF differs significantly from that of fractures in healthy bone, both in its origin and clinical presentation. Pain management plays a crucial role in improving patients’ subjective well-being and quality of life, thereby enhancing compliance with OP treatment.
Pain in OVF is multifactorial, commonly categorized into baseline bone pain, acute fracture pain, and chronic post-fracture pain. OP can cause pain even in the absence of fractures, with low back pain being a predominant complaint.8 Such pain often worsens during activities like turning over in bed, sitting up, or walking for prolonged periods, and may be accompanied by muscle spasms and restricted mobility.13 Clinical manifestations can include localized low back pain, radiating pain to the extremities, or low back pain with limb numbness.13 Notably, postmenopausal patients with OP frequently report moderate pain, with an average visual analogue scale (VAS) score of 4.33 prior to spinal compression fractures, often affecting sleep quality.9 Early-stage OP is characterized by active bone turnover, which may present as diffuse bone pain or discomfort. Chronic, poorly localized pain may occur even in the absence of overt fractures, particularly in states of high bone turnover. Mechanisms underlying this pain include bone marrow edema, increased intraosseous pressure from abnormal vasculature or marrow adiposity, microdamage accumulation, and the release of pro-inflammatory cytokines during accelerated bone remodeling.14
OVFs are a common complication in patients with OP, with acute vertebral fractures frequently causing sudden and severe back pain.15,16 Studies have shown that patients with back pain are generally older, have lower bone mineral density (BMD) at the lumbar spine and hip, and a greater number of prevalent vertebral fractures.17 Back pain is significantly associated with prevalent vertebral fracture (OR 4.60, P < 0.001), and OP (OR 2.14, P < 0.001).17 Chronic pain is usually defined as pain that persists for more than three months following OVF.18 Importantly, low back pain after vertebral fracture can persist for 24 weeks or even up to 48 weeks, likely due to nonunion, local spinal deformity, and deterioration of spinal alignment.19,20 Such chronic pain following OVF further impairs quality of life and increases the risk of subsequent fractures.
Molecular Mechanisms Linking Impaired Bone Metabolism to OVF Pain
Osteoclast Overactivation and Disrupted Bone Remodeling as Primary Triggers
Low BMD, the hallmark of OP, is the main risk factor for OVF. In patients with OVF, the fracture microenvironment exhibits significant local bone metabolic dysregulation, characterized by reduced osteoblastic activity and excessive osteoclastic resorption. This imbalance hinders fracture healing and perpetuates a pathological cycle. The resulting structural and neurological changes are key contributors to the development and persistence of pain after fracture.21 Emerging evidence suggests that chronic pain may develop as a result of ongoing changes in bone structure, joint involvement, and muscle strain associated with altered biomechanics and reduced bone density.22 These structural alterations continuously stimulate pain receptors and inflammatory mediators, thereby maintaining chronic pain perception.23
The ovariectomy (OVX) rodent model, which closely mimics key features of postmenopausal OP (PMOP), is characterized by accelerated bone loss and deterioration of bone microarchitecture. This is evidenced by reduced bone volume fraction (BV/TV), trabecular thickness (Tb.Th), and trabecular number (Tb.N), as well as increased trabecular separation (Tb.Sp), all of which are associated with pain-related behaviors.24,25 Estrogen deficiency in this model upregulates RANKL expression and activates the NF-κB/Akt signaling pathway, resulting in increased expression of NFATc1 and c-Fos and subsequent osteoclast hyperactivation.26 When OVX is combined with other pathologies, such as knee osteoarthritis (OA) in rats, there is a marked increase in subchondral bone damage in weight-bearing regions due to enhanced osteoclastic activity.27 Although OVX is known to contribute to baseline “bone ache”, one study paradoxically found that OVX does not always increase the severity of acute post-fracture pain compared to controls.7 This observation suggests that complex interactions, potentially involving compensatory mechanisms or differential regulation of acute and chronic pain pathways, may influence pain outcomes and merit further investigation.
Emerging evidence indicates that increased osteoclast activity plays a crucial role not only in bone matrix degradation but also in the direct sensitization of nociceptors at fracture sites.28,29 Recent studies show that heightened osteoclast infiltration promotes the abnormal sprouting of calcitonin gene-related peptide (CGRP)-positive sensory nerve fibers within fracture regions, which is associated with a marked decrease in mechanical pain thresholds.28 Elevated 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), which is an AMPK activator secreted by pre-osteoclasts, activates the AMPK/mTORC1/S6K signaling pathway, resulting in the transcriptional repression of CGRP. This process inhibits sensory nerve fiber outgrowth and reduces pain perception. Supporting this, studies using strontium hydrogen phosphate (SrHPO4)-coated magnesium alloy implants in OP rats demonstrated that, suppression of osteoclastogenesis led to reduced CGRP-positive nerve fiber density and alleviated post-fracture pain.28 It is important to note that current evidence is largely limited to cell and animal studies. Future clinical trials are needed to confirm the biocompatibility and efficacy of these materials. However, some animal studies show that systemic or local administration of receptor activator of nuclear factor kappa B ligand (RANKL) increases osteoclast formation but does not induce pain like behaviors in mice.30,31 This finding suggests that while osteoclast hyperactivity is necessary for the development of pain, it is not sufficient by itself. Additional factors, including disruption of bone matrix integrity or the presence of inflammation, are likely required to facilitate full nociceptor sensitization. For example, inflammatory mediators released in cancer related or inflammatory bone microenvironments, such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), can directly promote osteoclastogenesis. They can also increase osteoclast formation indirectly by upregulating RANKL.32
In addition, enhanced osteoclastic bone resorption leads to acidification of the extracellular microenvironment.31 This localized acidosis activates Acid-Sensing Ion Channel 3 (ASIC3), which is expressed on peripheral nociceptors and is most sensitive at approximately pH 7.2. Activation of ASIC3 significantly contributes to persistent musculoskeletal pain under acidic conditions by sensitizing nociceptors to additional stimuli (Figure 1). Preclinical studies underscore the critical role of ASIC3 in mediating pain associated with both inflammatory and non-inflammatory bone pathologies, including osteoporosis and osteoarthritis.31
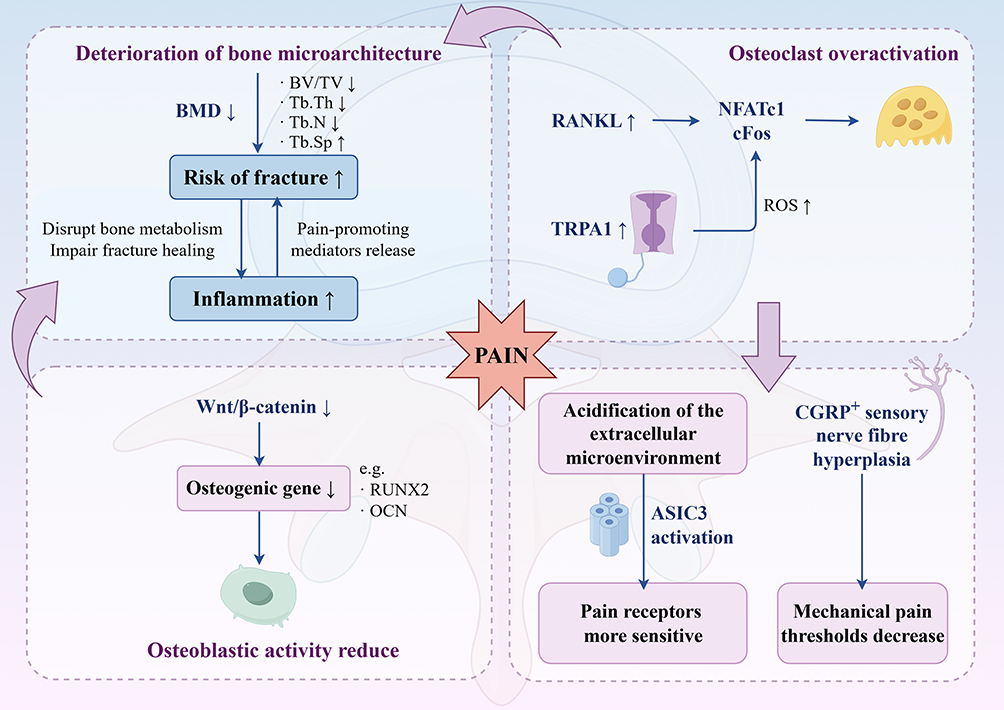 |
Figure 1 Partial mechanisms of pain in OVF. In OP, local Wnt signaling is reduced, while RANKL signaling and TRPA1 expression are increased. These changes activate downstream pathways and lead to excessive osteoclast activity and reduced osteoblast function. As a result, bone microarchitecture deteriorates and fracture risk increases, which can trigger acute fracture pain. Inflammation further accelerates these processes. Elevated osteoclast activity also lowers the mechanical pain threshold and increases nociceptor sensitivity. This figure was created using Home for Researchers. |
Intrinsic Defects in Bone Formation: Wnt Signaling and Beyond
Clinical studies confirm that people with OP often have an unfavourable bone metabolic profile. This is characterised by elevated bone resorption markers, such as CTX-1, and reduced bone formation markers, such as OCN.33,34 Impaired osteoblast function is a critical contributor to osteoporotic bone fragility and associated pain. Ionizing radiation (IR) shifts bone marrow stem cell (BMSC) differentiation toward adipogenesis at the expense of osteoblastogenesis.35 This alteration weaken bone strength and increase susceptibility to pathological fractures. Such fractures, as well as complications arising from poor blood supply to fragile bones, such as osteonecrosis and non-union, are a major cause of persistent chronic pain.
Clinical reports in children with autosomal dominant early-onset OP due to heterozygous WNT1 mutations provide clear evidence of these mechanisms.36 These patients exhibit severely reduced bone formation and extremely low BMD. They experience recurrent fragility fractures, including OVFs, as early as two to 12 years of age. They also suffer from significant back pain and reduced physical activity. Following fractures, they experience chronic mechanical pain. Treatment with zoledronate, which increases BMD, reduces fracture incidence, and alleviates pain, highlights the central role of dysregulated WNT/β-catenin signaling in the pathology. The Wnt/β-catenin pathway is a master regulator of BMSC commitment to osteogenesis. Canonical Wnt ligands bind to FZD/LRP5/6 receptors, leading to β-catenin stabilization, nuclear translocation, and activation of key osteogenic genes such as RUNX2, OSX, and OCN.37 Loss-of-function mutations in LRP5, as observed in OP-pseudoglioma syndrome (OPPG), disrupt the Wnt signalling pathway and reduce bone formation. By contrast, gain-of-function mutations in LRP5 (LRP5-High Bone Mass, LRP5-HBM) are linked to increased bone density.38 Restoring key osteogenic signalling pathways can increase the levels of OCN and RUNX2, thereby promoting bone healing and potentially alleviating structural pain.33
Additionally, dysregulation of the Hippo-YAP pathway component VGLL4, which normally facilitates osteogenesis by alleviating TEAD4-mediated repression of RUNX2, has been implicated in the development of PMOP bone pain.25 However, the precise mechanistic link between VGLL4 and osteoporotic bone pain remains to be fully elucidated.
Target Molecule in OVF and Related Pain
Recent findings have identified the transient receptor potential znkyrin 1 (TRPA1) cation channel as a key contributor to both OP pathophysiology and related pain.24 TRPA1 expression is significantly upregulated in osteoporotic bone and colocalizes with NFATc1, a master transcription factor involved in osteoclastogenesis. Mechanistically, TRPA1 activation during osteoclastogenesis increases intracellular Ca2⁺ levels and promotes the accumulation of reactive oxygen species (ROS). This rise in ROS induces endoplasmic reticulum (ER) stress, activating the PERK/eIF2α/ATF4/CHOP signaling pathway, which stabilizes and activates NFATc1, thereby facilitating osteoclast formation and bone loss.34 Notably, selective knockdown of TRPA1 in OP mice reduces pain-related behaviors, establishing a direct link between TRPA1 activity in bone and pain perception. Further investigation revealed a novel TRPA1-Sulfiredoxin-1 (SRXN1) regulatory axis.24 Inhibition of TRPA1 leads to upregulation of SRXN1, which reduces ROS and ER stress and subsequently suppresses NFATc1-driven osteoclastogenesis. In summary, TRPA1 functions as a central integrator of metabolic stress, osteoclast activation, and nociception within the osteoporotic bone microenvironment.
Studies have demonstrated that WWP1 expression is elevated in both TNF transgenic mice and aged mice, which is associated with impaired osteogenesis and exacerbated OP. Mechanistic investigations revealed that increased WWP1 promotes the degradation of key osteogenic genes, including RUNX2 and CXCR4. In contrast, upregulation of WWP2 enhances RUNX2 activity and subsequently promotes osteogenesis.39 WWP1 has been implicated in the pathogenesis of osteoporosis, delayed fracture healing, and tumor-induced bone metastasis. In comparison, WWP2 primarily supports cartilage and bone development, with its deficiency resulting in chondrodysplasia. Notably, inhibition of WWP1 using miR-142-5p, miR-19b, C3A, or WWP1-specific siRNA restores RUNX2 function, increases bone density, accelerates fracture healing, and shows potential for alleviating bone pain.39
In addition, clinical analyses have demonstrated significantly reduced STK11 expression in bone tissue from patients with OP.34 In contrast, overexpression of STK11 activates the AMPK/SIRT1/PGC1α signaling pathway, which enhances the activity of human mesenchymal stem cells (hMSCs) and reduces apoptosis by upregulating Bcl-2 and downregulating Bax. Additionally, overexpression of STK11 leads to a marked decrease in oxidative stress markers, including reactive oxygen species (ROS), malondialdehyde (MDA), inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX2). At the same time, activation of the Nrf2/HO-1 antioxidant pathway is observed. In experimental mouse models, STK11 overexpression restores mineralized nodule formation and reduces fat droplet accumulation in bone tissue.34 These findings suggest that STK11 overexpression can restore the osteogenic potential of hMSCs by activating the AMPK/SIRT1/PGC1α signaling axis, while simultaneously reducing apoptosis and oxidative stress. This mechanism may help prevent or alleviate glucocorticoid-induced osteoporosis and the associated localized pain.
Non-Coding RNA Networks Influencing Osteogenesis and Pain Risk
Similarly, dysregulated non-coding RNAs, particularly microRNAs (miRNAs), play influential roles. For instance, miR-18a-3p is significantly upregulated in the bone tissue of OP patients, particularly those with spinal fractures.40 Mechanistically, it suppresses human BMSC osteogenic differentiation by directly targeting and downregulating key factors glutamate AMPA receptor subunit 1 (GRIA1) and adenylate cyclase 5 (ADCY5).40,41 This repression of osteoblastogenesis exacerbates osteoporosis and micro-fracture propensity, indirectly amplifying the risk of structural pain. Conversely, miR-140-5p has been shown to inhibit osteogenesis and impair fracture healing in aged mice, potentially via targeting CXCR4, and its expression can be modulated by cyclic mechanical stretch via the lncRNA MEG3.42,43 Weighted gene co-expression network analysis (WGCNA) demonstrates significant enrichment of differentially expressed genes related to PMOP in “cytokine-cytokine receptor interaction” pathways, implicating miRNAs as a core pathological driver.44 The relevant miRNAs, such as miR-21 and miR-139-5p, and their associated targets, EIF5 and CCT5, could be used to develop new non-hormonal anti-OP and anti-analgesic therapeutics.
At present, miRNAs show stronger potential as clinical predictors.45,46 Serum miR-148a-3p levels in OVF patients were significantly higher than in healthy controls, and they gradually decreased over time after surgery. miR-148a-3p expression was also significantly higher in the delayed healing group than in the healing group. It showed potential diagnostic value for delayed healing (AUC = 0.859).45 Key challenges remain, including how to deliver miRNAs effectively and how to maintain their stability during delivery. These issues need to be addressed before miRNA based therapies can be widely and successfully applied in the future.
The Role of Inflammation in OVF Pain
Inflammation as a Critical Amplifier of Bone Fragility and Pain Hypersensitivity in OP
Chronic inflammation represents a pivotal pathologic driver in OP, synergistically exacerbating bone metabolic imbalance and nociceptive signaling to culminate in heightened fracture risk and debilitating pain. Estrogen deficiency, the hallmark trigger of postmenopausal osteoporosis, not only potentiates osteoclast activity but also induces a profound pro-inflammatory shift within the bone microenvironment. In murine postmenopausal osteoporosis models, localized elevations in pro-inflammatory cytokines, including TNF-α, IL-1β, IL-18, and IL-17, are consistently observed.8,33,47 Critically, these molecules exert dual detrimental effects: they potently activate osteoclasts to accelerate bone resorption while concurrently sensitizing sensory nerve terminals, thereby precipitating mechanical allodynia and spontaneous pain.33,47 This cytokine surge further reinforces osteolytic processes, as demonstrated by TNF-α and IL-6 acting to upregulate RANKL expression, thereby fostering a feed-forward loop that amplifies bone destruction and associated pain.33 Notably, analogous pathways are implicated in joint disease. IL-18 and TNF-α elevations similarly promote pain and cartilage breakdown in osteoarthritis.47
The nexus between systemic inflammation and osteoporosis extends beyond estrogen loss. Chronic inflammatory conditions—such as rheumatoid arthritis (RA), inflammatory bowel disease (IBD), periodontitis, and glucocorticoid-induced OP (GIOP)—promote inflammatory osteolysis through RANKL-dependent and independent pathways, leading to generalized bone loss and heightened fracture risk.32 Furthermore, aging significantly contributes to this inflammatory milieu through the accumulation of senescent bone cells, which secrete the senescence-associated secretory phenotype (SASP), encompassing factors like IL-6, TNF-α, and MMPs.32 This sustained local inflammation drives bone pain, a relationship underscored by findings that targeted clearance of senescent cells mitigates bone loss, reduces inflammation, and alleviates pain behaviors in aged models. Clinically, the link between inflammation and pain is substantiated by the positive correlation between serum IL-6 levels and pain intensity in osteoporotic patients, and the observation that IL-6 blockade via the IL-6 receptor inhibitor tocilizumab provides symptomatic relief, albeit with increased susceptibility to infections.47
Inflammation Causes Delayed Fracture Healing, Leading to Chronic Pain
OVF frequently underdiagnosed in the elderly (>65 years), instigate a potent acute inflammatory response critical to both immediate pain generation and the development of chronic pain states.48 Fracture-induced hematoma formation initiates the recruitment of neutrophils and macrophages, which release key pro-nociceptive mediators including IL-1β, TNF-α, IL-6, and prostaglandin E2 (PGE2).5,24 These mediators can directly facilitate peripheral sensitization, activating the pain-sensing cation channels TRPV1 and TRPA1, dramatically lowering activation thresholds and inducing hypersensitivity to mechanical and thermal stimuli.32 This acute phase is intrinsically linked to healing outcomes. Sustained inflammation, characteristic of aged or osteoporotic bone due to prolonged inflammatory cytokine release, disrupts bone metabolism and inhibits bone formation, resulting in delayed fracture consolidation. Consequently, non-union, instability, and deformity persist as chronic sources of nociceptor activation and mechanical stress, significantly extending the duration of pain.48
Psychological comorbidities further complicate this scenario, as depression and anxiety drive sympathetic activation and neutrophil-derived catecholamine release.49 This catecholaminergic environment inhibits osteogenesis through β-adrenergic receptor signaling on osteoblasts, thereby perpetuating poor healing, sustained nociception, and chronic pain vulnerability.
The Link Between Autophagy and Inflammation
Recent investigations highlight dysregulated autophagy as a crucial mechanism linking inflammation, impaired bone metabolism, and pain. Osteoporotic models exhibit diminished autophagic flux, reflected in reduced expression of autophagy markers Beclin-1 and LC3B, and accumulation of the substrate p62, alongside increased caspase-3/8 activity.47,50 This impairment in cellular clearance exacerbates inflammatory stress and bone cell dysfunction.
The AMPK/mTOR signaling axis serves as a central regulator of autophagy activation.51 Downregulation of AMPK coupled with upregulation of mTOR suppresses autophagosome initiation and lysosomal degradation (reducing Beclin-1, LC3II, and ULK1), intensifying NF-κB-mediated transcriptional upregulation of inflammatory cytokines (eg, IL-6, TNF-α, COX-2).50 Pharmacological AMPK activation reverses this deficit, restoring autophagic flux, dampening inflammatory cascades, reducing osteoclast hyperactivity, promoting osteoblast survival, and effectively alleviating bone loss and pain in postmenopausal osteoporosis models.50 Complementary research reveals that pulsed electromagnetic fields (PEMF) also offer benefit by activating the peroxisome proliferator-activated receptors (PPARγ) signaling pathway. PPARγ stimulation restores autophagic flux and mitigates chondrocyte apoptosis and inflammatory responses, thereby decelerating cartilage degeneration and bone loss in osteoporosis-related osteoarthritis.47,52
NF-κB emerges as the master transcriptional hub integrating inflammatory bone destruction and pain sensitization pathways. Its activation by cytokines (TNF-α, IL-1), DAMPs, or RANKL triggers downstream transcription of pro-inflammatory mediators, pro-resorptive factors (including NFATc1 and TRACP, critical for osteoclastogenesis), and pro-algesic molecules such as nerve growth factor (NGF).21,26,53 This profound regulatory role positions NF-κB inhibition as a promising therapeutic strategy for simultaneously targeting inflammation, bone loss, and pain.
Neural Regulation of Bone-Pain Pathways in Osteoporotic Fractures
Peripheral Sensory Neuropeptides
Chronic pain after OVF extends far beyond mere structural damage, involving active neuro-immune crosstalk, neural plasticity, and complex regulatory circuits at both the peripheral and central levels.54 The peripheral nervous system (PNS), particularly sensory and sympathetic fibers, plays a pivotal role in fracture healing and post-fracture pain modulation through the release of neuropeptides such as Substance P (SP) and Calcitonin Gene-Related Peptide (CGRP), as well as neurotrophic factors like nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF).54 CGRP, abundantly present in both the central and peripheral nervous systems, is not only a potent facilitator of pain transmission and neuronal sensitization, but also modulates macrophage polarization towards an osteogenic M2 phenotype.55 This macrophage reprogramming enhances secretion of osteoinductive cytokines—including BMP-2, BMP-6, WNT10b, and Oncostatin M—thereby directly linking neuropeptide activity to osteo-immunological healing at the fracture site.56,57 Similarly, SP supports osteogenesis and inhibits bone resorption, potentially via activation of Wnt/β-catenin signaling pathways.58 Both SP and CGRP, upon binding to receptors on sensory nerve terminals, can amplify neurogenic inflammation and peripheral sensitization, setting up a positive feedback loop that aggravates pain perception and disturbs metabolic equilibrium within the fracture microenvironment.21,57
The persistent sprouting of sensory and sympathetic nerve fibers in non-healed fractures is a hallmark of chronic skeletal pain, potentially converting normally non-noxious mechanical stimuli into painful sensations and contributing to neuropathic pain states.57 Targeting neurotrophic factors, such as NGF, has shown promising therapeutic effects. For instance, anti-NGF therapy significantly alleviates pain in murine fracture models without compromising bone repair, underscoring the translational potential of modulating specific neurotrophic pathways in OVF management.7
Autonomic Nervous System (ANS) and Stress-Induced Dysregulation
The ANS, particularly sympathetic hyperactivity, plays a crucial role in regulating adaptive immune responses and nociceptive sensitivity following a fracture. Modulating autonomic tone by suppressing sympathetic activity or enhancing parasympathetic function may attenuate maladaptive immune responses and the progression of chronic pain after skeletal injury.21,57 Chronic psychological stress has emerged as an independent risk factor for osteoporosis and fragility fractures.21,49 In animal models such as the chronic social defeat stress (CSDS) mouse, elevated numbers of TH⁺ Ly6G⁺ neutrophils have been observed in the bone marrow and fracture haematomas. These activated neutrophils release catecholamines, such as norepinephrine (NE), epinephrine (EPI), and dopamine (DOP), which potently inhibit osteogenesis.49 In vitro studies using the ATDC5 chondrogenic cell line have demonstrated that these catecholamines downregulate key pluripotency transcription factors (such as Sox2 and Nanog) and osteogenic genes (such as Cbfa1/RUNX2, Sp7/Osterix and Alpl) in a dose- and time-dependent manner.49 This suppresses the transition of chondrocytes to osteoblasts, delays callus mineralisation, and decreases tissue mineral density and bone volume. These findings refine our understanding of the potential mechanisms underlying chronic pain following non-union fractures. However, further validation and exploration of these mechanisms in OVF mice and clinical patients is required. Targeting specific components of neural regulation, such as neuropeptide signalling, neurotrophic control or autonomic modulation, represents a promising direction for future therapeutic interventions. The molecular mechanisms of osteoporotic fracture pain are summarized in Table 1.
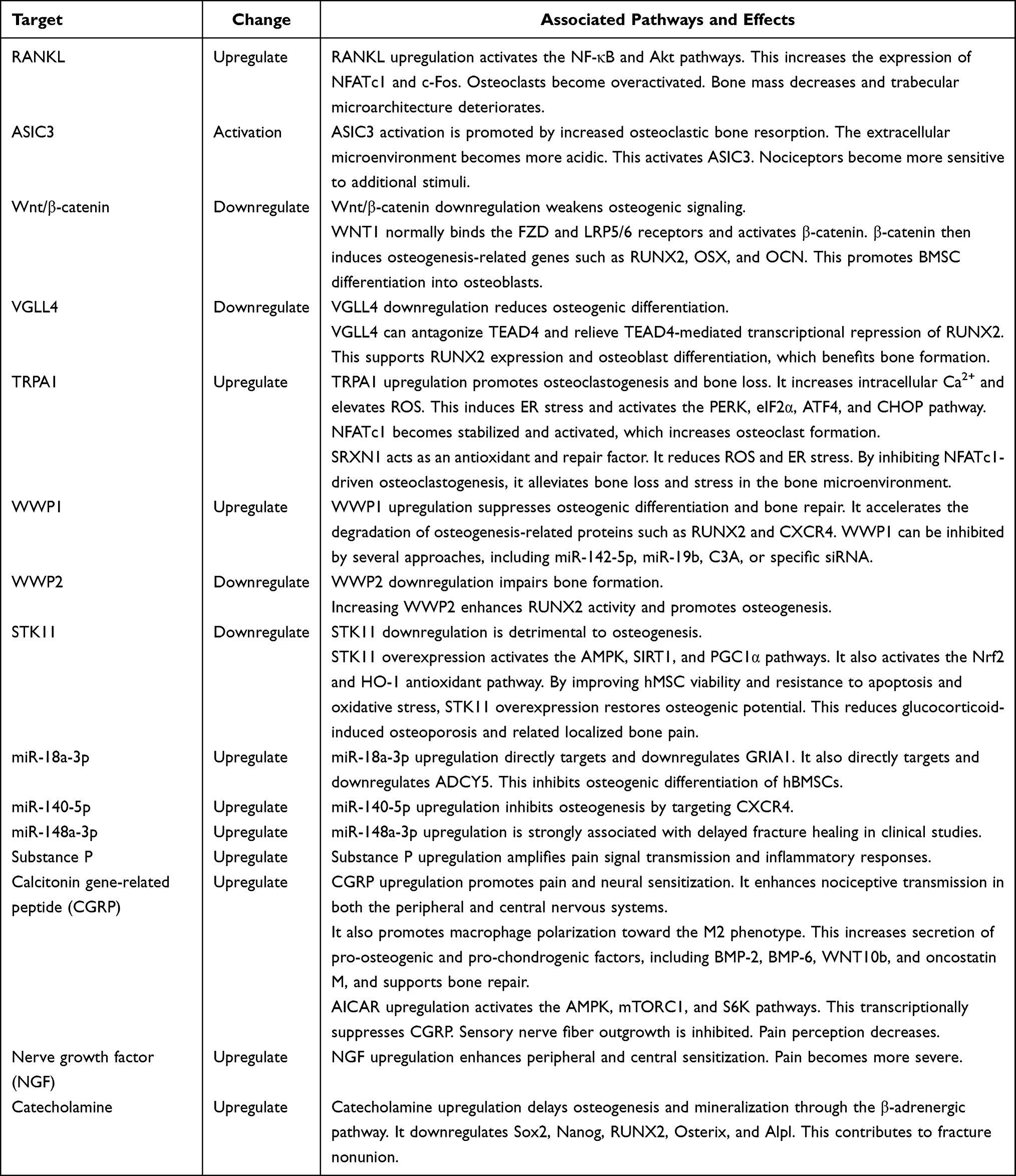 |
Table 1 Potential Molecular Mechanisms for Pain Following OVFs |
Pain Management in OVF
Optimizing pain management in OVF remains a clinical challenge and often requires a multimodal, individualized strategy. The European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO) recommended multimodal and multicomponent approaches for pain management.59 Combining non-pharmacological measures with currently available pharmacological options may better address the needs. Early administration of non-opioid analgesics is recommended to alleviate acute pain and potentially reduce the risk of chronic pain development. However, high-dose non-steroidal anti-inflammatory drugs (NSAIDs) should be avoided due to their potential to impair bone healing.60 Surgical intervention, particularly vertebral cement augmentation procedures such as balloon kyphoplasty and vertebroplasty, has been widely adopted to provide OVF patients with persistent symptoms with rapid pain relief and functional recovery.61–63 In a prospective randomized controlled trial including 90 patients with OVCFs, both the deflectable percutaneous kyphoplasty group and the bilateral percutaneous kyphoplasty group showed significant improvements in VAS scores at 1 year postoperatively compared with preoperative values (p < 0.001).63 However, risk factors for residual postoperative pain include low BMD, multilevel fractures, posterior fascial injury, inadequate cement filling, facet joint damage, and depression, warranting meticulous preoperative and postoperative assessment.62,64 Accordingly, the use of imaging tools such as CT or MRI, together with scoring systems such as the VAS, is recommended for risk stratification. Analgesics and surgery are effective treatments for relieving acute pain associated with OVF. The current strategy emphasises a combination of pharmacological interventions and anti-neurosensitisation therapies alongside lifestyle modifications, such as exercise, nutritional optimisation and vitamin D supplementation, to reduce pain synergistically, facilitate bone repair and improve functional outcomes (Table 2).
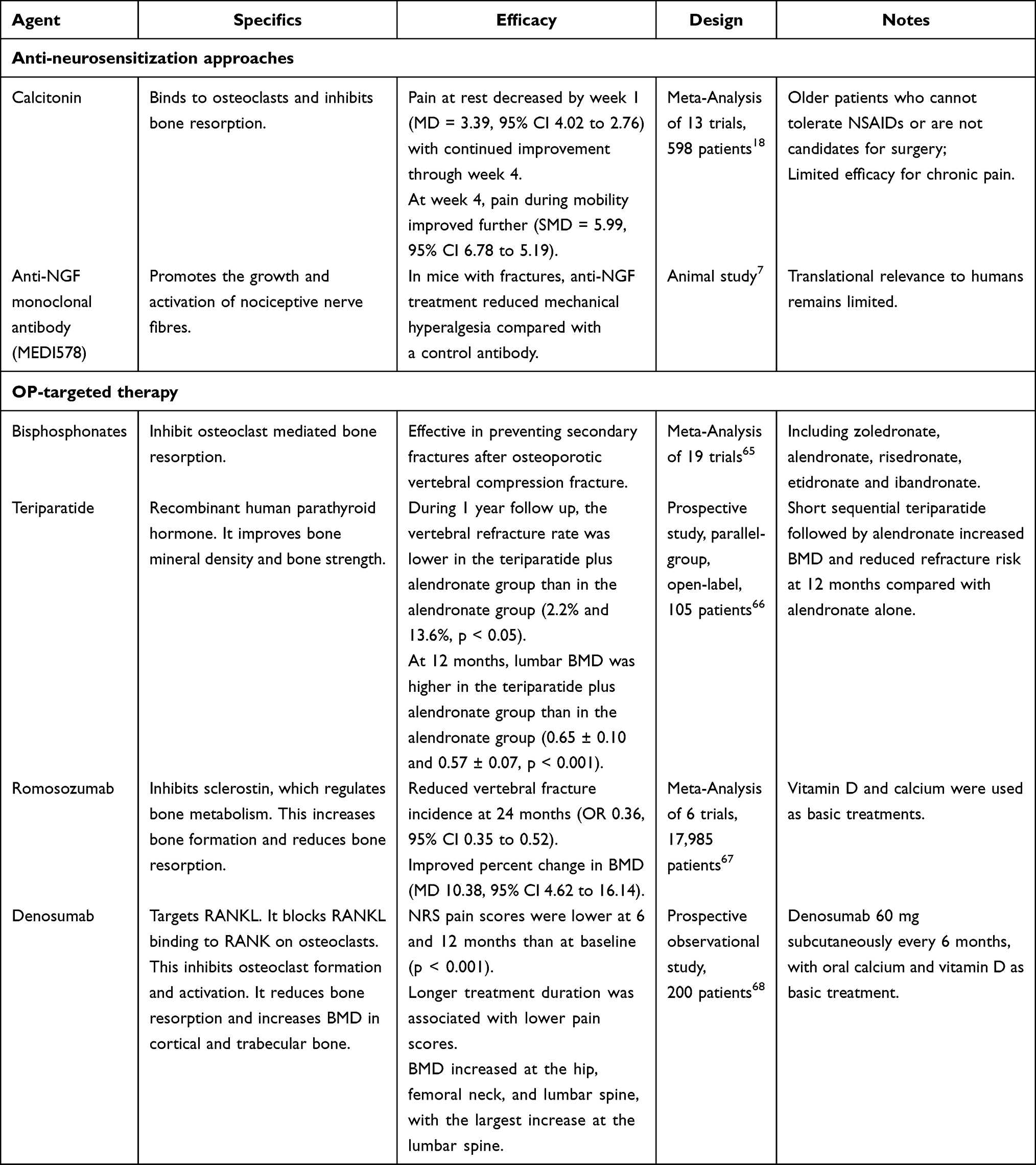 |
Table 2 Pharmacological Management of Pain Following OVFs |
Anti-Neurosensitization Approaches
Calcitonin is a peptide that is released by the parathyroid gland. It binds to osteoclasts, preventing them from inducing bone resorption. Due to its dual effects on osteoclast inhibition and neuromodulation, calcitonin is suitable for elderly patients who are intolerant to NSAIDs or surgery.69 Multiple randomized controlled trials and meta-analyses indicate that calcitonin significantly alleviates acute pain following OVF.18 The analgesic effect peaks around day 10 and persists up to day 45, with significant improvements in both VAS scores and electrical pain thresholds.5 However, its effectiveness in treating chronic pain is limited.
NGF is a key neurotrophic factor involved in peripheral and central sensitisation. It promotes the growth and activation of nociceptive nerve fibres and plays a significant role in chronic pain following fracture.70 Anti-NGF monoclonal antibody therapy effectively attenuates fracture-related pain behaviors in murine models without compromising bone healing.5 By neutralizing NGF and inhibiting TrkA-mediated nociceptive signaling, anti-NGF treatment prevents aberrant nerve sprouting and central sensitization, offering a promising, non-opioid analgesic strategy for managing chronic fracture pain.5,7
OP-Targeted Therapy
Persistent low back pain following OVF is closely related to non-union and local spinal deformities, with non-union serving as the most significant independent predictor of chronic pain.20,71 Thus, promoting fracture healing and preventing non-union are crucial therapeutic goals that may improve long-term prognosis more effectively than simply correcting deformity.
Pharmacologic treatment is central to both the management and prevention of osteoporotic fractures. Bisphosphonates, as first-line agents, inhibit osteoclast-mediated bone resorption, thereby reducing bone loss, minimizing subsequent fracture risk, and potentially decreasing inflammatory mediators contributing to pain.72,73 For example, alendronate has demonstrated substantial efficacy and safety in clinical studies, reducing the incidence of vertebral fractures by 44%-57%.60 In addition, anabolic agents, including teriparatide and romosozumab, accelerate callus maturation and fracture healing, as well as improving pain and function. Denosumab, administered biannually, increases BMD at key sites and has been associated with significant improvements in both pain and functional measures, especially in patients with a history of fragility fractures or who are new to osteoporosis therapy.68 Similarly, romosozumab, when used in high-risk postmenopausal women, significantly reduces vertebral fracture risk and increases BMD. However, its long-term safety profile requires further investigation.67 International guidelines recommend the early initiation of anti-OP treatment after a fracture. For patients at very high risk of fracture, sequential therapy involving anabolic agents followed by anti-resorptive therapy is advised.74
Summary and Prospect
The mechanisms underlying pain after OVFs involve bone metabolism, inflammation, neural processes, and genetic regulation, forming a complex and interconnected network. Both basic and clinical research targeting these pathways offer promising opportunities for precise management of OVF-related pain. Future studies should further define key regulatory nodes within the bone–neuro–immune axis, clarify the roles of novel molecular targets such as TRPA1 and STK11, and expand understanding of inflammatory and autophagic pathways. Future studies should use multi omics platforms, including genomics, transcriptomics, proteomics, and metabolomics, to identify patient specific molecular signatures. These data may enable molecular stratification of osteoporotic vertebral fracture patients, support biomarker discovery for early prediction of persistent pain and treatment response, and inform personalized therapeutic algorithms. Integrating multi omics data with translational research may further advance safer and more effective individualized therapies.
In clinical practice, multimodal management that combines pharmacologic, surgical, and neuromodulatory interventions with functional rehabilitation and psychological support may reduce chronic pain and improve outcomes. Multidisciplinary collaboration among orthopedics, pain medicine, and physiatry is essential for comprehensive pain management in osteoporotic vertebral compression fractures. The use of intelligent, automated imaging and follow-up tools could enable earlier detection of fractures and related complications. In summary, combining molecular insights with personalized therapeutic strategies has the potential to markedly enhance quality of life for OVF patients and lessen social and healthcare burdens. Ongoing multidisciplinary collaboration in both clinical and basic research will be crucial for further advances in this field.
Funding
This study was supported by the Key Laboratory Project of Jiaxing, China (Grant Agreement 2022-yzcsgtjzjz) and the Key Departments of Jiaxing, China (Grant Agreement 2023-ZC-012).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Anish RJ, Nair A. Osteoporosis management-current and future perspectives – a systemic review. J Orthop. 2024;53:101–14. doi:10.1016/j.jor.2024.03.002
2. Sözen T, Özışık L, Başaran N. An overview and management of osteoporosis. Eur J Rheumatol. 2019;4(1):46–56. doi:10.5152/eurjrheum.2016.048
3. Ponzano M, Tibert N, Brien S, et al. International consensus on the non-pharmacological and non-surgical management of osteoporotic vertebral fractures. Osteoporos Int. 2023;34(6):1065–1074. doi:10.1007/s00198-023-06688-9
4. Moretti A, Liguori S, Paoletta M, et al. Characterization of neuropathic component of back pain in patients with osteoporotic vertebral fractures. NeuroRehabilitation. 2022;51(2):325–331. doi:10.3233/nre-220040
5. Zhao Y, Zhang H, Li N, Li J, Zhang L. Chronic pain after bone fracture: current insights into molecular mechanisms and therapeutic strategies. Brain Sci. 2022;12(8):1056. doi:10.3390/brainsci12081056
6. Kutsal FY, Ergin Ergani GO. Vertebral compression fractures: still an unpredictable aspect of osteoporosis. Turk J Med Sci. 2021;51(2):393–399. doi:10.3906/sag-2005-315
7. Magnusdottir R, Gohin S, Ter Heegde F, et al. Fracture-induced pain-like behaviours in a femoral fracture mouse model. Osteoporos Int. 2021;32(11):2347–2359. doi:10.1007/s00198-021-05991-7
8. Okçu M, Erden Y, Tuncay F, Koçak FA, Kaya SS, Doğru YG. Does osteoporosis cause pain even without a fracture? An observational study. Somatosens Mot Res. 2023;40(3):110–115. doi:10.1080/08990220.2023.2188929
9. Sawicki P, Tałałaj M, Życińska K, Zgliczyński WS, Wierzba W. Comparison of the characteristics of back pain in women with postmenopausal osteoporosis with and without vertebral compression fracture: a retrospective study at a single osteoporosis center in Poland. Med Sci Monit. 2021;27:e929853. doi:10.12659/msm.929853
10. Chen R, Zhang P, Li K, Liu Q, Li G. Risk factors of costal pain of thoracic osteoporotic vertebral compression fractures: a multicenter retrospective analysis. Sci Rep. 2025;15(1):10739. doi:10.1038/s41598-025-88920-6
11. Li W-G, Zeng R, Lu Y, et al. The value of radiomics-based CT combined with machine learning in the diagnosis of occult vertebral fractures. BMC Musculoskelet Disord. 2023;24(1):819. doi:10.1186/s12891-023-06939-0
12. Imamudeen N, Basheer A, Iqbal AM, Manjila N, Haroon NN, Manjila S. Management of osteoporosis and spinal fractures: contemporary guidelines and evolving paradigms. Clin Med Res. 2022;20(2):95–106. doi:10.3121/cmr.2021.1612
13. Wang H, Huang J, Tao L, Liu D, Song C. Efficacy and safety of minodronate in the treatment of postmenopausal osteoporosis with low back pain: a single-centre, randomized and open-label controlled trial. Trials. 2024;25(1):534. doi:10.1186/s13063-024-08364-7
14. Noh M, Che X, Jin X, et al. Dimeric R25CPTH(1–34) activates the parathyroid hormone-1 receptor in vitro and stimulates bone formation in osteoporotic female mice. Elife. 2025;13. doi:10.7554/eLife.97579.5
15. Xiong Y, Zhang C, Chen X, et al. Prediction of subsequent vertebral fracture after acute osteoporotic fractures from clinical and paraspinal muscle features. Calcif Tissue Int. 2024;114(6):614–624. doi:10.1007/s00223-024-01209-0
16. Gutierrez-Gonzalez R, Royuela A, Zamarron A. Vertebral compression fractures: pain relief, progression and new fracture rate comparing vertebral augmentation with brace. BMC Musculoskelet Disord. 2023;24(1):898. doi:10.1186/s12891-023-07041-1
17. Kuroda T, Shiraki M, Tanaka S, Shiraki Y, Narusawa K, Nakamura T. The relationship between back pain and future vertebral fracture in postmenopausal women. Spine. 2009;34(18):1984–1989. doi:10.1097/BRS.0b013e3181b0c97a
18. Knopp-Sihota JA, Newburn-Cook CV, Homik J, Cummings GG, Voaklander D. Calcitonin for treating acute and chronic pain of recent and remote osteoporotic vertebral compression fractures: a systematic review and meta-analysis. Osteoporosis Int. 2012;23(1):17–38. doi:10.1007/s00198-011-1676-0
19. Iwata A, Kanayama M, Oha F, et al. Is bone nonunion, vertebral deformity, or spinopelvic malalignment the best therapeutic target for amelioration of low back pain after osteoporotic vertebral fracture? Spine. 2020;45(13):E760–e767. doi:10.1097/brs.0000000000003422
20. Inose H, Kato T, Ichimura S, et al. Factors contributing to residual low back pain after osteoporotic vertebral fractures. J Clin Med. 2022;11(6):1566. doi:10.3390/jcm11061566
21. Morris AJ, Parker RS, Nazzal MK, et al. Cracking the code: the role of peripheral nervous system signaling in fracture repair. Curr Osteoporos Rep. 2024;22(1):193–204. doi:10.1007/s11914-023-00846-y
22. Wawrzyniak A, Balawender K. Structural and metabolic changes in bone. Animal. 2022;12(15). doi:10.3390/ani12151946
23. Iaconisi GN, Mancini R, Ricci V, et al. Biochemical mechanisms and rehabilitation strategies in osteoporosis-related pain: a systematic review. Clin Prac. 2024;14(6):2737–2758. doi:10.3390/clinpract14060216
24. Zhu P, Tao H, Chen K, et al. TRPA1 aggravates osteoclastogenesis and osteoporosis through activating endoplasmic reticulum stress mediated by SRXN1. Cell Death Dis. 2024;15(8):624. doi:10.1038/s41419-024-07018-5
25. Li C, Liu X, Chen X, et al. Bu-Sui-Dan enhances osteoblast differentiation by upregulating VGLL4 to counteract TEAD4-mediated RUNX2 transcription suppression in ovariectomized rats. J Ethnopharmacol. 2024;335:118690. doi:10.1016/j.jep.2024.118690
26. Zhu M, Shan J, Xu H, et al. Glaucocalyxin A suppresses osteoclastogenesis induced by RANKL and osteoporosis induced by ovariectomy by inhibiting the NF-κB and Akt pathways. J Ethnopharmacol. 2021;276:114176. doi:10.1016/j.jep.2021.114176
27. Wada H, Aso K, Izumi M, Ikeuchi M. The effect of postmenopausal osteoporosis on subchondral bone pathology in a rat model of knee osteoarthritis. Sci Rep. 2023;13(1):2926. doi:10.1038/s41598-023-29802-7
28. Qi G, Jiang Z, Niu J, et al. SrHPO4-coated Mg alloy implant attenuates postoperative pain by suppressing osteoclast-induced sensory innervation in osteoporotic fractures. Mater Today Bio. 2024;28:101227. doi:10.1016/j.mtbio.2024.101227
29. Xu J, Wang J, Chen X, Li Y, Mi J, Qin L. The effects of calcitonin gene-related peptide on bone homeostasis and regeneration. Curr Osteoporos Rep. 2020;18(6):621–632. doi:10.1007/s11914-020-00624-0
30. de Clauser L, Santana‐Varela S, Wood JN, Sikandar S. Physiologic osteoclasts are not sufficient to induce skeletal pain in mice. Eur J Pain. 2021;25(1):199–212. doi:10.1002/ejp.1662
31. Jurczak A, Delay L, Barbier J, et al. Antibody-induced pain-like behavior and bone erosion: links to subclinical inflammation, osteoclast activity, and acid-sensing ion channel 3–dependent sensitization. Pain. 2022;163(8):1542–1559. doi:10.1097/j.pain.0000000000002543
32. Torres HM, Arnold KM, Oviedo M, Westendorf JJ, Weaver SR. Inflammatory processes affecting bone health and repair. Curr Osteoporos Rep. 2023;21(6):842–853. doi:10.1007/s11914-023-00824-4
33. Ge G, Yang S, Hou Z, et al. Theaflavin-3,3′-digallate promotes the formation of osteoblasts under inflammatory environment and increases the bone mass of ovariectomized mice. Front Pharmacol. 2021;12:648969. doi:10.3389/fphar.2021.648969
34. Xiao J, Li W, Li G, Tan J, Dong N. STK11 overexpression prevents glucocorticoid-induced osteoporosis via activating the AMPK/SIRT1/PGC1α axis. Hum Cell. 2022;35(4):1045–1059. doi:10.1007/s13577-022-00704-6
35. Wei F, Tuong ZK, Omer M, et al. A novel multifunctional radioprotective strategy using P7C3 as a countermeasure against ionizing radiation-induced bone loss. Bone Res. 2023;11(1):34. doi:10.1038/s41413-023-00273-w
36. Wang Q, Liu M, Cao B-Y, et al. Osteoporosis caused by monoallelic variant of WNT1 gene in four pediatric patients. Am J Med Genet A. 2025;197(5):e63987. doi:10.1002/ajmg.a.63987
37. Zhang DH, Shao J. Research progress of basing on Wnt/β-catenin pathway in the treatment of bone tissue diseases. Tissue Eng Part B Rev. 2025;31(6):555–565. doi:10.1089/ten.teb.2024.0170
38. Ren N, Lv S, Li X, et al. Clinical features, treatment, and follow-up of OPPG and high-bone-mass disorders: LRP5 is a key regulator of bone mass. Osteoporos Int. 2024;35(8):1395–1406. doi:10.1007/s00198-024-07080-x
39. Wang Y, Wu Z, Wang C, et al. The role of WWP1 and WWP2 in bone/cartilage development and diseases. Mol Cell Biochem. 2024;479(11):2907–2919. doi:10.1007/s11010-023-04917-7
40. Zhao M, Dong J, Liao Y, et al. MicroRNA miR-18a-3p promotes osteoporosis and possibly contributes to spinal fracture by inhibiting the glutamate AMPA receptor subunit 1 gene (GRIA1). Bioengineered. 2022;13(1):370–382. doi:10.1080/21655979.2021.2005743
41. Wang L, Dong J, Ma J, et al. By inhibiting ADCY5, miR-18a-3p promotes osteoporosis and possibly contributes to spinal fracture. Biochem Biophys Res Commun. 2021;550:49–55. doi:10.1016/j.bbrc.2021.02.118
42. Toury L, Frankel D, Airault C, Magdinier F, Roll P, Kaspi E. miR-140-5p and miR-140-3p: key actors in aging-related diseases? Int J Mol Sci. 2022;23(19):11439. doi:10.3390/ijms231911439
43. Zhu G, Zeng C, Qian Y, et al. Tensile strain promotes osteogenic differentiation of bone marrow mesenchymal stem cells through upregulating lncRNA-MEG3. Histol Histopathol. 2021;36(9):939–946. doi:10.14670/hh-18-365
44. Hao M-L, Zuo X-Q, Qiu Y, Li J. WGCNA identification of genes and pathways involved in the pathogenesis of postmenopausal osteoporosis. Int J Gen Med. 2021;14:8341–8353. doi:10.2147/ijgm.S336310
45. Li Y, Yang S, Wang G, Huang Y. MiR-148a-3p regulates the fracture healing process by targeting MAFB. J Orthop Surg Res. 2025;20(1):826. doi:10.1186/s13018-025-06223-y
46. Song G, Lin S, Zhang X, Pan H. The relationship between the expression of serum asprosin and miR-21 in patients with osteoporosis and delayed healing after OVCF surgery. J Orthopaedic Surg. 2025;33(1):10225536251331325. doi:10.1177/10225536251331325
47. Liu J, Zhou J, Huang X, et al. Protective effects of pulsed electromagnetic field therapy attenuates autophagy and apoptosis in osteoporotic osteoarthritis model rats by activating PPARγ. Electromagnetic Biol Med. 2024;43(1–2):61–70. doi:10.1080/15368378.2024.2314108
48. Khalid TY, Adamali H, Zahoor N, Drew S, Clark EM. Translation from English into Urdu of a clinical decision tool to screen older women with back pain for osteoporotic-related vertebral fragility fractures. BMC Musculoskelet Disord. 2025;26(1):691. doi:10.1186/s12891-025-08837-z
49. Tschaffon-Müller MEA, Kempter E, Steppe L, et al. Neutrophil-derived catecholamines mediate negative stress effects on bone. Nat Commun. 2023;14(1):3262. doi:10.1038/s41467-023-38616-0
50. Men Z, Huang C, Xu M, et al. Zhuanggu Zhitong capsule alleviates postmenopausal osteoporosis in ovariectomized rats by regulating autophagy through AMPK/mTOR signaling pathway. Ann Transl Med. 2022;10(16):900. doi:10.21037/atm-22-3724
51. Chen R, Yang C, Yang F, et al. Targeting the mTOR-autophagy axis: unveiling therapeutic potentials in osteoporosis. Biomolecules. 2024;14(11):1452. doi:10.3390/biom14111452
52. Kiani P, Khodadadi ES, Nikdasti A, et al. Autophagy and the peroxisome proliferator-activated receptor signaling pathway: a molecular ballet in lipid metabolism and homeostasis. Mol Cell Biochem. 2025;480(6):3477–3499. doi:10.1007/s11010-025-05207-0
53. Ding C, Wang H, Yang C, Hang Y, Zhu S, Cao Y. Radiofrequency field inhibits RANKL-induced osteoclast differentiation in RAW264.7 cells via modulating the NF-κB signaling pathway. Electromagn Biol Med. 2024;43(4):292–302. doi:10.1080/15368378.2024.2401554
54. Oo WM, Hunter DJ. Nerve growth factor (NGF) inhibitors and related agents for chronic musculoskeletal pain: a comprehensive review. BioDrugs. 2021;35(6):611–641. doi:10.1007/s40259-021-00504-8
55. Rees TA, Hendrikse ER, Hay DL, Walker CS. Beyond CGRP: the calcitonin peptide family as targets for migraine and pain. Br J Pharmacol. 2022;179(3):381–399. doi:10.1111/bph.15605
56. Zhang Q, Wu B, Yuan Y, et al. CGRP-modulated M2 macrophages regulate osteogenesis of MC3T3-E1 via Yap1. Arch Biochem Biophys. 2021;697:108697. doi:10.1016/j.abb.2020.108697
57. Parker RS, Nazzal MK, Morris AJ, et al. Role of the neurologic system in fracture healing: an extensive review. Curr Osteoporos Rep. 2024;22(1):205–216. doi:10.1007/s11914-023-00844-0
58. Wang X, Su N. Neurokinin-1-tachykinin receptor agonist promotes diabetic fracture healing in rats with type 1 diabetes via modulation of Wnt/β-catenin signalling axis. Saudi J Biol Sci. 2021;28(4):2139–2145. doi:10.1016/j.sjbs.2021.02.026
59. Veronese N, Cooper C, Bruyère O, et al. Multimodal multidisciplinary management of patients with moderate to severe pain in knee osteoarthritis: a need to meet patient expectations. Drugs. 2022;82(13):1347–1355. doi:10.1007/s40265-022-01773-5
60. Patel D, Liu J, Ebraheim NA. Managements of osteoporotic vertebral compression fractures: a narrative review. World J Orthop. 2022;13(6):564–573. doi:10.5312/wjo.v13.i6.564
61. Yeh K-L, Wu S-H, Liaw C-K, Hou S-M, Wu -S-S. Outcomes of different minimally invasive surgical treatments for vertebral compression fractures: an observational study. World J Clin Cases. 2021;9(31):9509–9519. doi:10.12998/wjcc.v9.i31.9509
62. Wang Z-W, Wang G-Y, Liu D-K, Zhang D-Z, Zhao C. Risk factors for residual back pain after PVP treatment for osteoporotic thoracolumbar compression fractures: a retrospective cohort study. World Neurosurg. 2023;180:e484–e493. doi:10.1016/j.wneu.2023.09.094
63. Shi X, Li P, Li J, Bao C, Xiang J, Lu Y. Comparative evaluation of an innovative deflectable percutaneous kyphoplasty versus conventional bilateral percutaneous kyphoplasty for osteoporotic vertebral compression fractures: a prospective, randomized and controlled trial. Spine J. 2023;23(4):585–598. doi:10.1016/j.spinee.2022.12.012
64. Seah SJ, Yeo MH, Tan J-H, Hey HWD. Early cement augmentation may be a good treatment option for pain relief for osteoporotic compression fractures: a systematic review and meta-analysis. Eur Spine J. 2023;32(5):1751–1762. doi:10.1007/s00586-023-07658-9
65. Jin Y-Z, Lee JH, Xu B, Cho M. Effect of medications on prevention of secondary osteoporotic vertebral compression fracture, non-vertebral fracture, and discontinuation due to adverse events: a meta-analysis of randomized controlled trials. BMC Musculoskelet Disord. 2019;20(1):399. doi:10.1186/s12891-019-2769-8
66. Yang D, Tan J, Long Y, et al. Sequential treatment of teriparatide and alendronate versus alendronate alone for elevation of bone mineral density and prevention of refracture after percutaneous vertebroplasty in osteoporosis: a prospective study. Aging Clin Exp Res. 2023;35(3):531–539. doi:10.1007/s40520-023-02342-w
67. Gao G, Cui J, Xie Y, Dong J. Effects of romosozumab combined with routine therapy on pain relief, disease progression and adverse reactions in patients with postmenopausal osteoporosis: a systematic review and meta-analysis. Front Med Lausanne. 2024;11:1440948. doi:10.3389/fmed.2024.1440948
68. Zheng M, Wang X-Y, Zhu K-Y, et al. The effects of denosumab treatment on pain and function beyond bone density in patients with postmenopausal osteoporosis: a prospective study. Orthop Surg. 2025;17(6):1644–1655. doi:10.1111/os.70032
69. Kaneb A, Berardino K, Hanukaai JS, Rooney K, Kaye AD. Calcitonin (FORTICAL, MIACALCIN) for the treatment of vertebral compression fractures. Orthopedic Rev. 2021;13(2):24976. doi:10.52965/001c.24976
70. Barker PA, Mantyh P, Arendt-Nielsen L, Viktrup L, Tive L. Nerve growth factor signaling and its contribution to pain. J Pain Res. 2020;13:1223–1241. doi:10.2147/jpr.S247472
71. Xin J, Liu X, Jing X, et al. Multifactor analysis of costal pain in osteoporotic fracture of thoracic vertebra. Pain Physician. 2021;24(6):E795–e802.
72. Hsu Y-H, Li -C-C, Liang F-W, et al. Reduced all-cause mortality with bisphosphonates among post-fracture osteoporosis patients: a nationwide study and systematic review. Clin Pharmacol Ther. 2022;112(3):711–719. doi:10.1002/cpt.2645
73. Xu Q, Zhan P, Li X, et al. Bisphosphonate-enoxacin inhibit osteoclast formation and function by abrogating RANKL-induced JNK signalling pathways during osteoporosis treatment. J Cell Mol Med. 2021;25(21):10126–10139. doi:10.1111/jcmm.16949
74. Oh YK, Moon NH, Shin WC. Management of osteoporosis medication after osteoporotic fracture. Hip Pelvis. 2022;34(4):191–202. doi:10.5371/hp.2022.34.4.191
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.