Back to Journals » The Application of Clinical Genetics » Volume 15
Molecular Genetic Screening of Neonatal Intensive Care Units: Hyperbilirubinemia as an Example
Authors Yang Y, Wang Y, Zhou L, Long W ![]() , Yu B
, Yu B ![]() , Wang H
, Wang H ![]()
Received 15 February 2022
Accepted for publication 10 May 2022
Published 18 May 2022 Volume 2022:15 Pages 39—48
DOI https://doi.org/10.2147/TACG.S362148
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Yuqi Yang,1,* Yu Wang,2,* Lingna Zhou,1 Wei Long,2 Bin Yu,1 Huaiyan Wang2
1Department of Medical Genetics, Changzhou Maternal and Child Health Care Hospital, Changzhou, Jiangsu Province, People’s Republic of China; 2Department of Neonatology, Changzhou Maternal and Child Health Care Hospital, Changzhou, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bin Yu; Huaiyan Wang, Email [email protected]; [email protected]
Objective: To explore the clinical value of newborn genomic screening (nGS) for neonatal intensive care units (NICU) infants (taking neonatal hyperbilirubinemia as an example).
Methods: Dried blood spots (DBSs) were collected after 72 hours of birth. The tandem mass spectrometry (TMS) screening and Angel Care genomic screening (GS, based on Targeted next-generation sequencing) were performed at the same time.
Results: Ninety-six hyperbilirubinemia newborns were enrolled in this study and none was identified with inborn errors of metabolism (IEM) by TMS, while 6 infants (6.25%, 6/96) were suspected to have a genetic disorder by Angel Care, including 2 cases of glucose-6-phosphate dehydrogenase deficiency (G6PD), and 1 case of maple syrup urine disease type 1B (MSUD1B), autosomal recessive deafness 1A (DFNB1A), Leber hereditary optic neuropathy (LHON), thyroid dyshormonogenesis 6 (TDH6) each. In addition, 44 infants (45.8%) were detected having at least one variant which conferred a carrier status for a recessive childhood-onset disorder. A total of 33 out of 60 variants (55.0%) reported for carrier status were pathogenic (P), 24 (40.0%) were likely pathogenic (LP), and 3 variants were variant of uncertain significance (VUS). Top six common genes of carrier status were GJB2, DUOX2, PRODH, ATP7B, SLC12A3, SLC26A4. Two newborns showed abnormalities in elementary screening of TMS, but were confirmed as false positive after recall. Their results of Angel Care did not found abnormality.
Conclusion: Using neonatal hyperbilirubinemia as an example, genome sequencing screening can find more evidence of genetic variation in NICU newborns, and “Angel Care” is an effective method.
Keywords: neonatal intensive care units, neonatal hyperbilirubinemia, newborn screening, genomic sequencing, next-generation sequencing
Introduction
Recently, newborn genomic sequencing (nGS) brings new opportunities to further expand newborn screening (NBS), which is an important public health project.1 From 2013, BabySeq,2 NBSeq,3 NC NEXUS,4 STATseq,5 NESTS6 and NeoSeq7 successively confirmed that nGS could further expand the genetic diseases that could not be found by traditional screening methods and was considered as another innovation in the field of NBS. However, there are still many problems to be discussed and solved before large-scale clinical practice. Among them, the applicable population is one of the focuses of discussion. What is the reasonable population for nGS? Is the high-risk population (such as neonatal intensive care units (NICU)8) or a general newborn population?2 Some authoritative and representative projects include the BabySeq2,9 Project and NC NEXUS.4 Their main subjects range is from ill newborns of the NICU to healthy newborns. For example, Ceyhan-Birsoy et al2 reported the results of 159 newborns (32 from NICU and 127 healthy newborns). Fifteen newborns were identified with variants that conferred disease risk. Among them, 10 (7.87%) cases were from the healthy newborns and 5 cases (14.29%) in the NICU. These projects indicate that newborns from NICU usually have a higher risk of genetic variation and successfully demonstrate the advantages and prospects of nGS in newborn screening.
It is known that newborns of NICU are involved in many diseases, and their causes are also complicated. The current studies about nGS are still limited. A full understanding of the impact of nGS application in NICU common diseases will help us to determine the strategy of NBS. In NICU, jaundice is the most common abnormal finding in neonates, of which the etiology can be physiological or pathological.10 Without standardized monitoring and effective treatment, jaundice may rapidly progress to severe hyperbilirubinemia. Neonatal hyperbilirubinemia is a major issue worldwide.11 Untreated severe hyperbilirubinemia often leads to other critical conditions, such as acute bilirubin encephalopathy (ABE) and nuclear jaundice (kernicterus),12 resulting in permanent damage to the central nervous system, especially hand-foot hyperactivity or dystonic paralysis.
So far, the exact pathogenesis of neonatal hyperbilirubinemia is not fully understood in the majority of cases. It has been confirmed that its etiology was multifactorial and known causes included ABO incompatibility, glucose-6-phosphate dehydrogenase deficiency (G6PD), other antibody incompatibility and hereditary spherocytosis.13 Recently, more and more medical scholars agreed that both genetic and environmental factors are involved in the occurrence and development of hyperbilirubinemia.14 Increasing studies also suggest that hyperbilirubinemia could be caused by polymorphisms and mutations of genes. At present, a majority of research is focused on related pathogenic genes, mainly including uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1), organic anion transporter polypeptide 2 (OATP2), heme oxygenase 1 (HO-1), and biliverdin reductase A (BLVRA). UGT1A1 is the most studied gene, and more than 150 mutations have been reported. Zhou et al15 identified the genetic risk factors for neonatal hyperbilirubinemia in a case–control study. They reported that G211 mutation in UGT1A1 and GT repeat in HO-1 were the main risk factors for neonatal hyperbilirubinemia in Southeastern China. Wu et al16 also found that UGT1A1 211G>A (G211A) mutation was a risk factor in Chinese Han populations, but (TA)7 insertion mutation was not. The incidence rates of homozygous, heterozygous, and wild-type variation at UGT1A1 211G>A were 3.7%, 27.1%, and 66.1% in the infants with hyperbilirubinemia, respectively; however, in the control group, the incidence rates were 0, 16.3% and 83.7%, respectively. Similar results have been demonstrated in population studies from other regions,17,18 although differences remain.19,20 For instance, SLCO1B1 polymorphism, a gene encoding OATP2, is associated with jaundice, but such association varies across regions and populations. For the Chinese population, 388G>A and 521T>C mutations of SLCO1B1 are associated with an increased and decreased risk of neonatal jaundice, respectively,21,22 but no such correlation is found in Indonesia,23 white, Thai, Brazilian or Malaysian populations.24 Some studies have shown that HO-1 gene polymorphism is associated with the risk of jaundice.15,25 Although these studies have revealed the etiological complexity of neonatal hyperbilirubinemia, our understanding of its genetic characteristics is still considered limited. With the development of molecular biology technologies, more and more related candidate genes will be identified, which will provide new insights into the pathogenesis and treatment of hyperbilirubinemia.
Nevertheless, for neonatal hyperbilirubinemia, genomic sequencing is not normally performed on the dried blood spots (DBSs) in patient NICU; rather targeted variant detection of specific genes is adopted with whole-genome sequencing (WGS), exome sequencing (ES) or gene panel sequencing. These methods have great technical advantages and contributed to discover the pathogenic genes of neonatal jaundice. However, they also have many limitations such as complex experimentation, difficulty in standardization, and high cost.
Here, we report on the initiation of a new project called “Angel Care”, which was based on the next-generation sequencing (NGS) technology and can be applied to genomic sequencing in newborn screening with DBSs for critical conditions. The project involved target capture sequencing of 159 disease-related genes with a sequencing depth of >100X. In relation to other newborn screening strategies, the project demonstrates the following benefits of utilizing NGS: 1) easy-to-operate experimental process; 2) straightforward interpretation of results; 3) well-controlled cost. Until now, how to effectively carry out nGS is not very clear. It is still debatable which newborns are suitable for nGS. Therefore, similar to the Babyseq project, we firstly carried out the clinical study of high-risk groups (neonatal hyperbilirubinemia in NICU as an example) receiving Angel Care. We hope to provide an effective method of nGS for NICU infants. By the way, Angel Care is not specifically developed for neonatal hyperbilirubinemia, and the purpose is not for specific populations. We took neonatal hyperbilirubinemia as an example to show the necessity of nGS in NICU.
Materials and Methods
Study Design and Participants
This was a retrospective study conducted at the Department of Medical Genetics, Changzhou Maternal and Child Health Care Hospital. From January 2019 to June 2020, 96 hyperbilirubinemia newborns were enrolled in our study, including 53 males and 43 females. Of them, 13 cases were ABO incompatibility.26 The average birth weight and gestational week of delivery were 3349.60 ± 450.60g and 38.37 ± 1.43 weeks, respectively. According to the level of total serum bilirubin (TSB),27 82 (85.4%) cases were defined as severe hyperbilirubinemia (>342 µmol/L), and 7 cases as extreme hyperbilirubinemia (>427 µmol/L) and 2 cases as hazardous hyperbilirubinemia (>510 µmol/L). Infants with a birth weight lower than 2000g and/or gestational age <35 weeks, and infants with other congenital malformations were excluded from the study. The baseline demographic and clinical characteristics are shown in Table 1.
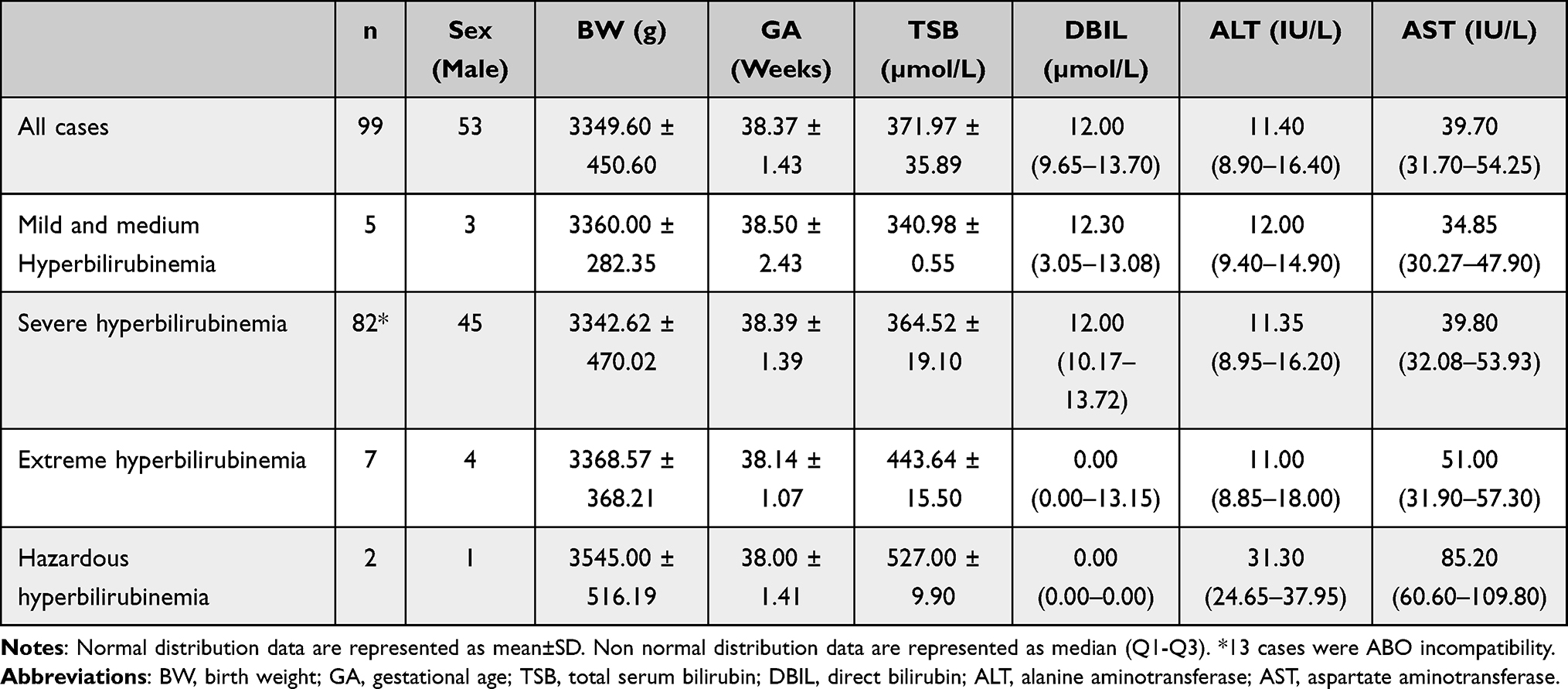 |
Table 1 Baseline Demographic and Clinical Characteristics |
Newborn Screening with Tandem Mass Spectrometry
As described in our previous study,28 dried blood spots (DBSs) were collected from all infants on a 903 filter paper (Wallace Oy, Turku, Finland) after 72 hours of birth. All DBSs were analyzed with MS/MS using the NeoBase™ Non-derivatized MS/MS Kit (PerkinElmer, Turku, Finland). Infants with positive results were brought in for further assessment, including clinical manifestations, individualized assistant examination, and gene testing.
Angel Care Panel Design
The Angel Care panel (BGI) covered the coding sequence of 159 disease-associated genes, including inherited metabolic disorders (eg, amino acid, organic acid and fatty acid metabolism disorders, lysosomal storage diseases and glucose metabolism disorders), endocrine disorders, hearing loss, neuromuscular disorders, hematologic disorders, and immune disorders (Supplementary Table 1).
Targeted Next-Generation Sequencing
A standard procedure of genomic DNA extraction was performed with MagPure Blood & Tissue DNA KF Kit B (Magen, China). The extracted DNA was fragmented by restriction enzyme (Universal Plus Fragmentation Module, VAHTS), followed by magnetic-bead sieving of 200–250bp fragments, adapter ligation, amplification by PCR and eventually purification to construct a DNA library for each sample. After the concentration level of these libraries was verified with a multimode microplate reader (FLUOstar, Omega), IDT xGen Lockdown probes were used to capture the targeted regions of the sequences through hybridization, followed by library pooling and quantification. Single-strand circularization and rolling-circle replication were carried out in the pooled library. Finally, after DNA-nanoball preparation, sequencing was performed with MGISEQ-2000 (PE100+10) to generate raw sequencing data.
Data Analysis
Sequencing data were analyzed with an automated pipeline (BGI). Quality filtering reads were aligned to the human reference genome (GRCh37/hg19) using Burrows-Wheeler Aligner (BWA) software, and SNV and small indels less than 20 bp were called by Genome Analysis Toolkit (GATK). Copy number variants (CNVs) of DMD exons, common CNVs related to thalassemia and SMN1 exon 7 deletions of were detected via read-depth analysis developed by BGI.29
The reported variants included 1) 10,136 variants recorded in a neonate-specific database (V2021.6, BGIPhoenix Database), including pathogenic and likely pathogenic variants classified according to ACMG standards and guidelines for the interpretation of sequence variants; 2) null variants with low frequency not recorded in the database (null variants refer to frameshift, stop-loss and splice donor/acceptor variants as well as variants with a loss of ≥2 exons; rare variants refer to variants with a frequency of ≤1% in both GnomAD and 1000G databases).
When the reported variants in a sample followed the specific inheritance pattern of related disorders which suggested an increased risk in the subject, the sample was classified as suspicious in genetic screening: one or more heterozygous variants in autosomal dominant disorders; homozygous variants or 2 heterozygous variants in trans in autosomal recessive disorders; hemizygous or heterozygous variants in X-linked dominant disorders, hemizygous, homozygous variants or 2 heterozygous variants in trans in X-linked recessive disorders; variants in mitochondrial disorders. If the reported variants did not follow a specific inheritance pattern, the sample was classified as negative in genetic screening.
Results
A total of 96 subjects fulfilled our criteria and were enrolled in this study, and they accepted Angel Care and TMS newborn screening at 72 hours after birth. No infant was identified with inborn errors of metabolism (IEM) by TMS, while 6 infants (6.25%, 6/96) were suspected to have a genetic disorder by Angel Care. Meanwhile, 45.8% (44/96) of participants were detected as a carrier status for a genetic disorder.
Of 96 hyperbilirubinemia newborns, 6 were identified with pathogenic variants in disease-related genes by Angel Care screening; however, no observable abnormality was found in their TMS results. Five genetic disorders were subsequently confirmed in these 6 infants: 2 cases of Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD), and 1 case in each of Maple Syrup Urine Disease Type 1B (MSUD1B), Autosomal Recessive Deafness 1A (DFNB1A), Leber Hereditary Optic Neuropathy (LHON), thyroid dyshormonogenesis 6 (TDH6) (Table 2). Case 1 (female) and case 3 (female) were confirmed with G6PD and a heterogynous likely pathogenic variant (c.1388G>A and c.1024C>T) was detected in G6PD gene. Their clinical phenotype was analogous and mild. They were born in normal gestational weeks (38 weeks) and their weight was normal (2770g and 2680g) at birth. These 2 female infants developed jaundice on 3 days after birth which gradually aggravated. Their TSB levels were 366.4μmol/L and 443.5μmol/L. Both have achieved satisfactory outcomes after treatment, and the phototherapy time was 42.5h and 50.0h. Case 1 has been followed up for 12 months. Her indicators were normal (height: 74.7cm; weight: 9kg; head circumference: 44cm) and her hemoglobin level is 135g/L. Within the follow-up period of 6 months, case 3 did not show any developmental anomalies. One infant (case 2) diagnosed with Autosomal Recessive Deafness 1A (DFNB1A), was found to carry two pathogenic mutations (c.109G>A, c.95G>A) in GJB2 gene by Angel Care. She also began to jaundice on 3 days after birth, and her TSB level was 395.8μmol/L at admission. She also presented mild intraventricular hemorrhage. After 4 times of phototherapy (75h), she achieved significant improvement. In addition, she received bilateral brainstem auditory-evoked potentials, and both ears passed the auditory test. Unfortunately, she lost follow-up due to the floating population. One infant was diagnosed as having Maple Syrup Urine Disease Type 1B (MSUD1B) by Angel Care and was heterozygous for a variant of uncertain significance (c.1159C>T) and a deletion of 7–9 exons in BCKDHB gene. He developed jaundice on 2 days after birth and the TSB levels were 358.5μmol/L. He had no observable clinical manifestations such as vomiting or feeding difficulties. His leucine (LEU+ILE+PRO-OH) and valine (VAL) levels were 135.15μmol/L and 134.0μmol/L in TMS first screening, which did not meet the criteria of MSUD1B. The boy was born full-term (40 weeks) and his birth weight was 3360g. After 48 hours of phototherapy, hyperbilirubinemia was well controlled. He has been followed up for 8 months, and there was no obvious abnormality in his growth and development, and his latest hemoglobin level was 130 g/L. Similarly, case 4 and case 6 were diagnosed with LHON and TDH6, respectively. However, there was no significant clinical manifestation related to the disorders. The therapeutic outcomes for hyperbilirubinemia were also satisfactory in case 4 and case 6.
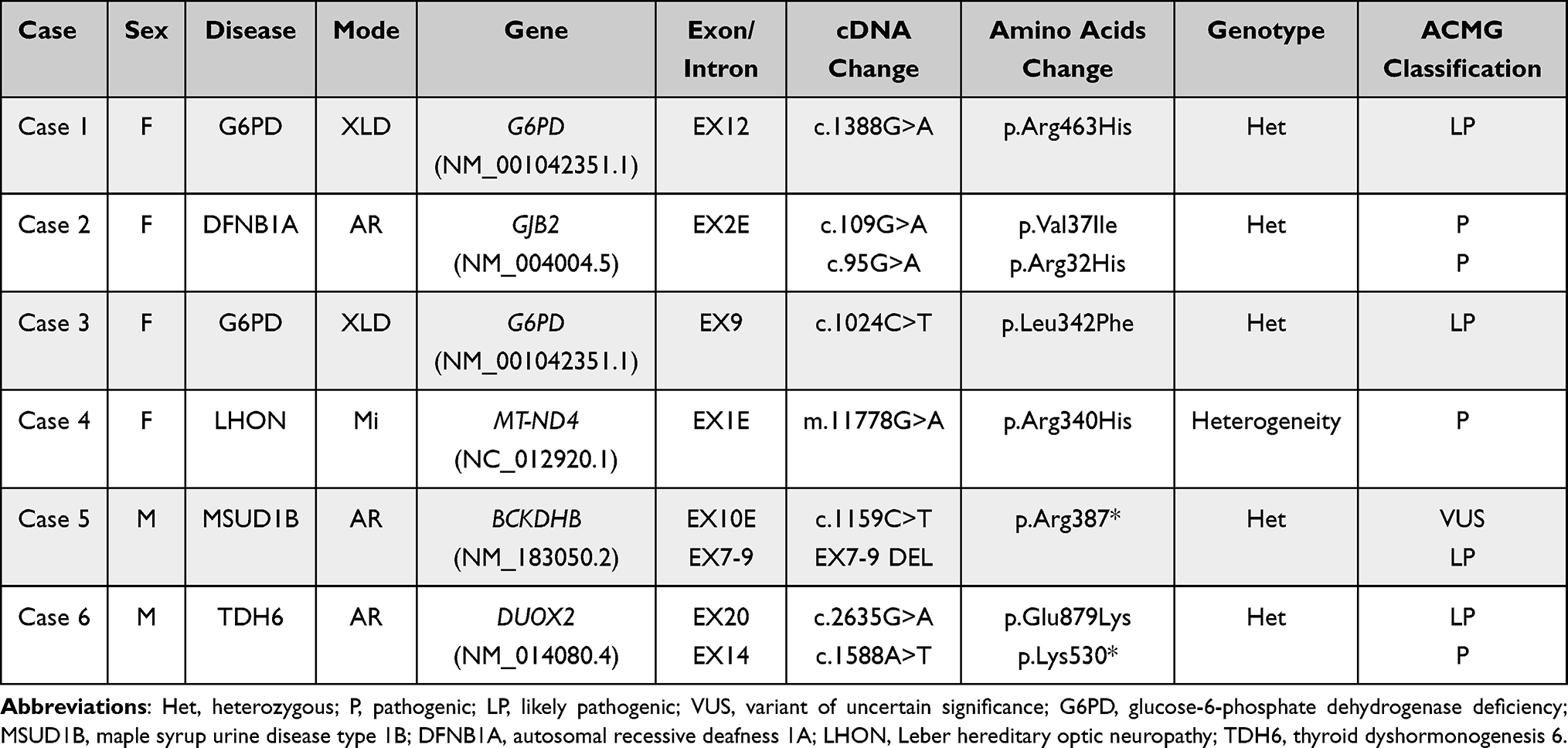 |
Table 2 Genetic Test Results of 6 Positive Cases |
In addition, 44 infants were detected having at least one variant, which confers a carrier status for a recessive childhood-onset disorder. The detection rate for carrier-status is 45.8%. The number of carrier findings in hyperbilirubinemia newborns range from 1 to 4 variants, and the median is 1.4 variants per individual. There was one variant in 30 infants (68.2%) and two variants in 13 infants (29.5%). A total of 32 genes and 60 variants were reported in this study. According to the American College of Medical Genetics and Genomics guidelines,30 33 (55.0%) out of 60 variants reported for carrier status are pathogenic (P), 24 (40.0%) are likely pathogenic (LP), and 3 variants are variants of uncertain significance (VUS). The top six common genes and variants of carrier status are listed in Table 3 and Figure 1. Six individual genes and thirteen specific variants were detected in more than three infants. The most frequently identified variant was the c.109G>A (p.Val37Ile) variant in GJB2 gene. Surprisingly, all 9 newborns detected carried the same variant.
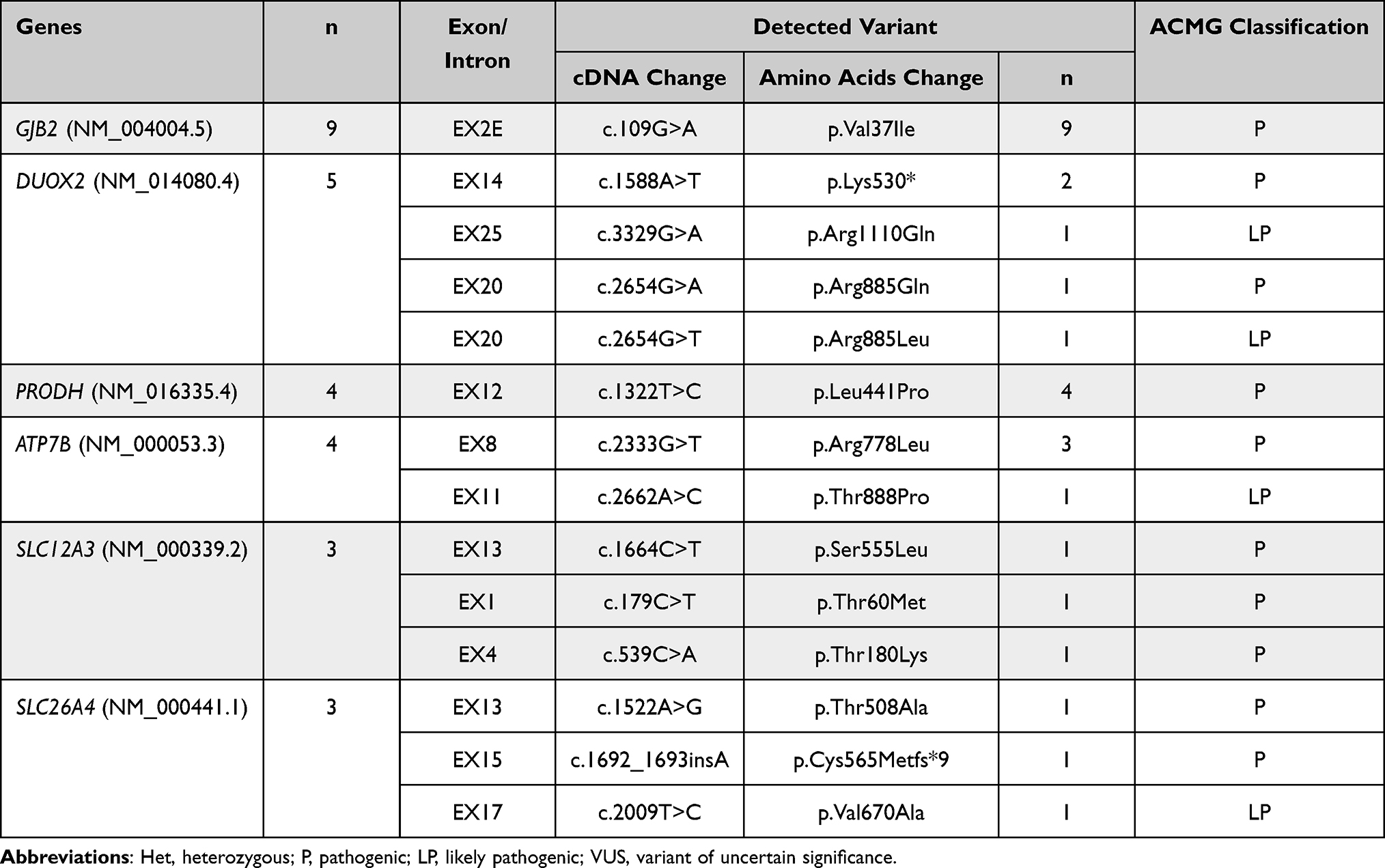 |
Table 3 Top Six Pathogenic Genes and Variants in Present Study |
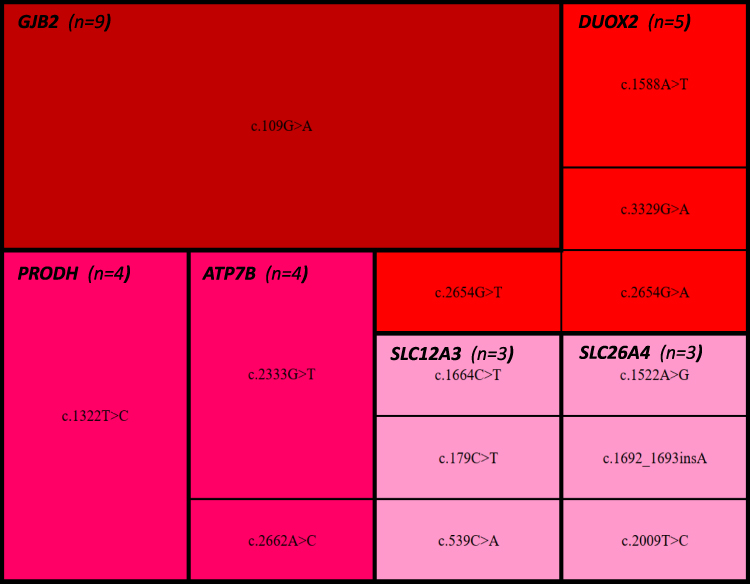 |
Figure 1 Heatmap of top six pathogenic genes and variants. |
In this study, we performed TMS screening and genomic sequencing (GS) screening on dried blood spots at the same time. TMS screening did not identify any newborns with inborn errors of metabolism. Only two newborns showed abnormalities in elementary screening, including a heightened tyrosine (TYR) level (447.61μmol/L) and a raised C3 level=4.54μmol/L respectively. After recalling and retesting these indicators returned to normal levels and obvious growth and development anomalies were observed in the follow-up of the two newborns. Meanwhile, six newborns of genetic disorders and 44 newborns as a carrier were detected by GS screening; there was also no obvious abnormality in their TMS screening results. Moreover, no significant correlation was found between genetic variation and clinical severity of hyperbilirubinemia, either in confirmed patients or carriers.
Discussion
Growing attention has been paid to the relationship between neonatal hyperbilirubinemia and genetics. Due to technological limitations, previous studies could only focus on the detection of polymorphisms of one or several related genes. In recent years, genomic sequencing has gradually entered the clinic and has been successfully applied to newborn screening, especially for patients in NICU. Genomic sequencing is considered an effective method, which can not only definitively diagnose a disease as soon as possible but also help to uncover the new cause(s) of the disease. Here, we adopted a new genomic sequencing project (“Angel Care”) based on NGS technology, which involved the detection of 159 disease-associated genes. Our results suggest that the main advantages of “Angel Care” include: (1) Hyperbilirubinemia is closely related to genetic variation, and neonatal gene screening is helpful to comprehensively explain its genetic characteristics; (2) compared to WGS and WES technologies, “Angel Care” is simpler, faster and cheaper. It is a method suitable for population screening.
Hyperbilirubinemia is one of the most common neonatal diseases. Recent studies have shown that certain pathogenic gene polymorphisms are closely related to the occurrence and development of diseases. However, there has been no study on large-scale genome sequencing screening so far. With the promotion of neonatal genome screening project, the patients from NICU constitute a suitable population as in Neoseq,37 Newbie Seq,3 NC NEXUS,4 BabySeq.9 A high incidence of genetic disorders is often found in this population, and so is a high false-positive rate of TMS screening. Gene screening programs can contribute to a clear diagnosis as soon as possible.38 Although there is no specific study for hyperbilirubinemia, gene sequencing in children in NICU has already achieved good results. For example, Dimmock’s group carried out a randomized, controlled trial of genome sequencing in the children with conditions of unknown etiology in NICUs.39 A total of 51 (24%) of the 213 enrolled infants were identified with genetic conditions by genomic sequencing, and their clinical management has significantly improved. The authors came to the conclusion that there is high clinical utility with rapid genomic sequencing in children from NICU with conditions of unknown etiology. Another multicenter study explored the feasibility of ultra-rapid exome sequencing in critically ill pediatric patients. Fifty-five patients (51%) received the molecular diagnoses in 3.3 days, with a 3-fold reduction in the meantime from hospital admission to diagnosis.40 Instead of WES, we used targeted next-generation sequencing to study hyperbilirubinemia, which is the main etiology in NICU patients. The results suggest that there is a high risk of genetic variation in neonatal hyperbilirubinemia. A 6.25% of cases were suspected to have genetic disorders and 45.8% cases were detected with carrier status. All patients received a molecular diagnostic report within 7 days. We believe that by taking neonatal hyperbilirubin as the starting point, we should pay more attention to nGS in NICU.
Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD) is an important cause of neonatal hyperbilirubinemia. It could cause neonatal hyperbilirubinemia, acute hemolysis, and chronic hemolysis. Because acute hemolysis is caused by exposure to an oxidative stressor in the form of an infection, oxidative drug, or fava beans, treatment is geared toward avoidance of these and other stressors. Neonatal hyperbilirubinemia due to acute hemolysis may require phototherapy or exchange transfusion to prevent bilirubin encephalopathy. However, part of these children with G6PD deficiency also may be asymptomatic in the neonatal period. So, early screening and prevention of risk factors are important for avoiding the possible severe hemolysis. It is well known that G6PD deficiency is heterogeneous in different regions and nationalities. Many areas in southern China have carried out newborn screening programs for G6PD. Current data showed that the frequencies of G6PD deficiency ranged from 0% to 11.6% in China, and it is especially high in southern China.41 Compared to healthy newborns, there is a higher risk of hyperbilirubinemia in the patients with G6PD deficiency.42 G6PD screening has not been routinely carried out in Jiangsu Province, so the importance of G6PD in neonatal hyperbilirubinemia may be underestimated. Through newborn genome screening, it was found that two of 96 newborns with hyperbilirubinemia were affected by G6PD deficiency, with an incidence rate of 2.1%. Therefore, newborn genomic screening, particularly in patients with neonatal hyperbilirubinemia, is of great importance. According to previous reports, the most frequently occurring G6PD variants were c.1376G>T, c.1388G>A, c.95A>G, c.1024C>T and c.871G>A.43,44 Two cases in this study were detected carrying one likely pathogenic mutation (c.1388G>A and c.1024C>T) in G6PD gene, which are common variants in Chinese population.
Meanwhile, newborn genome screening provides valuable carrier information, as 45.8% neonatal hyperbilirubinemia cases were detected with a carrier status for a genetic disorder in present study. It is well known that the mutations in GJB2 (Cx26) are the main cause of hereditary deafness, with more than 220 different mutations accounting for more than 50% of autosomal recessive nonsyndromic hearing loss.31 Different from European origin,32 c.109G>A is the most frequent mutation in populations of China in this study, similar to other reports.33,34 This study suggested that the rate of carrier-status of pathogenic variants in GJB2 gene is 9.4% (9/96) in the newborn with hyperbilirubinemia, which is close to the report of Yang33 but higher than Zeng’s report. In their studies, their subjects were healthy newborns. DUOX2 is another gene that deserves the attention in Chinese population studies. A previous study indicated that DUOX2 was the most common gene mutation in congenital hypothyroidism (CH) infants in China,35 and high phenotypic heterogeneity was observed.36 Currently, the carrying rate of DUOX2 in newborns is still not clear. Our results show that the carrier rate was about 5.2% (5/96) in the hyperbilirubinemia infants. The mutations are also well distributed among subjects, which c.1588A>T (p.Lys530*) may be slightly common.
Although such information does not improve the treatment and prevention of hyperbilirubinemia in patients per se, it plays a role in future health management, especially for patients’ future reproductive plans and reproductive plans. Indeed, there are still disputes about whether it is necessary to report carrier information to newborn parents.
This study also has the following limitations. For one, the targeted sequencing panel we audited is not specifically designed for hyperbilirubinemia, so it does not contain high-frequency pathogenic genes associated with hyperbilirubinemia, such as UGT1A1, OATP2, HO-1. The sample size of this study is not statistically sufficient, so we could not analyze the correlation between harmful variants and TSB level or phototherapy effect. The follow-up time of positive cases could be lengthened, and the variant verification in family was not carried out. In addition, newborns from NICU have a higher risk of genetic variation and seem that they are more necessary for nGS. However, we should remind ourselves that the genetic diseases of NICU newborns are more complex. Whether the technology based on NGS panel is enough? Is the effect of WES technology better? This reminds us that different screening techniques might be selected for nGS in different populations. However, the current clinical practice is still very limited, which requires us to study more deeply.
In conclusion, taking neonatal hyperbilirubinemia as an example, we confirmed that NICU newborns are closely related to genetic variation. “Angel Care” is an effective method of genomic sequencing for newborn screening. It can identify most conditions traditionally included in TMS-NGS screening, while demonstrating a drastic reduction in false-positive rate. Compared to WGS and WES technologies, it may be a method suitable for population screening.
Data Sharing Statement
The datasets presented in this article are not readily available because of Regulations on the management of human genetic resources in China. Requests to access the datasets should be directed to Prof. Bin Yu. [email protected].
Ethics Approval and Consent to Participate
The study design and protocol were reviewed and approved by the ethics committee of Changzhou Maternal and Child Health Care Hospital. Written informed contents were obtained from the newborns’ parents before screening.
Acknowledgments
The authors would like to acknowledge BGI for their provision of data and technical support. We also thank all of the project participants for their contributions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by a project supported by Jiangsu Maternal and Children Health Care Key Discipline, Changzhou Social Development Science and Technology Support Project (CE20205035).
Disclosure
The authors declare that they have no competing interests.
References
1. Moreno MA. Newborn screening. JAMA Pediatr. 2016;170(6):628. doi:10.1001/jamapediatrics.2015.2519
2. Ceyhan-Birsoy O, Murry JB, Machini K, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq project. Am J Hum Genet. 2019;104(1):76–93. doi:10.1016/j.ajhg.2018.11.016
3. Adhikari AN, Gallagher RC, Wang Y, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med. 2020;26(9):1392–1397. doi:10.1038/s41591-020-0966-5
4. Roman TS, Crowley SB, Roche MI, et al. Genomic sequencing for newborn screening: results of the NC NEXUS project. Am J Hum Genet. 2020;107(4):596–611. doi:10.1016/j.ajhg.2020.08.001
5. Willig LK, Petrikin JE, Smith LD, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3(5):377–387. doi:10.1016/S2213-2600(15)00139-3
6. Hao C, Guo R, Hu X, et al. Newborn screening with targeted sequencing: a multicenter investigation and a pilot clinical study in China. J Genet Genom. 2021;49:13–19. doi:10.1016/j.jgg.2021.08.008
7. Wang H, Yang Y, Zhou L, Wang Y, Long W, Yu B. NeoSeq: a new method of genomic sequencing for newborn screening. Orphanet J Rare Dis. 2021;16(1):481. doi:10.1186/s13023-021-02116-5
8. Borghesi A, Mencarelli MA, Memo L, et al. Intersociety policy statement on the use of whole-exome sequencing in the critically ill newborn infant. Ital J Pediatr. 2017;43(1):100. doi:10.1186/s13052-017-0418-0
9. Wojcik MH, Zhang T, Ceyhan-Birsoy O, et al. Discordant results between conventional newborn screening and genomic sequencing in the BabySeq project. Genet Med. 2021;23:1372–1375. doi:10.1038/s41436-021-01146-5
10. Abbey P, Kandasamy D, Naranje P. Neonatal jaundice. Indian J Pediatr. 2019;86(9):830–841. doi:10.1007/s12098-019-02856-0
11. Olusanya BO, Teeple S, Kassebaum NJ. The contribution of neonatal jaundice to global child mortality: findings from the GBD 2016 study. Pediatrics. 2018;141(2). doi:10.1542/peds.2017-1471
12. Amin SB, Smith T, Timler G. Developmental influence of unconjugated hyperbilirubinemia and neurobehavioral disorders. Pediatr Res. 2019;85(2):191–197. doi:10.1038/s41390-018-0216-4
13. Sgro M, Campbell D, Shah V. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. CMAJ. 2006;175(6):587–590. doi:10.1503/cmaj.060328
14. Rets A, Clayton AL, Christensen RD, Agarwal AM. Molecular diagnostic update in hereditary hemolytic anemia and neonatal hyperbilirubinemia. Int J Lab Hematol. 2019;41(Suppl 1):95–101. doi:10.1111/ijlh.13014
15. Zhou J, Yang C, Zhu W, et al. Identification of genetic risk factors for neonatal hyperbilirubinemia in Fujian Province, Southeastern China: a case-control study. Biomed Res Int. 2018;2018:7803175. doi:10.1155/2018/7803175
16. Wu XJ, Zhong DN, Xie XZ, Ye DZ, Gao ZY. UGT1A1 gene mutations and neonatal hyperbilirubinemia in Guangxi Heiyi Zhuang and Han populations. Pediatr Res. 2015;78(5):585–588. doi:10.1038/pr.2015.134
17. Halis H, Ergin H, Köseler A, Atalay E. The role of UGT1A1 promoter polymorphism and exon-1 mutations in neonatal jaundice. J Matern Fetal Neonatal Med. 2017;30(22):2658–2664. doi:10.1080/14767058.2016.1261105
18. Tiwari PK, Bhutada A, Agarwal R, Basu S, Raman R, Kumar A. UGT1A1 gene variants and clinical risk factors modulate hyperbilirubinemia risk in newborns. J Perinatol. 2014;34(2):120–124. doi:10.1038/jp.2013.140
19. Carvalho CG, Castro SM, Santin AP, de Azevedo LA, Pereira ML, Giugliani R. Polymorphic variants of UGT1A1 in neonatal jaundice in southern Brazil. J Trop Pediatr. 2010;56(5):366–367. doi:10.1093/tropej/fmp131
20. Long J, Zhang S, Fang X, Luo Y, Liu J. Neonatal hyperbilirubinemia and Gly71Arg mutation of UGT1A1 gene: a Chinese case-control study followed by systematic review of existing evidence. Acta Paediatr. 2011;100(7):966–971. doi:10.1111/j.1651-2227.2011.02176.x
21. Bai J, Luo L, Liu S, et al. Combined effects of UGT1A1 and SLCO1B1 variants on Chinese adult mild unconjugated hyperbilirubinemia. Front Genet. 2019;10:1073. doi:10.3389/fgene.2019.01073
22. Min J, Jie L, Caiyun Y, Ying L, Xuefang Y. Gene mutation in neonatal jaundice - mutations in UGT1A1 and OATP2 genes. Indian J Pediatr. 2016;83(7):723–725. doi:10.1007/s12098-016-2064-8
23. Amandito R, Rohsiswatmo R, Halim M, Tirtatjahja V, Malik A. SLCO1B1 c.388A > G variant incidence and the severity of hyperbilirubinemia in Indonesian neonates. BMC Pediatr. 2019;19(1):212. doi:10.1186/s12887-019-1589-1
24. Liu J, Long J, Zhang S, Fang X, Luo Y. The impact of SLCO1B1 genetic polymorphisms on neonatal hyperbilirubinemia: a systematic review with meta-analysis. J Pediatr. 2013;89(5):434–443. doi:10.1016/j.jped.2013.01.008
25. Kaplan M, Wong RJ, Stevenson DK. Heme oxygenase-1 promoter polymorphisms: do they modulate neonatal hyperbilirubinemia? J Perinatol. 2017;37(8):901–905. doi:10.1038/jp.2017.6
26. Urschel S, West LJ. ABO-incompatible heart transplantation. Curr Opin Pediatr. 2016;28(5):613–619. doi:10.1097/MOP.0000000000000398
27. American Academy of Pediatrics. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114(1):297–316. doi:10.1542/peds.114.1.297
28. Yang Y, Wang L, Wang B, Liu S, Yu B, Wang T. Application of next-generation sequencing following tandem mass spectrometry to expand newborn screening for inborn errors of metabolism: a multicenter study. Front Genet. 2019;10:86. doi:10.3389/fgene.2019.00086
29. Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159. doi:10.1016/j.ebiom.2017.08.015
30. Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–257. doi:10.1038/s41436-019-0686-8
31. Guan Q, Balciuniene J, Cao K, et al. AUDIOME: a tiered exome sequencing-based comprehensive gene panel for the diagnosis of heterogeneous nonsyndromic sensorineural hearing loss. Genet Med. 2018;20(12):1600–1608. doi:10.1038/gim.2018.48
32. Koohiyan M, Koohian F, Azadegan-Dehkordi F. GJB2-related hearing loss in central Iran: review of the spectrum and frequency of gene mutations. Ann Hum Genet. 2020;84(2):107–113. doi:10.1111/ahg.12354
33. Yang H, Luo H, Zhang G, Zhang J, Peng Z, Xiang J. A multiplex PCR amplicon sequencing assay to screen genetic hearing loss variants in newborns. BMC Med Genomics. 2021;14(1):61. doi:10.1186/s12920-021-00906-1
34. Lin YF, Lin HC, Tsai CL, Hsu YC. GJB2 mutation spectrum in the Taiwanese population and genotype-phenotype comparisons in patients with hearing loss carrying GJB2 c.109G>A and c.235delC mutations.. Hear Res. 2020;413:108135. doi:10.1016/j.heares.2020.108135
35. Yu B, Long W, Yang Y. Newborn screening and molecular profile of congenital hypothyroidism in a Chinese population. Front Genet. 2018;9:509. doi:10.3389/fgene.2018.00509
36. Long W, Zhou L, Wang Y, Liu J, Wang H, Yu B. Complicated relationship between genetic mutations and phenotypic characteristics in transient and permanent congenital hypothyroidism: analysis of pooled literature data. Int J Endocrinol. 2020;2020:6808517. doi:10.1155/2020/6808517
37. de Castro MJ, González-Vioque E, Barbosa-Gouveia S, et al. Rapid phenotype-driven gene sequencing with the NeoSeq panel: a diagnostic tool for critically ill newborns with suspected genetic disease. J Clin Med. 2020;9(8):2362. doi:10.3390/jcm9082362
38. Bamborschke D, Özdemir Ö, Kreutzer M, et al. Ultra-rapid emergency genomic diagnosis of Donahue syndrome in a preterm infant within 17 hours. Am J Med Genet A. 2021;185(1):90–96. doi:10.1002/ajmg.a.61917
39. Dimmock DP, Clark MM, Gaughran M, et al. An RCT of rapid genomic sequencing among seriously ill infants results in high clinical utility, changes in management, and low perceived harm. Am J Hum Genet. 2020;107(5):942–952. doi:10.1016/j.ajhg.2020.10.003
40. Lunke S, Eggers S, Wilson M, et al. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA. 2020;323(24):2503–2511. doi:10.1001/jama.2020.7671
41. Zheng Y, Wang J, Liang X, et al. Epidemiology, evolutionary origin, and malaria-induced positive selection effects of G6PD-deficient alleles in Chinese populations. Mol Genet Genom Med. 2020;8(12):e1540. doi:10.1002/mgg3.1540
42. Liu H, Liu W, Tang X, Wang T. Association between G6PD deficiency and hyperbilirubinemia in neonates: a meta-analysis. Pediatr Hematol Oncol. 2015;32(2):92–98. doi:10.3109/08880018.2014.887803
43. Liu Z, Yu C, Li Q, et al. Chinese newborn screening for the incidence of G6PD deficiency and variant of G6PD gene from 2013 to 2017. Hum Mutat. 2020;41(1):212–221. doi:10.1002/humu.23911
44. Gao J, Lin S, Chen S, et al. Molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Shenzhen population. Hum Hered. 2020;85(3–6):110–116. doi:10.1159/000516808
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.