")
Back to Journals » International Journal of General Medicine » Volume 14
Molecular Mechanism of Sphingosine-1-Phosphate Receptor 1 Regulating CD4+ Tissue Memory in situ T Cells in Primary Sjogren’s Syndrome
Authors Yang XX, Yang C, Wang L , Zhou YB, Yuan X, Xiang N, Wang YP, Li XM
Received 7 July 2021
Accepted for publication 25 August 2021
Published 27 September 2021 Volume 2021:14 Pages 6177—6188
DOI https://doi.org/10.2147/IJGM.S327304
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Xiao-Xiao Yang,1,2 Chao Yang,2 Li Wang,2 Ying-Bo Zhou,2 Xiang Yuan,2 Nan Xiang,2 Yi-Ping Wang,3 Xiao-Mei Li1,2
1School of Life Sciences, University of Science and Technology of China, Hefei, Anhui, People’s Republic of China; 2The First Affiliated Hospital of USTC, Department of Rheumatology and Immunology, University of Science and Technology of China, Hefei, People’s Republic of China; 3Westmead Institute for Medical Research, University of Sydney, Sdyney, NSW, 2145, Australia
Correspondence: Xiao-Mei Li Email [email protected]
Objective: Although extensive research has been carried out on CD4+T cells infiltrating the labial glands in patients with primary Sjögren’s Syndrome (pSS), it is still unclear how CD4+T cells remain in the labial gland tissue and develop into tissue resident cells. The aim of this study was to investigate the molecular mechanism by which CD4+T reside in labial glandular tissue of pSS patients.
Methods: Lymphocyte infiltration in labial salivary glands (LSG) of pSS patients was detected by H&E staining. Expression of sphingosine-1-phosphate receptor 1 (S1PR1) in LSG was examined by Immunohistochemistry. Immunofluorescence analyses were utilized to detect the co-expression of CD4, CD69 and S1PR1 in T cells of LSG of pSS patients. Expression of gene S1pr1 in peripheral blood CD4+T cells of healthy controls and pSS patients was detected by quantitative real-time PCR (QPCR). QPCR was used to examine the expression of gene S1pr1, Klf2, and Cd69 in the CD4+T cells that were co-cultured in vitro with cytokines TNF-α, TGF-β, and IL-33.
Results: S1PR1 was expressed in the infiltrating monocytes in LSG of pSS patients, and S1PR1 was weakly or even not expressed in cytoplasm of CD4+CD69+TRM cells of LSG in patients with pSS. Expression of gene S1pr1 in peripheral blood CD4+T cells of pSS patients was about three-fifths of that of healthy controls (P < 0.05). Expression of genes S1pr1 (P < 0.001) and Klf-2 (P < 0.001) was significantly decreased, and the expression of gene Cd69 (P < 0.05) was significantly increased in peripheral blood CD4+T cells of pSS patients co-cultured in vitro with cytokines TNF-α, TGF-β, and IL-33.
Conclusion: Our study suggests that the decrease of S1pr1 gene expression may provide a molecular basis for promoting the tissue retention and development of CD4+CD69+TRM cells.
Keywords: primary Sjogren’s syndrome, S1PR1, CD4+TRM
Introduction
Primary Sjogren’s syndrome (pSS) is a common systemic autoimmune disease without other connective tissue diseases, characterized by dysfunction of exocrine glands, especially salivary and lacrimal glands.1,2 Clinically, the hallmark of the disease is mucosal surface dry symptoms that include xerostomia and xerophthalmia as a result of lymphocytic infiltration of the lacrimal and salivary glands and even also arise lung, liver, kidney, nervous system, and other important visceral lesions. About 5% of patients with pSS will develop into non-Hodgkin malignant lymphoma, an additional severe complication.3–5 Epidemiological data from a meta-analysis showed that ratio of female to male patients with pSS was 9:1, and peak of onset was around 50 years old.6,7 But recently it has been shown that the ratio of female to male patients with pSS was 20:1.8 The 20:1 female-to-male ratio was considerably different from what has been commonly described in many previous studies.8
It has been reported that the monocytes infiltrated by exocrine glands are mainly CD4+T cells, but other cells are also involved including CD8+T cells, dendritic cells, and plasma cells.9 Evidence suggests that the infiltrate composition differs according to lesion severity. T cells, CD4+T cells, and B lymphocytes were significantly different among minor salivary gland (MSG) tissues with mild, intermediate, or severe inflammatory lesions. T cell, CD4+T cell, and T/B cell ratio were negative with lesion severity, whereas B cell was positively correlated with infiltration grade and biopsy focus score.10 CD4+T cells play a significant role in the pathogenesis of pSS. CD4+T cells are activated by antigens presented by MHC class II (MHCII) molecules. Activated T cells promote the further development of the disease by producing pro-inflammatory cytokines and inducing the activation of B cells to produce autoantibodies. In inflammatory gland tissue, there are many subsets of CD4+T cells, including T-helper 1 (Th1), T-helper 17 (Th17), T follicular helper cells (Tfh), and regulatory T cells (Treg).11 Recent evidence suggests that a large proportion of T cells in the infiltrating monocytes of salivary glands are tissue resident memory T cells (TRM cells).3 TRM cells are an essential part of local anti-infection immunity in the body, and TRM cells have memory function in mucosal tissues and permanently exist in tissues.12 When it accumulates, overactivates, or abnormally activates, pro-inflammatory mediators are produced and alert surrounding tissues to upregulate defense mechanisms.13 At present, CD8+TRM cells are the most reported TRM cells, but previous studies have reported that CD4+ memory T cells reside in lungs,13–15 reproductive organs,16 salivary glands,17,18 and skin.19–21 All TRM cells express the membrane protein CD69 including CD4+TRM, which is against the tissue exit mediated by the sphingosine-1-phosphate receptor 1 (S1PR1) and prevents TRM cells from entering circulation.22,23
S1PR1, a G-protein-coupled receptor, interacts with sphingosine-1-phosphate (S1P), a bioactive sphingosine-mediator, to regulate thymus and peripheral lymphocyte migration.24 The specificity and efficiency of S1P and S1PR1 binding to control lymphocyte migration are determined by the concentration of S1P and the expression level of S1PR1. S1P maintains a low concentration in thymus and lymphatic organs, and a high concentration in blood which can inhibit the expression of S1PR1 in circulating T cells. When the T cells enter non-inflamed lymphoid tissues, S1PR1 can reappear. However, in inflammatory lymphoid tissue, CD69 is expressed on the surface of lymphocytes and leads to the internalization and degradation of S1PR, thus delaying the egress. After several rounds of division, newly generated effector T cells upregulate S1PR1 and lose the expression of CCR7, then enter circulation.25 In a word, S1PR1 was required for lymphocytes to exit the inflamed tissue and that CD69 lowered its levels, thus inhibiting lymphocyte egress. In the process of delayed exit, under the microenvironment of inflammatory tissue, including cytokines, the transcription profile of lymphocyte changes, such as the decreased expression of klf-2 and s1pr1, it will form tissue resident memory T cells and permanently reside in the tissue, further mediating tissue inflammation.26
The critical role of S1PR1 signaling in lymphocyte trafficking is well recognized, and some related studies have reported the contribution of S1PR1 signaling in autoimmunity and cancer. It has been reported that S1P-S1PR1 signaling is primarily considered as a catalyst of inflammation and induces osteoclastogenesis.27 Myeloid cell-specific S1PR1 overexpression mice demonstrated that myeloid cell S1P1 directly contributed to severity of neuroinflammation.28 Sphingosine 1-phosphate receptor 1 (S1PR1) is an integral component of tumor progression and maintains an activated state of STAT3.29 In the colorectal cancer liver metastasis mouse models, the level of IL-6 and the associated myeloid-derived suppressor cells stimulated by the S1PR1–STAT3 were correlated with the number of liver metastatic nodes. A mutual activation loop between S1PR1 and STAT3 can enhance colorectal cancer cell proliferation, migration, and invasion in vitro and in vivo.30 Therefore, targeted drugs targeting S1PR1 have very important therapeutic prospects. Fingolimod (FTY720) is an approved drug for multiple sclerosis based on large clinical trials.31 And Fingolimod acts as a functional antagonist for S1PR1,29 because it reduces hyperalgesia in models of peripheral inflammatory and neuropathic pain.32 In addition, a new class of small molecules, sphingosine-1-phosphate (S1P) receptor modulators, has recently shown efficacy in inflammatory bowel disease (IBD). These modulators downregulate S1P receptors expressed in lymphocytes and are capable of preventing lymphocyte trafficking to the site of inflammation, making them likely candidates for the treatment of chronic inflammatory disorders, including IBD, multiple sclerosis, and rheumatoid arthritis, which are currently under clinical investigation.33
In autoimmune diseases, recent studies have shown that inhibiting the expression of S1PR1/3 can regulate the function of synovial fibroblasts, thus inhibiting inflammation in rheumatoid arthritis.34 It was also reported that the expression of S1PR1 increased with the grade of lymphocytic infiltration in the labial gland of patients with pSS, and the signal of S1P and S1PR1 was related to apoptosis.35
In our previous study, we found that CD8+TRM cells mediated submandibular gland tissue damage in pSS mice.3 Therefore, this study focuses on exploring the molecular mechanism of S1PR1 in CD4+TRM in labial gland of patients with pSS.
Materials and Methods
Patients
The peripheral blood and labial gland organization of pSS patients and the peripheral blood of healthy controls were obtained from the first affiliated hospital of University of Science and Technology of China (USTC). The diagnostic criteria of all included patients with pSS was according to the report of 2002 revised American–European criteria. A total of 48 patients (47 females/1 male, mean age: 52.27±1.64) diagnosed with pSS who did not have other connective tissue diseases and 43 age-matched healthy controls (all females, mean age: 53.26 ± 1.57) were included in the study. The study was approved by the Ethics Committee of the first affiliated hospital of USTC, and informed consent was obtained from all patients and healthy people. And our research was conducted in accordance with the Declaration of Helsinki. The detailed clinical characteristics of the patients with pSS are indicated in Table 1.
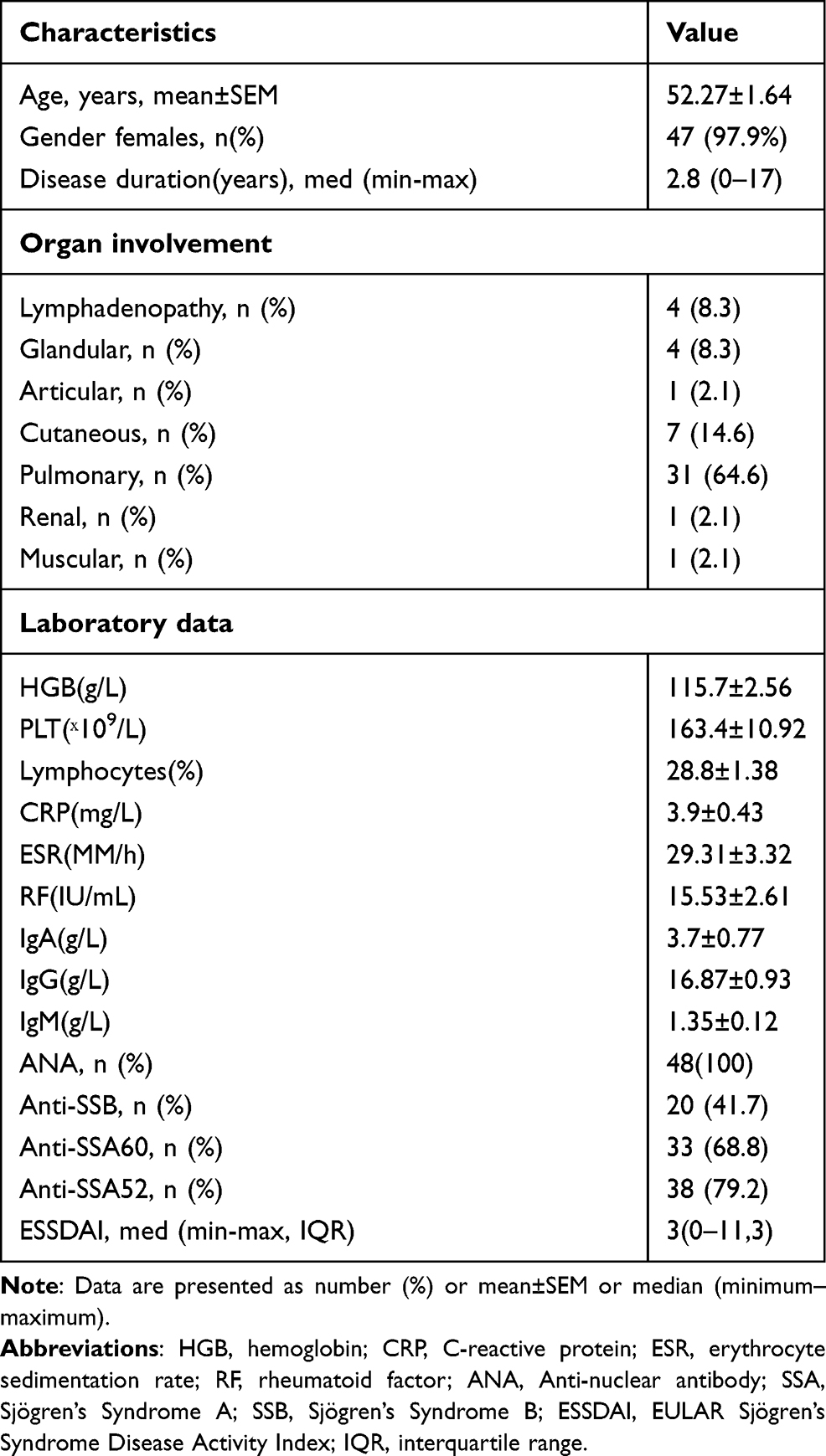 |
Table 1 Demographic and Clinical Characteristics of Primary Sjögren’s Syndrome |
Histological and Immunohistochemistry Analyses
Human labial glandular tissues were fixed with 4% paraformaldehyde (Servicebio, G1101), embedded in paraffin. The labial glandular tissue was cut into 4 micron slices, deplasticized to water, stained with hematoxylin and eosin (H&E) (Servicebio, G1003), dehydrated and sealed, examined under microscope (Nikon DS-U3), and analyzed by image acquisition.
For immunohistochemistry analysis, the tissue sections were placed in a repair box filled with citric acid (PH6.0) (Servicebio, G1202) antigen retrieval buffer for antigen retrieval. The sections were placed in 3% hydrogen peroxide (Pharmaceutical Group Chemical Reagent Co., LTD, 10011208) and incubated at room temperature in darkness for 25 min. Three percent BSA (Servicebio, G5001) was added to the circle to evenly cover the tissue, and the tissues were sealed for 30 min at room temperature. The primary antibody S1PR1 (Abcam, ab11424) prepared with PBS (PH7.4) in 1:100 dilution was added to the sections, and the sections were placed flat in a wet box and incubated overnight at 4°C. After the sections were washed and dried, the tissues were covered goat anti-rabbit labeled by HRP at 1:200 dilution and incubated at room temperature for 50 min. DAB color developing solution (Servicebio, G1211) newly prepared was added in the circle after the sections were washed and slightly dried. Rinsing the sections with tap water to stop the reaction, the sections were then counterstained with hematoxylin stain solution (Servicebio, G1004). Sections were visualized using a microscope (Nikon E100).
Immunofluorescence
Incubated slides with the first primary antibody CD69 (Thermo, PA5-102562) were prepared with PBS (PH7.4) in 1:1000 dilution overnight at 4°C and placed in a wet box containing a little water. The following are steps: 1) Cover objective tissue with secondary antibody goat anti-rabbit labeled by HRP (Servicebio, GB23301) at 1:500 dilution; 2) incubate at room temperature for 50 min in dark condition; 3) Wash slides three times with PBS (pH 7.4) in a Rocker device, 5 min each; 4) Incubate slides with CY3-TSA solution (Servicebio, G1223) at 1:2000 dilution with TBST appropriately for 10 min in dark condition; 5) Immerse the slides in EDTA antigen retrieval buffer (pH 8.0) (servicebio, G1206) to remove the primary antibodies and secondary antibodies combined with tissue; 6) Incubate slides with primary antibody S1PR1 (Abcam, ab11424) and CD4 (Thermo, 14-2444-82) at 1:100 dilution overnight at 4°C and placed in a wet box containing a little water. 7) Cover objective tissue with FITC goat anti-rabbit fluorescent (Servicebio, GB22303) and CY5 goat anti-mouse fluorescent (GB27301, GB27301), and incubate at room temperature for 50 min in dark condition at 1:100 dilution; 8) Incubate with DAPI solution (Servicebio, G1012) at room temperature for 10 min and kept in dark place; 9) Eliminate obvious liquid, incubate slides with spontaneous fluorescence quenching reagent (Servicebio, G1221) for 5 min, and then wash slides under flowing water for 10 min. Finally, the slides were visualized using a slice scanner (Pannoramic MIDI:3Dhistech).
Cell Isolation
The blood collected with heparin sodium or EDTA sampling vessel was mixed with an equal volume of 1×PBS. And peripheral blood mononuclear cells (PBMCs) were isolated by the separation solution of human peripheral blood mononuclear cells (TBD LDS1075).
The following are steps: 1) Determine cell number by Countess® Cell Counting Chamber Slides (Invitrogen, C10228); 2) Centrifuge cell suspension at 300×g for 10 min; 3) Aspirate supernatant completely; 4) Resuspend cell pellet in 80 µL of buffer per 107 total cells.; 5) Add 20 µL of CD4 MicroBeads (Miltenyi Biotec, 130-045-101) per 107 total cells; 6) Mix well and incubate for 15 min in 4°C the refrigerator. Finally, CD4+T cells were obtained by magnetic bead sorting on LS column (Miltenyi Biotec, 130-042-401).
In vitro Coculture
As previous studies,26 CD4+T cells were collected and added to 24-well flat-bottomed plates at a density of 106 cells per well in 400 µL complete RPMI medium in the presence of various combinations of cytokines, IL-33 (100ng/mL RD 3625-IL-010), TNF-α (125ng/mL RD 210-TA-005), and TGF-β (10ng/mL RD 240-B-002). At 48 h of culture, cells were stained for Quantitative Real-time PCR.
Quantitative Real-Time PCR
Total RNA from CD4+T cells were extracted by using RNeasy Mini Kit (Qiagen 74104). HiScript II Q RT SuperMix for qPCR (Vazyme R223-01) was used for reverse transcription. Gene expression was assessed with QuantStudioTM 5 system, and amplification was detected with QuantiNovaTM SYBR Green PCR Kit (Qiagen 208054) º Data analysis was the cycling threshold values of the target gene minus the control gene encoding GAPDH, with results calculated using the 2−∆∆CT method. Primers used to amplify specific gene fragments as follows: GAPDH: TGCACCACCAACTGCTTAGC (forward) and GGCATGGA CTGTGGTCATGAG (reverse); CD69: GAGCTGGACTTCAGCCCAAA (forward) and CCA CTTCCATGGGTGACCAG (reverse); S1PR1: GAAAACCAAGAAATTCCACCGA (forward) and TTTCAGCATTGTGATATAGCGC (reverse); KLF-2: AGACCT ACACCAA GAGTTC GC (forward) and GATCGGAGCGCGAGAAGG (reverse); TGF-β: CGA CTCGCCAGAGT GGTTAT (forward) and TAGTGAACCCGTTGATGTCCA (reverse); IL-33: GCTTTGCCTTT GGTATATCAGG (forward) and CTGATTCATTTGAGGGGTGTTG (reverse); TNF-α: TGG CGTGGAGCTGAGAGATAACC (forward) and CGATGCGGCTGATGGTGTGG (reverse).
Statistical Analyses
Statistical analyses were performed using Student’s two-tailed unpaired t-tests, Dunnett’s multiple comparison test, and Spearman test for correlation analysis in GraphPad Prism version 6. Results are expressed as mean ± SEM and correlation coefficient. And p value less than 0.05 was considered statistically significant.
Results
The Expression of S1PR1 in CD4+CD69+TRM Cells of the Labial Salivary Glands of Patients with pSS
Different degrees of lymphocytic infiltration of labial salivary glands (LSGs) in patients with pSS were stained with hematoxylin and eosin. We found that little lymphocyte infiltrated in the stroma of the LSG1, LSG2 have a large number of lymphocyte infiltration and formed obvious lymphocyte infiltration foci (Figure 1A). And according to the Chisholm and Mason classification,36 LSG1 with slight infiltration was rated as grad1, LSG2 displayed more than one focus per 4 mm3 was assessed as a grade 4. Immunohistochemical results showed that with the increase in lymphocyte infiltration, the expression of S1PR1 also showed an increased trend (Figure 1B). Co-expression of CD4, CD69, and S1PR1 was detected in immunofluorescence of LSG2 with a large number of lymphocytes infiltrating and forming an infiltrating foci (Figure 1C), indicating that strong CD69 fluorescence in CD4+T cells, S1PR1 has weak fluorescence or even no expression in the cytoplasm. Therefore, our results indicated that S1PR1 was weakly or even not expressed in cytoplasm of CD4+CD69+TRM cells of the labial salivary glands of patients with pSS.
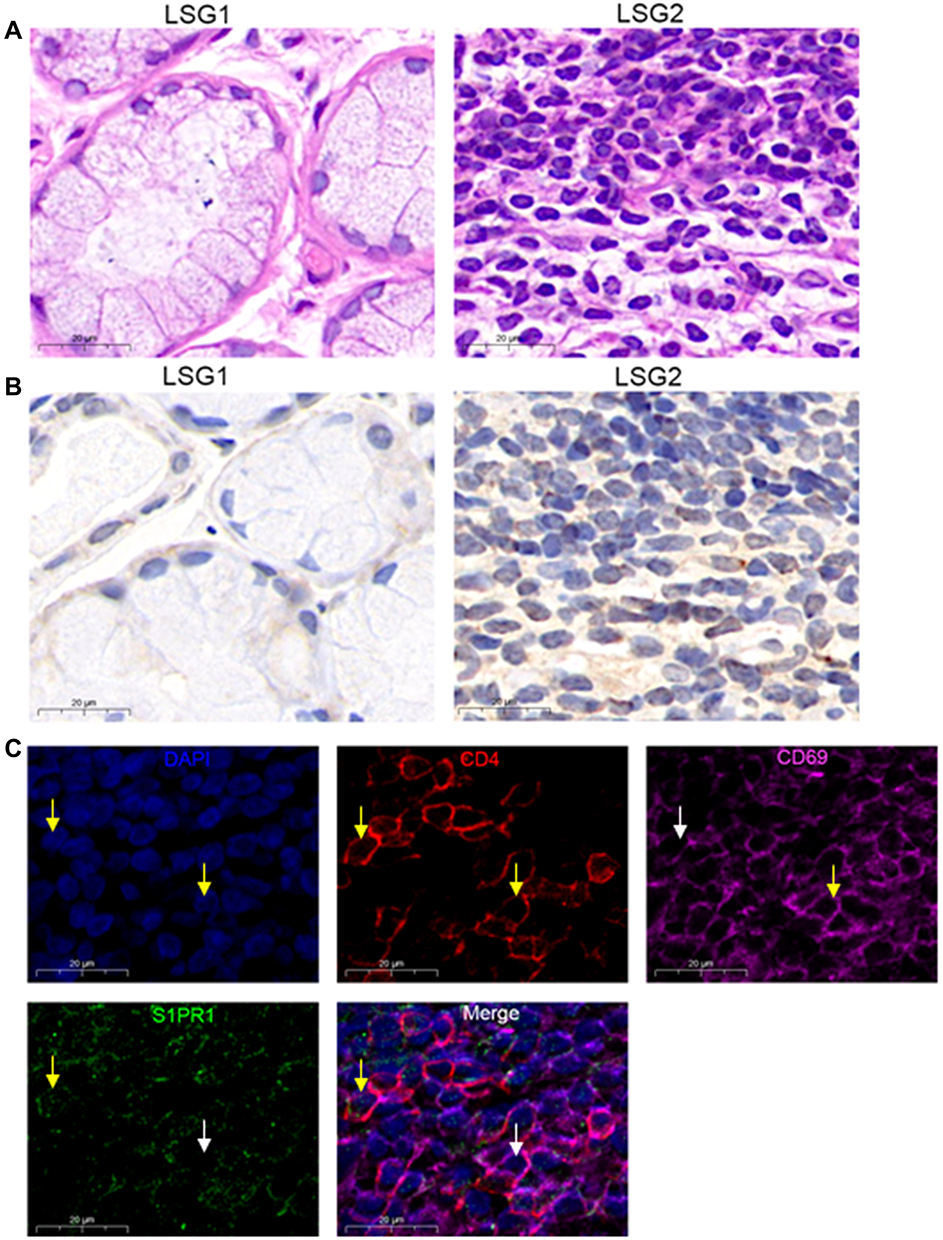 |
Figure 1 S1PR1 expression in CD4+CD69+ TRM cells. (A) Lymphocyte infiltration was detected by H&E staining of the labial salivary glands(LSG) of patients with pSS. (B) Expression of S1PR1 was detected by immunohistochemistry in labial salivary glands (LSG) of patients with pSS. (C) Immunofluorescence was performed to detect the expression of CD4, CD69 and S1PR1 in LSG samples with lymphocyte infiltration foci. Bar=20μm. DAPI was used to stain the nuclei and glowed blue. CD4 was pink light, CD69 was red light, and S1PR1 was green light. The yellow arrows point to positive fluorescence results and the white arrows point to negative fluorescence results in the DAPI, CD4, CD69 and S1PR1 image. In the in Merge image, the yellow arrows represent only S1PR1 expression in CD4+T cells and no surface expression of CD69, the white arrows point to the CD4+CD69+T cells without the expression of S1PR1. |
The Decreased Expression of Gene S1pr1 in CD4+T Cells of Patients with pSS
The peripheral blood CD4+T cells of healthy controls and pSS patients were selected by magnetic beads. The mRNA relative expression of S1pr1, Klf-2, and Cd69 were detected by qPCR. As shown in Figure 2A, the mRNA relative expression of S1pr1 in peripheral blood CD4+T cells of pSS patients was significantly lower than that of healthy controls. There was no significant difference in the gene expression of Klf-2 (Figure 2B) and Cd69 (Figure 2C) between healthy controls and pSS patients. In our study, the expression of S1pr1 in pSS-CD4+T cells was lower than that in Health-CD4+T cells, indicating that more lymphocytes in the blood of pSS patients may migrate to lymphatic organs or non-lymphoid tissue structures than that in healthy control.
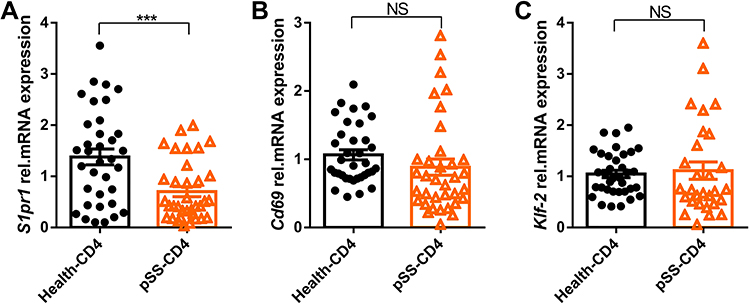 |
Figure 2 Gene S1pr1 is downregulated in pSS-CD4+T cells. The relative expression levels of gene S1pr1 (A), Klf-2 (B) and Cd69 (C) in peripheral blood CD4+T cells of healthy group (n=35) and pSS patients (n=35) were detected by Quantitative RT-PCR. Data represent the mean with SEM; ***P<0.001 by student-T test. Abbreviation: NS, not significant. |
Correlation Between S1pr1 Expression and Clinical Characteristics of Patients with pSS
Spearman correlation analysis showed that the relative expression of gene S1pr1 was positively correlated with the absolute percentage of basophil (r=0.3504, p=0.0391) (Figure 3D) and negatively correlated with C-reactive protein (CRP, r=−0.03535, p=0.0436) (Figure 3F). However, the data indicated that (Figure 3A–C, E and G–J) the expression of S1pr1 was not correlated with white blood cells (WBC), hemoglobin (HGB), ESR, IgG, IgM, IgA, lymphocyte percentage, and ESSDAI. Therefore, the correlation between the relative expression of S1pr1 and the serological characteristics of pSS was not high.
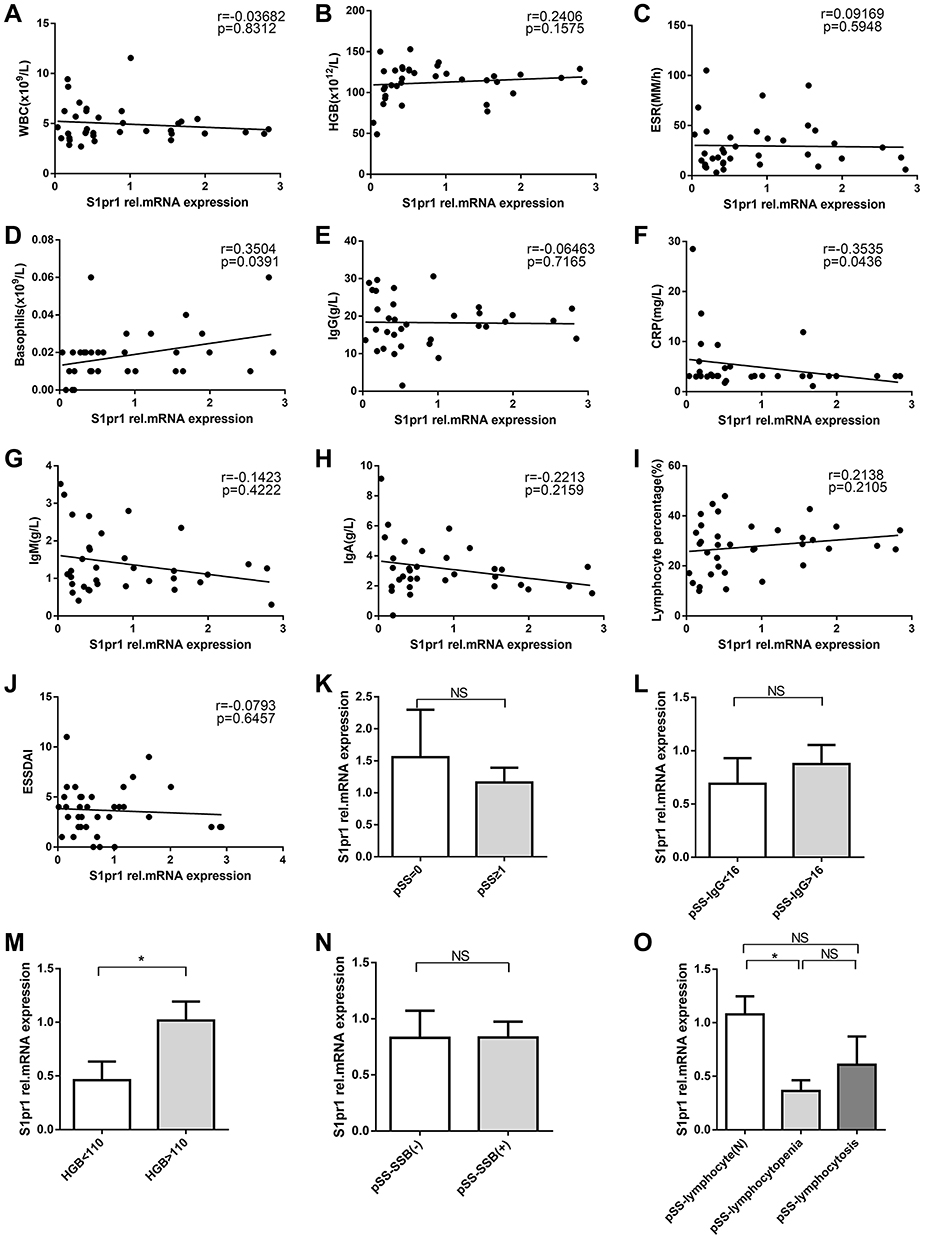 |
Figure 3 Correlation with clinical characteristics of patients with pSS. (A–J).Correlation analysis of relative expression level of gene S1pr1 and clinical indicators of pSS. (K).The expression of gene S1pr1 in peripheral blood CD4+T cells of patients with pSS in different pathological groups divided according to the number of lymphocytic foci. pSS=0 represented lymphocyte infiltration, but there is no formation of lymphocyte infiltration focus; pSS≥1 indicates at least one lymphocytic infiltrate of patients with pSS. (L–O).According to the clinical indicators of pSS, patients with pSS in the cohort were divided into groups to compare the relative expression of gene S1pr1 in peripheral blood CD4+T cells. Spearman test for correlation analysis. *P<0.05, by Student’s 2-tailed unpaired t-tests. Abbreviation: NS, not significant. |
However, according to the number of lymphocytic foci of LSG of patients with pSS, IgG, HGB, anti-SSB, lymphocyte percentage (Figure 3K–O), the patients with pSS were divided into different subgroups to compare the relative expression of S1pr1 mRNA in peripheral blood CD4+T cells and the results indicated that the expression of gene S1pr1 in peripheral blood CD4+T cells of pSS patients with lymphopenia and low hemoglobin was significantly lower than that of normal subgroup, and there was no significant difference in other subgroups. Lymphopenia is a marker of pSS disease activity.4 Therefore, the decreased expression of gene S1pr1 may affect cellular anemia and peripheral blood lymphopenia and participate in the development of pSS disease.
Downregulation of S1pr1/Klf-2 and Upregulation of Cd69 Expression in CD4+T Cells by Cytokines
We investigated whether these cytokines could induce the downregulation of S1pr1/Klf-2 in CD4+T cells to obtain CD4+CD69+ TRM cell phenotypes by co-culture with peripheral blood CD4+T cells from patients with pSS and healthy people selected by magnetic beads for 48 h in vitro.26,37 The results showed that TGF-β, IL-33, and TNF-α downregulated the expression of S1pr1 and Klf-2 and upregulated the expression of Cd69 in peripheral blood CD4+T cells of patients with pSS (Figure 4A–C) and healthy controls (Figure 4D–F) after coculture with cytokines in vitro. However, pSS patients had a higher gene expression of IL-33 (Figure 4H) and Tnf-α (Figure 4I), and lower S1pr1 gene expression than healthy controls, although the expression of gene Tgf-β was a significant decrease (Figure 4G).
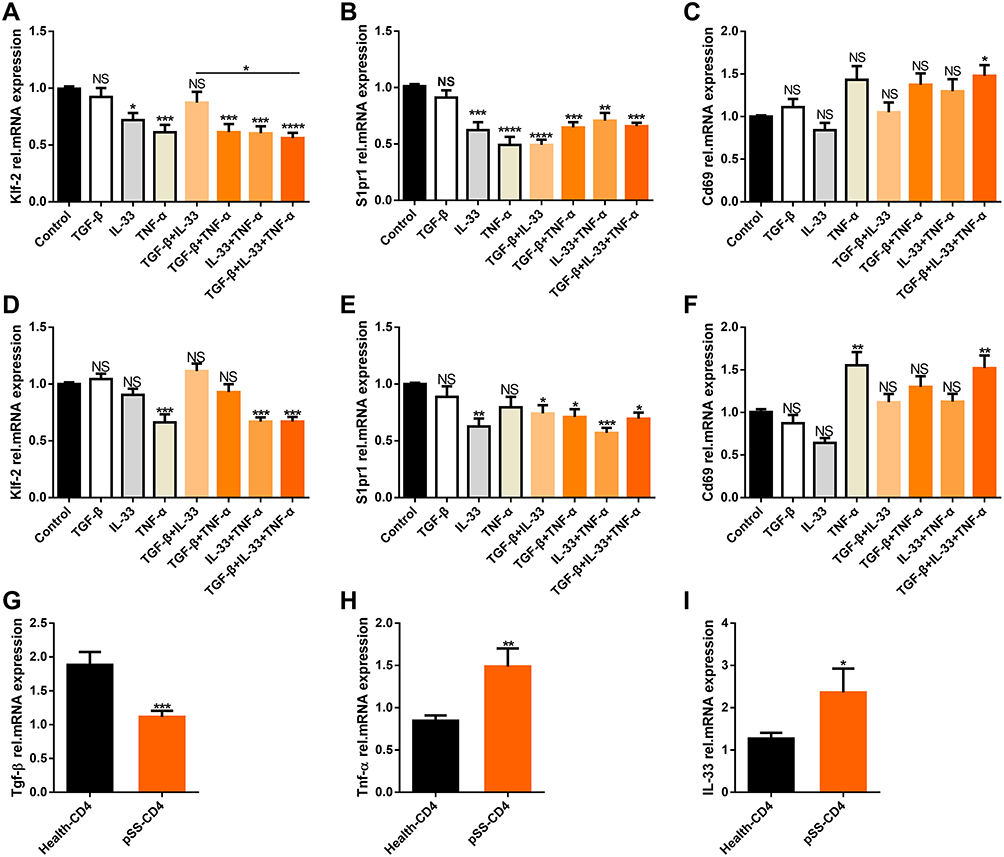 |
Figure 4 Cytokines induce downregulation of Klf2/S1pr1 in CD4+T cells and upregulate the expression of Cd69. Magnetic bead sorting (A–C) the CD4+T cells of peripheral blood of patients with pSS (n=13) and (D–F) healthy people (n=8), and cytokines TGF-β, IL-33 and TNF-α were co-cultured in vitro for 48 hours. The relative expression of genes Klf-2, S1pr1, Cd69 were detected by QPCR experiment. Magnetic beads were used to sort CD4+T cells from the peripheral blood of healthy people (n=35) and pSS patients (n=38) through QPCR to detect relative expression of genes Tgf-β (G) (P=0.0002), Tnf-α (H) (P=0.0057), IL-33 (I) (P=0.0175). Bars show the mean ± SEM in (A–I).*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by Dunnett’s multiple comparisons test or Student’s 2-tailed unpaired t-tests. Abbreviation: NS, not significant. |
Discussion
In rheumatoid arthritis, it has been reported that S1PR1 had pro-inflammatory effect in synovium and anti-inflammatory effect by inhibiting its expression.34 It was reported in the literature that S1P1 was expressed in the cytoplasm of inflammatory mononuclear cells, vascular endothelial cells, and salivary gland epithelial cells in LSG biopsy specimens. The expression of S1PR1 gradually increased with the increase in labial gland pathological grade, because the signal of S1PR1 and S1P was related to the apoptosis of epithelial cells in salivary gland.35 We also found that with the increase in lymphocyte infiltration, the expression of S1PR1 in pSS labial gland also showed an increasing trend. Result revealed that S1PR1 may participate in the development of inflammation in salivary glands of pSS.
In addition, S1PR1 was weakly or even not expressed in cytoplasm of CD4+CD69+TRM cells of the labial salivary glands of patients with pSS, because the expression of S1PR1 in CD4+T cells in salivary glands of patients with pSS was inhibited and degraded by CD69, resulting in the decrease in expression and delayed output of CD4+T cells. However, after several cycles of division, the expression of S1PR1 in newly generated T cells will increase to promote its output, but it will decrease in peripheral blood, so that cells can sense other signals to migrate from peripheral blood to other tissues. Our study also showed that the expression of S1pr1 in CD4+T cells in peripheral blood is significantly lower than that in healthy people. These results suggest that the migration of CD4+T cells is stronger in pSS patients.
The decrease in S1pr1 gene expression had no correlation with the percentage of peripheral blood lymphocytes in patients with pSS. However, when the pSS patients were grouped according to the percentage of peripheral blood lymphocytes, the results showed that compared with the normal lymphocyte percentage group, the expression of S1pr1 gene in pSS-CD4+T cells with lymphopenia was significantly reduced. This is similar to the significant reduction of S1pr1 gene expression in pSS-CD4+T cells. This suggests that decreased expression of S1pr1 gene may play a role in the reduction of lymphocytes in pSS blood. This is supported by literature that the decrease in the number of CD4+T cells in the blood of patients with pSS is associated with idiopathic CD4+T lymphopenia.38 CD4+T cell lymphopenia is a biological predictor of the development of non-Hodgkin’s malignant lymphoma.39 Studies have also shown that the decrease in the number of CD4+T cells in the blood is related to the increase in the number of lymphocytes in target tissues, such as the CD4+T cells in the minor salivary glands (MSGs).4 This supports the hypothesis that the reduction of CD4+T lymphocytes in the blood is due to the migration of CD4+T cells to inflammatory tissues.11 This hypothesis is consistent with our finding that the expression of the S1pr1 gene in pSS-CD4+T cells is reduced, and the CD4+T lymphocytes in the blood tend to migrate to inflammatory tissue.
Skon et al showed that downregulation of Klf-2 and S1pr1 expression played an important role in the tissue residence and development of CD8+TRM cells, in which the cytokines TGF-β, IL-33 and TNF-α together lead to significant downregulation of Klf-2.26 These cytokine combinations have been shown to promote putative tissue-resident precursor cells to acquire tissue resident cell phenotypic characteristics, such as up-regulation of CD103 expression. CD103 is an integrin that allows tissue-resident cells to persist in non-lymphoid organs.40,41 Therefore, the cytokine microenvironment may cultivate the development of tissue resident cells by guiding the transcriptional profile of tissue resident precursor cells.37 In our study, peripheral blood CD4+T cells of pSS patients were co-cultured with cytokines TGF-β, IL-33 and TNF-α in vitro due to the small labial gland tissue and difficulty in obtaining it. The results showed that TGF-β, IL-33 and TNF-α induced downregulation of Klf-2 and S1pr1 expression and upregulation of Cd69 expression in peripheral blood CD4+T cells of pSS patients and healthy people after coculture with cytokines in vitro. However, the expression of IL-33 and Tnf-α genes is higher, and the expression of S1pr1 gene is lower in pSS patients. This seems to be similar to the molecular mechanism of activated T cell and cytokine in vitro culture to obtain tissue-resident phenotype. Therefore, it can be concluded that the cytokines TGF-β, IL-33, and TNF-α can cause the downregulation of Klf-2 and S1pr1 expression and the upregulation of Cd69 expression in CD4+T cells at the gene level, and CD4+T cells have the tendency of tissue retention and development at the gene level. According to existing research reports, TGF-β, IL-33, and TNF-α cytokines are expressed in the salivary glands of pSS patients, and they play an important role in the development of pSS.42–51 Studies have reported that IL-33 levels were significantly increased in the serum of patients with pSS, and IL-33 was detected in the acinar cells of the salivary glands of patients with pSS and some cells infiltrating the connective tissue.42 IL-33 was released by endothelial cells and activated mast cells after cell injury in the acute phase of pSS, participates in the polarization of T cells to T-helper 2(Th2) cells and induces some adaptive immune Th2 cytokines, which play a pro-inflammatory effect.43,44 IL-33 together with IL-23 and IL-12 induce the secretion of interferon gamma (IFN-γ). IFN-γ is a typical T-helper 1(Th1) cytokine. IFN-γ and TNF-α are mainly secreted by infiltrating Th1 cells and CD8+T cells in the LSG of pSS patients, and are involved in gland damage and secretory dysfunction.22,42 In addition, overexpression of TNF-α not only cause inflammation but also cause acinar cell atrophy.45 According to a previous report, TNF-α induced an increase in the expression of Ro/SSA and La/SSB on the surface of human keratinocytes, which may cause apoptosis and impaired secretion of SGs in Sjogren’s syndrome (SS).52 The expression level of TGF-β in the serum, salivary glands and saliva of pSS patients was increased,47 and it cooperated with IL-6 to mediate the differentiation of helper Th17 cells to activate the pro-inflammatory response.48 However, TGF-β has a dose-dependent pleiotropic effect in the process of immune regulation. The pro-inflammatory or anti-inflammatory effect of TGF-β in the pathogenesis of pSS still needs further research. Nevertheless, in vitro experiments have shown that the fibrosis of pSS minor salivary glands is mediated by TGF-β signaling, and inflammation at the lymphocyte infiltration site stimulates TGF-β1-mediated epithelial-mesenchymal transition, which causes fibrosis near lymphocyte aggregation.49–51 There is a molecular basis for the hypothesis that CD4+CD69+TRM progener cells in labial gland tissues of pSS patients develop into CD4+CD69+TRM cells through down-regulation of Klf-2 and S1pr1 expression and up-regulation of Cd69 expression in labial gland tissues caused by the interaction of cytokines TGF-β, IL-33 and TNF-α. The temporal and spatial relationship between the residence and development of CD4+CD69+TRM cells in the tissues and the expression of TGF-β, TNF-α and IL-33 in the labial glands of pSS patients still needs further research.
In conclusion, our study suggests that the expression of S1PR1 in CD4+CD69+TRM cells in labial glands of pSS patients was weak, and the decrease of S1pr1 gene expression may provide a molecular basis for promoting the tissue retention and development of CD4 +CD69+TRM cells.
Acknowledgment
This work was supported by key research and development projects of Anhui Province (1804b06020354) and the National Natural Science Foundation of China (81871271).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Borchers AT, Naguwa SM, Keen CL, et al. Immunopathogenesis of Sjögren’s syndrome. Clin Rev Allergy Immunol. 2003;25(1):89–104. doi:10.1385/CRIAI:25:1:89
2. Peri Y, Agmon-Levin N, Theodor E, et al. Sjögren’s syndrome, the old and the new. Best practice & research. Clin Rheumatol. 2012;26(1):105–117.
3. Gao C-Y, Yao Y, Li L, et al. Tissue-Resident Memory CD 8+ T Cells Acting as Mediators of Salivary Gland Damage in a Murine Model of Sjögren’s Syndrome. Arthritis Rheumatol. 2019;71(1):121–132. doi:10.1002/art.40676
4. Mingueneau M, Boudaoud S, Haskett S, et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjögren’s signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol. 2016;137(6):1809–1821.e12. doi:10.1016/j.jaci.2016.01.024
5. Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat Rev Rheumatol. 2013;9(9):544–556. doi:10.1038/nrrheum.2013.110
6. Qin B, Wang J, Yang Z, et al. Epidemiology of primary Sjögren’s syndrome: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(11):1983–1989. doi:10.1136/annrheumdis-2014-205375
7. Mariette X, Criswell LA. Primary Sjögren’s Syndrome. N Engl J Med. 2018;378(10):931–939. doi:10.1056/NEJMcp1702514
8. Chatzis L, Pezoulas VC, Ferro F, et al. Sjögren’s syndrome: the clinical spectrum of male patients. J Clin Med. 2020;9(8):2620. doi:10.3390/jcm9082620
9. Stefanski A-L, Tomiak C, Pleyer U, et al. The diagnosis and treatment of Sjögren’s syndrome. Dtsch Arztebl Int. 2017;114(20):354–361.
10. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos.Characteristics HM. of the minor salivary gland infiltrates in Sjögren’s syndrome. J Autoimmun. 2010;34(4):400–407. doi:10.1016/j.jaut.2009.10.004
11. Verstappen GM, Kroese FGM, Bootsma H. T cells in primary Sjögren’s syndrome: targets for early intervention. Rheumatology. 2019; 60(7):3088.
12. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41(6):886–897. doi:10.1016/j.immuni.2014.12.007
13. Zundler S, Becker E, Spocinska M, et al. Hobit- and Blimp-1-driven CD4 tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol. 2019;20(3):288–300. doi:10.1038/s41590-018-0298-5
14. Teijaro JR, Turner D, Pham Q, et al. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187(11):5510–5514. doi:10.4049/jimmunol.1102243
15. Turner DL, Bickham KL, Thome JJ, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014;7(3):501–510. doi:10.1038/mi.2013.67
16. Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. 2014;346(6205):93–98. doi:10.1126/science.1257530
17. Smith CJ, Caldeira-Dantas S, Turula H, et al. Murine CMV infection induces the continuous production of mucosal resident T cells. Cell Rep. 2015;13(6):1137–1148. doi:10.1016/j.celrep.2015.09.076
18. Thom JT, Weber TC, Walton SM, et al. The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep. 2015;13(6):1125–1136. doi:10.1016/j.celrep.2015.09.082
19. Collins N, Jiang X, Zaid A, et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun. 2016;7:11514. doi:10.1038/ncomms11514
20. Glennie ND, Yeramilli VA, Beiting DP, et al. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212(9):1405–1414. doi:10.1084/jem.20142101
21. Mackay LK, Kallies A. Transcriptional Regulation of tissue-resident lymphocytes. Trends Immunol. 2017;38(2):94–103. doi:10.1016/j.it.2016.11.004
22. Yao Y, Ma J-F, Chang C, et al. Immunobiology of T Cells in Sjögren’s Syndrome. Clin Rev Allergy Immunol. 2021;60(1):111–131. doi:10.1007/s12016-020-08793-7
23. Kumar BV, Ma W, Miron M, et al. Human Tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20(12):2921–2934. doi:10.1016/j.celrep.2017.08.078
24. Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi:10.1038/nature02284
25. Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11(6):403–415. doi:10.1038/nri2974
26. Skon CN, Lee J-Y, Anderson KG, et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14(12):1285–1293. doi:10.1038/ni.2745
27. Xiao L, Zhou Y, Friis T, et al. S1P-S1PR1 Signaling: the “Sphinx” in Osteoimmunology. Front Immunol. 2019;10:1409. doi:10.3389/fimmu.2019.01409
28. Tsai H-C, Nguyen K, Hashemi E, et al. Myeloid sphingosine-1-phosphate receptor 1 is important for CNS autoimmunity and neuroinflammation. J Autoimmun. 2019;105:102290. doi:10.1016/j.jaut.2019.06.001
29. Lankadasari MB, Aparna JS, Mohammed S, et al. Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine. Theranostics. 2018;8(14):3824–3840. doi:10.7150/thno.25308
30. Lin Q, Ren L, Jian M, et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid-derived suppressor cells induced by the S1PR1-STAT3 signaling pathway. Cell Death Dis. 2019;10(10):693. doi:10.1038/s41419-019-1922-5
31. Kappos L, Radue E-W, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi:10.1056/NEJMoa0909494
32. Doolen S, Iannitti T, Donahue RR, et al. Fingolimod reduces neuropathic pain behaviors in a mouse model of multiple sclerosis by a sphingosine-1 phosphate receptor 1-dependent inhibition of central sensitization in the dorsal horn. Pain. 2018;159(2):224–238. doi:10.1097/j.pain.0000000000001106
33. Nielsen OH, Li Y, Johansson-Lindbom B, et al. Sphingosine-1-phosphate signaling in inflammatory bowel disease. Trends Mol Med. 2017;23(4):362–374. doi:10.1016/j.molmed.2017.02.002
34. Wang R-H, Dai X-J, Wu H, et al. Anti-Inflammatory Effect of Geniposide on Regulating the Functions of Rheumatoid Arthritis Synovial Fibroblasts via Inhibiting Sphingosine-1-Phosphate Receptors1/3 Coupling Gαi/Gαs Conversion. Front Pharmacol. 2020;11:584176. doi:10.3389/fphar.2020.584176
35. Sekiguchi M, Iwasaki T, Kitano M, et al. Role of sphingosine 1-phosphate in the pathogenesis of Sjögren’s syndrome. J Immunol. 2008;180(3):1921–1928. doi:10.4049/jimmunol.180.3.1921
36. Ramos-Casals M, Font J. Primary Sjögren’s syndrome: current and emergent aetiopathogenic concepts. Rheumatology. 2005;44(11):1354–1367. doi:10.1093/rheumatology/keh714
37. Zajac AJ, Harrington LE. Tissue-resident T cells lose their S1P1 exit visas. Cell Mol Immunol. 2014;11(3):221–223. doi:10.1038/cmi.2014.7
38. Mandl T, Bredberg A, Jacobsson LT, et al. CD4+ T-lymphocytopenia–a frequent finding in anti-SSA antibody seropositive patients with primary Sjögren’s syndrome. J Rheumatol. 2004;31(4):726–728.
39. Nocturne G, Mariette X. Sjögren Syndrome-associated lymphomas: an update on pathogenesis and management. Br J Haematol. 2015;168(3):317–327. doi:10.1111/bjh.13192
40. Casey KA, Fraser KA, Schenkel JM, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188(10):4866–4875. doi:10.4049/jimmunol.1200402
41. Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–1301. doi:10.1038/ni.2744
42. Awada A, Nicaise C, Ena S, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary Sjogren’s syndrome. Ann Rheum Dis. 2014;73(6):1259–1263. doi:10.1136/annrheumdis-2012-203187
43. Conti P, Stellin L, Caraffa A, et al. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: promising Inhibitory Effect of IL-37. Int J Mol Sci. 2020;21(12):12. doi:10.3390/ijms21124297
44. McCarthy PC, Phair IR, Greger C, et al. IL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol Cell Biol. 2019;97(1):54–71. doi:10.1111/imcb.12200
45. Limaye A, Hall BE, Zhang L, et al. Targeted TNF-α overexpression drives salivary gland inflammation. J Dent Res. 2019;98(6):713–719. doi:10.1177/0022034519837240
46. Patrussi L, Capitani N, Martini V, et al. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res. 2015;75(19):4153–4163. doi:10.1158/0008-5472.CAN-15-0986
47. Katsifis GE, Rekka S, Moutsopoulos NM, et al. Systemic and local interleukin-17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am J Pathol. 2009;175(3):1167–1177. doi:10.2353/ajpath.2009.090319
48. Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi:10.1038/nature04753
49. Sisto M, Lorusso L, Tamma R, et al. Interleukin-17 and −22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren’s syndrome. Clin Exp Immunol. 2019;198(2):261–272. doi:10.1111/cei.13337
50. Sisto M, Lorusso L, Ingravallo G, et al. The TGF-β 1 Signaling pathway as an attractive target in the fibrosis pathogenesis of Sjögren’s syndrome. Mediators Inflamm. 2018;2018:1965935. doi:10.1155/2018/1965935
51. Leehan KM, Pezant NP, Rasmussen A, et al. Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin Exp Rheumatol. 2018;36 Suppl 112(3):80–88.
52. Dörner T, Hucko M, Mayet WJ, et al. Enhanced membrane expression of the 52 kDa Ro(SS-A) and La(SS-B) antigens by human keratinocytes induced by TNF alpha. Ann Rheum Dis. 1995;54(11):904–909. doi:10.1136/ard.54.11.904
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.