Back to Journals » Drug Design, Development and Therapy » Volume 11
Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer
Authors Yadav DK ![]() , Kumar S, Saloni, Singh H
, Kumar S, Saloni, Singh H ![]() , Kim M
, Kim M ![]() , Sharma P
, Sharma P ![]() , Misra S, Khan F
, Misra S, Khan F
Received 19 December 2016
Accepted for publication 1 May 2017
Published 22 June 2017 Volume 2017:11 Pages 1859—1870
DOI https://doi.org/10.2147/DDDT.S130601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Dharmendra K Yadav,1 Surendra Kumar,2 Saloni,1 Harpreet Singh,3 Mi-hyun Kim,1 Praveen Sharma,4 Sanjeev Misra,4 Feroz Khan5
1Department of Pharmacy, College of Pharmacy, Gachon University, Yeonsu-gu, Incheon, Republic of Korea; 2Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Babu Banarasi Das Northern India Institute of Technology, Lucknow, 3Department of Bioinformatics, Indian Council of Medical Research, New Delhi, 4Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur, 5Metabolic & Structural Biology Department, CSIR– Central Institute of Medicinal & Aromatic Plant, Lucknow, India
Abstract: Withanolides are a group of pharmacologically active compounds present in most prodigal amounts in roots and leaves of Withania somnifera (Indian ginseng), one of the most important medicinal plants of Indian traditional practice of medicine. Withanolides are steroidal lactones (highly oxygenated C-28 phytochemicals) and have been reported to exhibit immunomodulatory, anticancer and other activities. In the present study, a quantitative structure activity relationship (QSAR) model was developed by a forward stepwise multiple linear regression method to predict the activity of withanolide analogs against human breast cancer. The most effective QSAR model for anticancer activity against the SK-Br-3 cell showed the best correlation with activity (r2=0.93 and rCV2 =0.90). Similarly, cross-validation regression coefficient (rCV2=0.85) of the best QSAR model against the MCF7/BUS cells showed a high correlation (r2=0.91). In particular, compounds CID_73621, CID_435144, CID_301751 and CID_3372729 have a marked antiproliferative activity against the MCF7/BUS cells, while 2,3-dihydrowithaferin A-3-beta-O-sulfate, withanolide 5, withanolide A, withaferin A, CID_10413139, CID_11294368, CID_53477765, CID_135887, CID_301751 and CID_3372729 have a high activity against the Sk-Br-3 cells compared to standard drugs 5-fluorouracil (5-FU) and camptothecin. Molecular docking was performed to study the binding conformations and different bonding behaviors, in order to reveal the plausible mechanism of action behind higher accumulation of active withanolide analogs with β-tubulin. The results of the present study may help in the designing of lead compound with improved activity.
Keywords: ADMET, breast cancer, QSAR, molecular docking, withanolides
Introduction
Breast cancer, a heterogeneous group of tumors, is the most commonly diagnosed cancer in women globally. In spite of improvements in early diagnosis and development of several targeted therapeutic methods, breast cancer-related morbidity still remains high. The existing therapeutic approaches are associated with high toxicity, low efficacy, therapeutic resistance and therapy-related morbidity. Recent studies have reported that the natural products owing to their cancer preventive potential will pay a way for more effective, non-endocrine, nontoxic therapeutic approaches for anticancer therapies. Historically, phytomolecules have played a key role in the discovery and development of novel anticancer agents.1,2 Withania somnifera (commonly known as ashwagandha or Indian winter cherry) is one such medicinal plant, whose all parts are used as ayurvedic remedies for healing various diseases, including inflammation, arthritis, asthma and hypertension.3–5 The root extract of W. somnifera is known as withanolide, which is composed of 14 bioactive compounds.6,7 Preclinical experimental data indicates that W. somnifera leaf and root extracts have anticancer potential.8,9 For example, chemically induced tumorigenesis in the stomach and skin of mice was inhibited significantly following administration of W. somnifera root.9 Although no controlled clinical trials of ashwagandha have been reported for any indication, it appears to have a relatively low toxicity profile based on a single human study10 and toxicological studies in mice.11 In ayurvedic medicine, ashwagandha has also been claimed as an effective agent against arthritis, anxiety, insomnia and stress.12 W. somnifera is currently regulated in the US and Europe as a dietary supplement. The first withanolide to be characterized was withaferin A (WFA), isolated in 1965,13 with its cytotoxic activities being the focus of interest.14,15 The noncytotoxic, anti-inflammatory16 and immunomodulatory mechanisms17 of WFA have however thus far remained rather than poorly considered.
WFA, an important lactone of the withanolide class of phytomolecules, is a highly oxygenated steroidal prototype found in W. somnifera and its related Solanacea species, and exhibits potential anticancer activities.18 It has been reported to have shown decreases in mammary tumors and pulmonary metastasis in an MMTV-neu transgenic model and is associated with increased apoptosis.19 WFA-induced apoptosis involves the production of reactive oxygen species20 and induction of FOXO3a and Bim,21 while WFA effectively inhibits oncogenic transcription factors such as STAT3,22 resulting in growth inhibition. It has been reported that WFA promotes Notch signaling, plays an oncogenic role and is also often hyperactive in breast cancer cells.23 Herein, we report the development of a quantitative structure activity relationship (QSAR) model for the antiproliferative activity of withanolide analogs against human breast (SK-Br-3 and MCF7/BUS) cancer cell lines. Moreover, druggability of the compounds was evaluated using Lipinski’s “rule of five” and a series of absorption, distribution, metabolism, excretion and toxicity (ADMET) properties. Molecular docking simulations of these compounds against β-tubulin were also conducted to characterize their binding affinity and interactions.
Materials and methods
Molecular modeling
Molecular modeling studies of withanolide analogs were carried out using Sybyl-X 2.1 (Tripos International, St Louis, MO, USA). The biological activity of all the withanolide analogs measured as inhibitory concentration (GI50) was converted to negative logarithmic concentration in moles (pGI50). Drawing of structures and geometry optimization was performed using ChemBioOffice Suite Ultra v12.0 (2012; Cambridge Soft Corp., Cambridge, UK).24,25 The Surflex-Dock module in Sybyl was used to construct the binding orientation of withanolide analogs.26,27
QSAR model development
A QSAR model was developed to screen potential leads against human breast cancer cell lines SK-Br-3 and MCF7/BUS within a training set (Tables S1 and S2).28 A total of 52 physicochemical descriptors were calculated for each compound using Scigress Explorer v.7.7.0.47 (Fujitsu Ltd., Poland; Table S3). The dataset division into training and test was based on structural/pharmacophore or chemical class similarity in order to include only diverse compounds. Similarly, to select the best subset of physicochemical properties, highly correlated chemical descriptors were excluded through covariance analysis using a correlation matrix (Tables S4 and S5). Finally, a robust QSAR model equation was derived by using multiple linear regression; irrelevant chemical descriptors were removed through a forward stepwise approach leading to a selection of 4 out of 52 descriptors in the final QSAR regression equation. The resulting QSAR model equation showed high regression coefficients (r2) of 0.93 and 0.91 with activity data from the SK-Br-3 and MCF7/BUS cell lines, respectively. The developed QSAR model was validated by predicting the experimental activity of compounds in a test set. Furthermore, the robustness of the developed models was accessed by cross-validation regression coefficient (rCV2) using leave-one-out, test set prediction and randomization methods.29–31
Molecular docking
Molecular docking simulations were performed to predict the bioactive conformations and explore the binding interactions of withanolide analogs using the Surflex-Dock module of Sybyl X 2.1. The crystal structure of β-tubulin (Protein Data Bank [PDB] code 4IHJ)32 was used as the potential anticancer drug target, with the dataset of compounds as ligands. The Surflex-Dock scoring function was used to assess the strength of the ligand–protein interactions. This scoring function includes energy terms to account for hydrophobicity, polarity, repulsiveness, entropy and solvation. Docking simulations were conducted allowing ligand flexibility but maintaining the protein structure rigid, while all docking parameters were set to default values.
QSAR model validation
A reliable and predictive QSAR model should 1) be statistically significant and robust; 2) provide accurate predictions for external datasets not used during the model development and 3) have its application boundaries defined.
Cross-validation techniques
To explore the reliability of QSAR models, a leave-one-out procedure was performed. The outcome from the cross-validation procedure was cross-validation regression coefficient (rCV2), which was used as a criterion of both robustness and predictive ability of the model. It was calculated using the following equation:
|
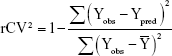
In the above equation, Y– means the average activity value of the training dataset, while Yobs and Ypred represent the observed and predicted activity values. A high rCV2 (>0.5) suggests a reasonably robust model.
Y-randomization test
In this technique, the dependent variable (biological activity) is randomly shuffled, and a new QSAR model is developed using the original independent variable. The new QSAR models (after several trials) are expected to have low r2 and rCV2 values; if the reverse happens, then an acceptable QSAR model cannot be obtained for the specific modeling method and data. These techniques ensure the robustness of a QSAR model.
Estimation of the predictive ability of a QSAR model
To ensure the high predictive power of a QSAR model, it should be estimated by an external test set of compounds that are not used in building of the QSAR model. The external validation or predictive capacity of the derived model was judged by predictive r2 (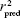 ) as shown in the following equation:
) as shown in the following equation:
|
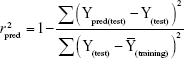
where Ypred (test) and Y(test) indicate the predicted and observed activity values, respectively, for test set compounds and  indicates the average bioactivity of compound in the training set. An acceptable predictive power of a QSAR model () should be >0.6 for the test set molecules.33–35
indicates the average bioactivity of compound in the training set. An acceptable predictive power of a QSAR model () should be >0.6 for the test set molecules.33–35
Defining the model – applicability domain (AD)
AD was calculated by the using 23 and 24 inhibitors of MCF7BUS and Sk-Br-3 cell lines used in bioassay model building. It was designed in the form of range values of Lipinski’s parameters, connectivity index (order 0, standard), dipole vector X (debye) and molar refractivity shape index (basic kappa, order 2) in Figure 1 and the connectivity index (order 0, standard), dipole vector X (debye) and shape index (basic kappa, order 2) inhibition in Figure 2. The input parameters for queried compounds were screened through AD decided by the training set.
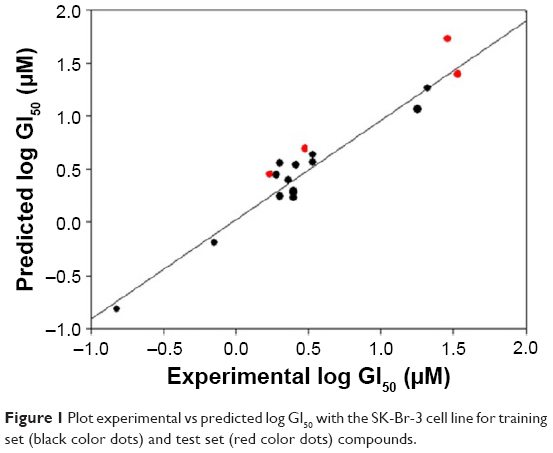 | Figure 1 Plot experimental vs predicted log GI50 with the SK-Br-3 cell line for training set (black color dots) and test set (red color dots) compounds. |
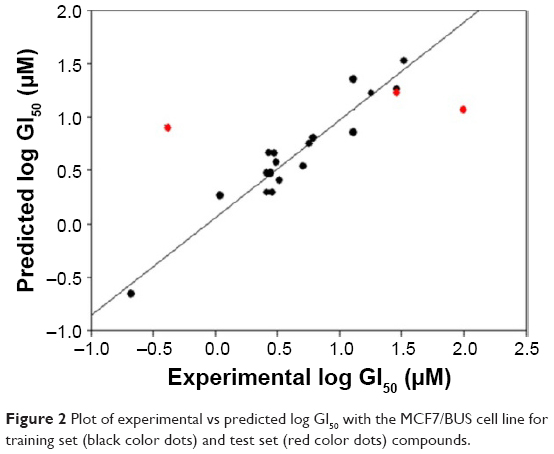 | Figure 2 Plot of experimental vs predicted log GI50 with the MCF7/BUS cell line for training set (black color dots) and test set (red color dots) compounds. |
Prediction of pharmacokinetic (PK) parameters
It would be advantageous in the process of drug discovery, if the ADMET properties of drug molecules were predicted earlier. This information helps the chemist to assess the PK profile of molecules. The PK properties depend on chemical descriptors of drugs, which determine their ADMET. The PK parameters were calculated by QikProp v3.236 module of Schrödinger Suite 2011. A number of mathematical predictive ADMET models for different PK parameters were available, namely, aqueous solubility, apparent Caco-2 and MDCK permeability, log Kp for skin permeability, blood–brain barrier (BBB; log BB), the volume of distribution and plasma protein binding (shown as log Khsa for serum protein binding), which were used to quantitatively predict the ADMET properties of the withanolide analogs. Similarly, the drug-likeness (DL) parameters, such as molecular weight (MW) of ≤500 Da, a logP value of ≤5, hydrogen bond donor ≤5 and hydrogen bond acceptor site (N and O atoms) ≤10, and Veber’s parameters, namely, topological polar surface area (TPSA) (≤140 Å2), which assess the passive molecular transport through membranes and number of rotatable bonds (≤10), used for flexibility measurement were calculated.35,37–39
Results and discussion
QSAR model for antiproliferative activity against the SK-Br-3 cell line
All the withanolide analogs were evaluated for their predictivity through the developed QSAR models. A forward stepwise variable selection strategies on 52 physicochemical properties (descriptors) using 23 known drugs/compounds in the training set (Table S1) identified four chemical descriptors, connectivity index (order 0, standard), dipole vector X (debye), molar refractivity and shape index (basic kappa, order 2), responsible for the anticancer activity against the SK-Br-3 cell line.
QSAR model equation 1
|

|
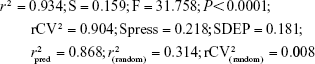
Spress is the standard predictive residual sum of squares; SDEP is the standard deviation of error of prediction.
The derived QSAR equation 1 showed a significant relationship between log GI50 (dependent variable) and the chemical descriptors (independent variables). The value of the regression coefficient (r2=0.934) indicates the existence of ~93% correlation between the activity and the chemical descriptors in the training dataset, while the value of the cross-validation regression coefficient (rCV2=0.903) suggests ~90% prediction accuracy of this QSAR model (Figure 1). A Y-randomization of 100 trials with original descriptor producing an average value of 0.314 and 0.008 for r2 and rCV2, respectively, suggests the robustness and significant accuracy of the developed QSAR model (Table S6). Furthermore, test set predictivity of 0.868 supported the robustness and accuracy of the model. It is evident among the molecular descriptors that molar refractivity is positively correlated with activity, that is, if molar refractivity increases, the biological activity also increases. However, the connectivity index (order 0, standard), dipole vector X (Debye) and shape index (basic kappa, order 2) are inversely correlated showing an increase in the biological activity with the decrease in the magnitude of these descriptors and vice versa. Based on contributing descriptors, the predictivity of 29 withanolide analogs through a QSAR model indicated that 2,3-dihydrowithaferin A-3-beta-O-sulfate, withanolide 5, withanolide A, withaferin A, CID_10413139, CID_11294368, CID_53477765, CID_135887, CID_301751 and CID_3372729 showed significant anticancer in vitro activity compared to the reference drug camptothecin (CPT) against the SK-Br-3 cell line (Table S7 and Figure S1).
QSAR model for antiproliferative activity against MCF7/BUS
The QSAR model for antiproliferative activity against the MCF7/BUS cell line was developed with a training set of 24 drugs/compounds. A total of 52 chemical descriptors were considered during model development (Table S2), and forward stepwise variable search techniques resulted in four parametric models that included all atom count, dielectric energy (kcal/mole), total energy (hartree) and heat of formation (kcal/mole) correlating significantly against the MCF7/BUS cell line.
QSAR model equation 2
|
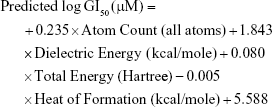
|
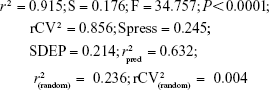
This QSAR equation shows the relationship between in vitro experimental activities (log GI50) as the dependent variable and the four chemical descriptors mentioned earlier as independent variables. The value of the regression coefficient (r2=0.91) shows the existence of ~91% correlation between the activities and the chemical descriptors of the training dataset compounds, while the value of cross-validation regression coefficient (rCV2=0.85) points to the 85% prediction accuracy of the QSAR model (Figure 2). A Y-randomization of 100 trials with the original descriptor producing an average value of 0.236 and 0.004 for r2 and rCV2, respectively, suggest the robustness of the developed QSAR model (Table S6). Furthermore, test set predictivity of 0.632 supported the robustness of the model. In the QSAR model, the molecular descriptors, namely, atom count (all atoms), dielectric energy (kcal/mole) and total energy (hartree), correlate positively with activity, that is, if any of these variables increase, the biological activity against the breast cancer also increases. Moreover, the heat of formation (kcal/mol) is negatively correlated with activity, showing that the biological activity decreases if the value of this descriptor increases. Analyzing the most active analogs, CID_73621 and CID_435144 possess higher anti-proliferative activity than the reference drugs 5-fluorouracil (5-FU) and CPT (Table S8 and Figure S1).
Binding affinity and interactions with β-tubulin
Molecular docking simulations were carried out to elucidate the likely binding affinity and binding interactions of withanolide analogs with anti-cancer target β-tubulin receptor protein. The predictions of the docking simulations are summarized in Table 1. Withanolide analogs were found to bind to the same active site as found for X-ray crystal structure. The reference compounds 5-FU and CPT were predicted to have significant binding affinity to β-tubulin with total docking scores of 2.5304 and 4.1837, respectively, while withanolide active analogs CID_11294368 and CID_53477765 showed higher binding affinities with total docking scores of 6.4885 and 6.1862, respectively. The binding interactions of these withanolide analogs within the active site of β-tubulin receptor protein are shown in Figure 3. By contrast, 2,3-dihydrowithaferin A-3-beta-O-sulfate, withanolide 5, withanolide A, withaferin A, CID_73621, CID_435144, CID_10413139, CID_135887 and CID_301751 were predicted to have moderate binding affinities to β-tubulin with total docking scores of 5.6272, 5.958, 5.6691, 5.4153, 5.3516, 5.9275, 5.0615, 5.2200 and 5.2417, respectively. The withanolide analog CID_3372729 was predicted to have low binding affinity with β-tubulin showing a total docking score of 4.6965. The chemical nature of binding amino acid residues in β-tubulin that were predicted to interact with the various withanolide analogs was hydrophobic (eg, Ile-165, Pro-162, Met-166 and Val-257), polar acidic (eg, Asp-163 and Asp-199), polar basic (eg, Arg-158, Arg-164 and Arg-253), polar uncharged (eg, Thr-198) and polar amide (eg, Asn-197). Consequently, the interactions of all analogs with β-tubulin involved predominantly strong hydrophobic interactions, leading to high affinities and likely high anticancer activity. The active analogs also formed one to three H-bonds with β-tubulin, contributing to the strength of their interactions, which may lead to a high inhibitory activity of withanolide analogs with β-tubulin.
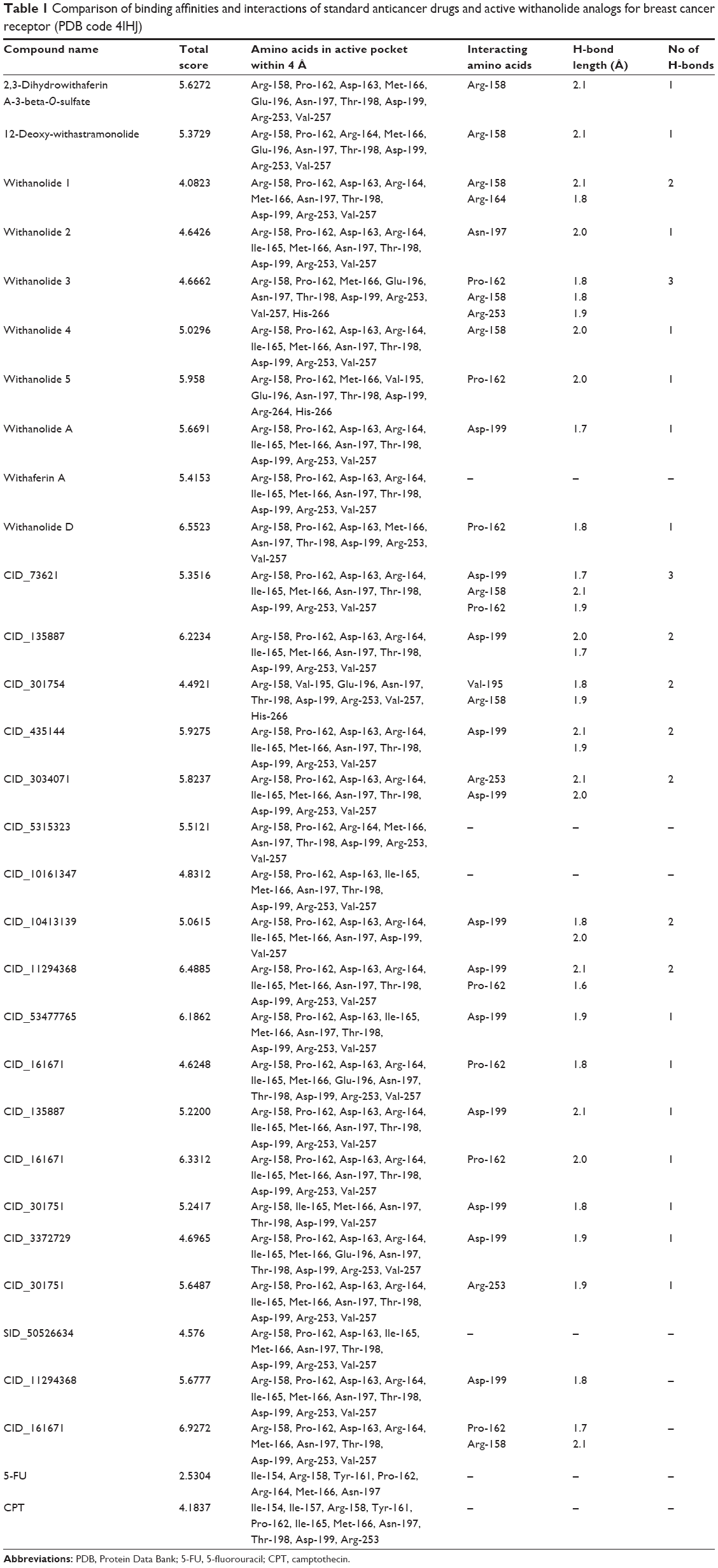 | Table 1 Comparison of binding affinities and interactions of standard anticancer drugs and active withanolide analogs for breast cancer receptor (PDB code 4IHJ) |
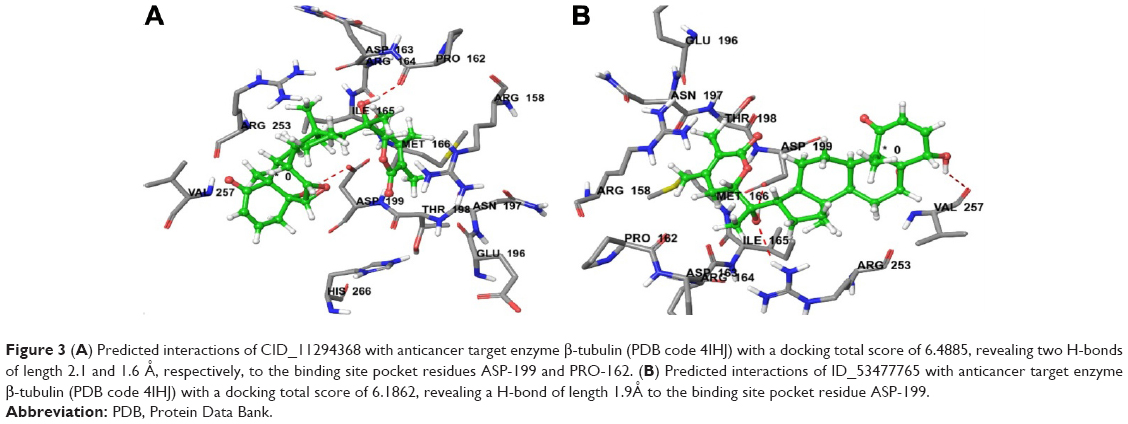 | Figure 3 (A) Predicted interactions of CID_11294368 with anticancer target enzyme β-tubulin (PDB code 4IHJ) with a docking total score of 6.4885, revealing two H-bonds of length 2.1 and 1.6 Å, respectively, to the binding site pocket residues ASP-199 and PRO-162. (B) Predicted interactions of ID_53477765 with anticancer target enzyme β-tubulin (PDB code 4IHJ) with a docking total score of 6.1862, revealing a H-bond of length 1.9Å to the binding site pocket residue ASP-199. |
The most active compound, CID_301751, was predicted to have a significant binding affinity as evidenced by a total docking score of 5.2417 and form a hydrogen bond of length 1.8 Å to the polar acidic residue Asp-199. The amino acid residues binding within a radius of 3 Å with ligand were polar acidic residue Asp-199; polar uncharged residue Thr-198; nonpolar hydrophobic residues Ile-165, Met-166 and Val-257; polar amide Asn-197 and polar basic Arg-158. The presence of these strong hydrophobic and polar interactions explains the high affinity of this molecule and its anticancer activity (Figure 4A).
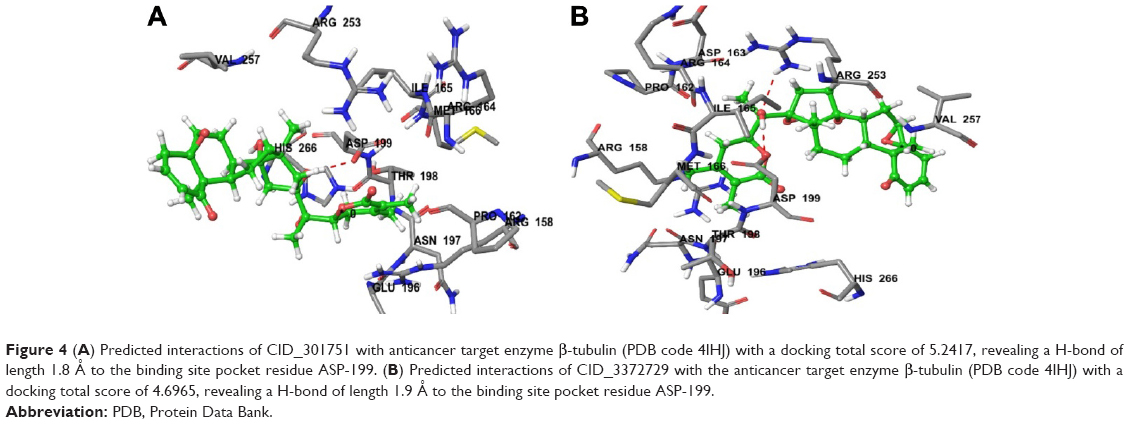 | Figure 4 (A) Predicted interactions of CID_301751 with anticancer target enzyme β-tubulin (PDB code 4IHJ) with a docking total score of 5.2417, revealing a H-bond of length 1.8 Å to the binding site pocket residue ASP-199. (B) Predicted interactions of CID_3372729 with the anticancer target enzyme β-tubulin (PDB code 4IHJ) with a docking total score of 4.6965, revealing a H-bond of length 1.9 Å to the binding site pocket residue ASP-199. |
Docking of compound CID_3372729 against β-tubulin resulted in a predicted total docking score of 4.6965 and form a hydrogen bond of length 1.9 Å to the polar acidic residue Asp-199. The binding site amino acid residues within a radius of 3 Å with ligand were hydrophobic Ile-165, Pro-162, Met-166 and Val-257; polar uncharged Thr-198; polar basic Arg-158, Arg-164 and Arg-253; polar acidic Asp-163, Asp-199 and Glu-196 and polar amide Asn-197. The presence of these strong hydrophobic and polar interactions with β-tubulin explains the high affinity of this molecule and its anticancer activity (Figure 4B).
PK properties, DL and bioavailability
All the withanolide analogs were predicted for their ADME properties. The predicted properties have shown that all withanolide analogs satisfy the Lipinski’s rule of five for oral bioavailability. The logP values were found to be in the range (logP<5) for all the compounds; however, withanolide 3, CID_73621 and CID_435144 showed low membrane permeability due to somewhat high MW (Table S9). The high value of their MWs in fact slightly violates one of Lipinski’s rule of five, since it affects the drug excretion (elimination from the body). Moreover, previous studies have suggested that molecules with intermediate lipophilicity are more likely to arrive to the receptor site.37,38 Predictions related to MDCK cell, skin permeability (Kp), Caco-2, CNS activity, metabolic reactions, log GI50 for HERG K+ channel blockage and human oral absorption in the gastrointestinal tract showed that these parameters for the active analogs fall within the standard ranges normally observed for drugs (Table 2). On the other hand, BBB and intestinal absorption penetration were predicted by developing an ADME model using 2D descriptors polar surface area and ALogP98 that define 95% and 99% confidence ellipses. These ellipses define the regions where well-absorbed compounds are expected to be found. All withanolide analogs were predicted to possess ~95% confidence levels for human intestinal absorption and BBB penetration, except active compounds CID_73621, CID_135887 and CID_435144, which showed 99% confidence level for intestinal absorption and 95% confidence level for BBB penetration, indicating good intestinal absorption and BBB penetration ability. The values and plot of polar surface area and ALogP for the withanolide analogs are shown in Table S10 and Figure 5, respectively.
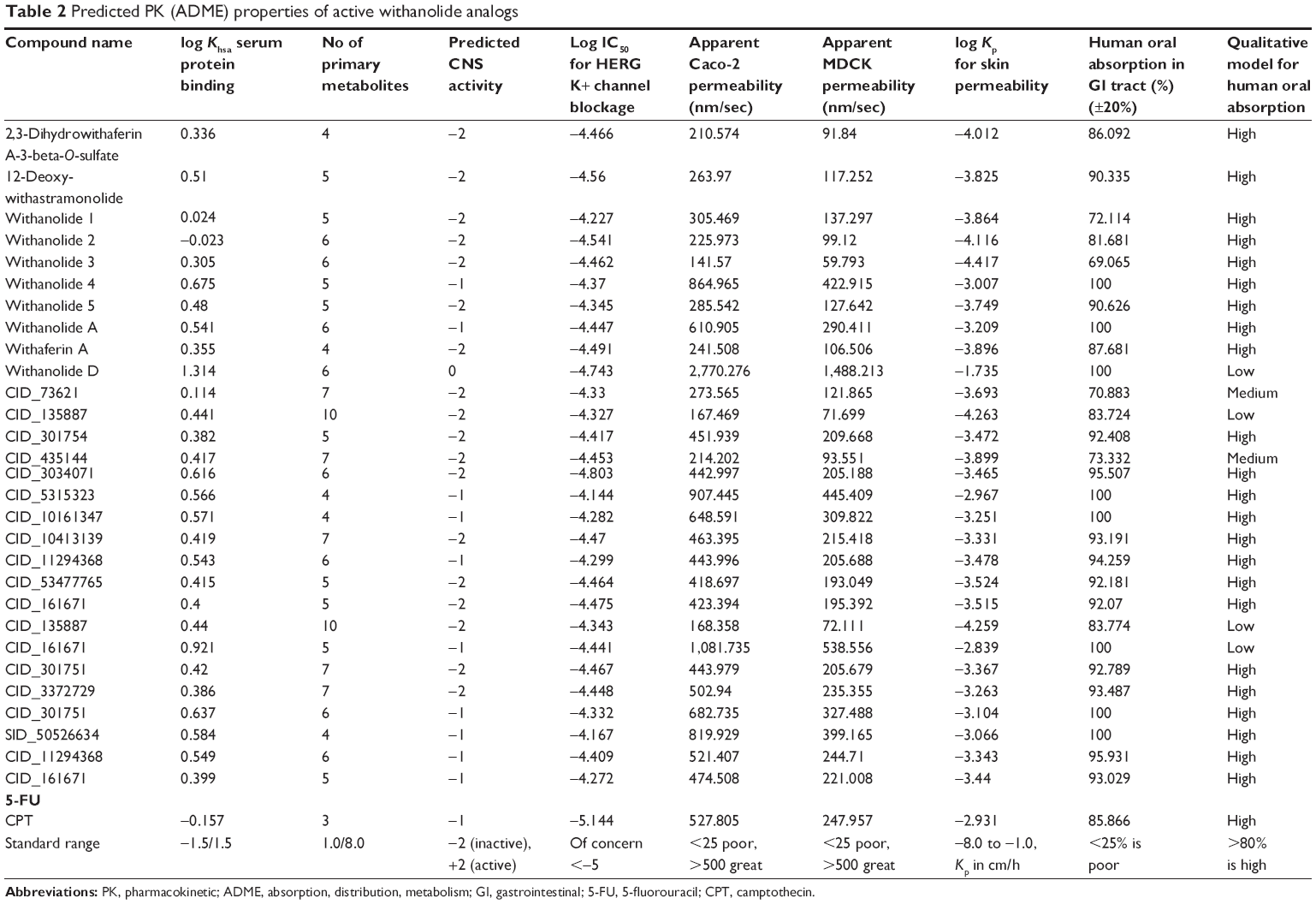 | Table 2 Predicted PK (ADME) properties of active withanolide analogs |
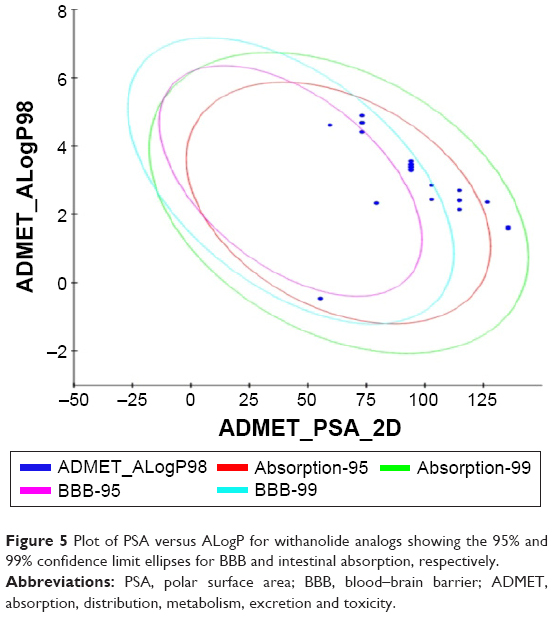 | Figure 5 Plot of PSA versus ALogP for withanolide analogs showing the 95% and 99% confidence limit ellipses for BBB and intestinal absorption, respectively. |
Toxicity risk assessment
The US Food and Drug Administration standard toxicity risk predictor software Osiris Toxicity Properties Calculator locates fragments within the structure of a molecule that indicates a potential toxicity risk.33–35 Toxicity risks parameters such as mutagenicity (MUT), tumorigenicity (TUMO), irritation (IRRI) and reproductive or developmental toxicity were computed for all the withanolide analogs. All compounds possess no risk of TUMO and developmental toxicity, with the exception of withanolide 2, which possesses high-risk developmental or reproductive toxicity. All active withanolide analogs were predicted to have high-risk MUT, except for compounds 2,3-dihydrowithaferin A-3-beta-O-sulfate, 12-deoxy-withastramonolide, withanolide 3, withanolide 5, withaferin A and CID_135887, which were predicted to have medium-risk toxicity. All active analogs were predicted to have high-risk skin IRRI, except for compounds 2,3-dihydrowithaferin A-3-beta-O-sulfate, 12-deoxy-withastramonolide, withanolide 1, withanolide 2, withanolide 3, withanolide 5, withaferin A and CID_135887, which showed medium-risk toxicity potential at high doses or long-term therapeutic use in human beings. The overall drug score (DS), which combines DL, hydrophilicity and toxicity risk parameters, was calculated for all compounds and was within acceptable limits (Table 3). The overall DS for all the active compounds predicted to be moderate to good, compared to reference anticancer compounds 5-FU and CPT.
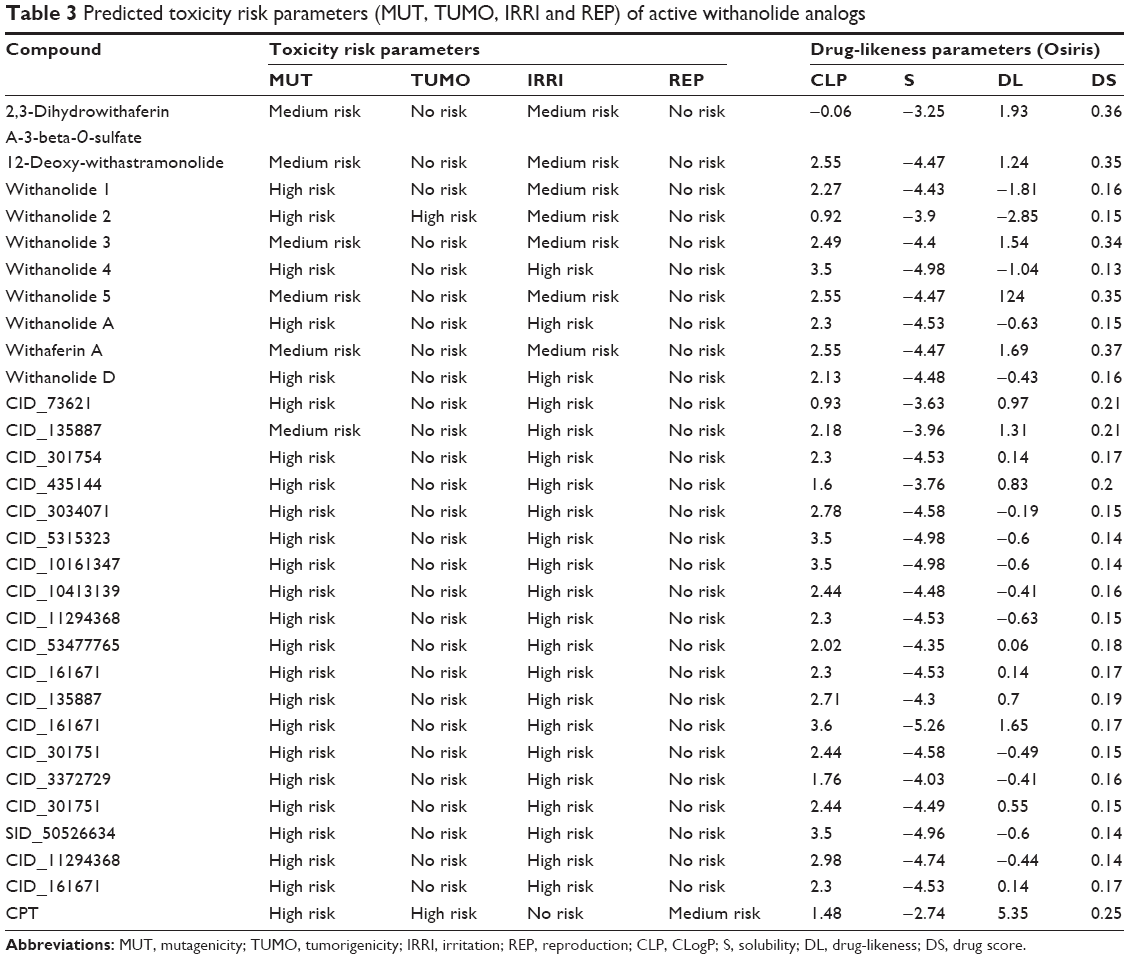 | Table 3 Predicted toxicity risk parameters (MUT, TUMO, IRRI and REP) of active withanolide analogs |
Conclusion
There are numerous statistically proven examples, where combined approaches of QSAR and molecular docking-based prediction have been applied successfully in the field of drug design and discovery. The present work of QSAR and molecular docking-based prediction of withanolide analogs showed that 2,3-dihydrowithaferin A-3-beta-O-sulfate, withanolide 5, withanolide A, withaferin A, CID_10413139, CID_11294368, CID_53477765, CID_135887, CID_301751 and CID_3372729 against Sk-Br-3 and CID_73621, CID_435144, CID_301751 and CID_3372729 possess a significant anticancer activity against the MCF7/BUS. The QSAR results for SK-Br-3 suggested that connectivity index (order 0, standard), dipole vector X (Debye), molar refractivity, shape index (basic kappa, order 2), whereas for MCF7/BUS, atom count (all atoms), dielectric energy (kcal/mole), total energy (hartree) and heat of formation (kcal/mole) correlated well with the activity. In docking studies, active withanolide analogs showed high binding affinity against β-tubulin receptor protein. The withanolide analogs CID_301751 and CID_3372729 showed good predicted activity and binding affinity to β-tubulin receptor protein. The docking results showed that the major influencing factors of molecular interactions between withanolide analogs and β-tubulin were H-bonds and hydrophobic and electrostatic interactions. The in silico prediction of oral bioavailability (rule of five) and ADMET risk profiling were within their acceptable limit for active analogs. These compounds have rationalized the structural requirement and need further lead optimization for designing of novel β-tubulin inhibitors.
Acknowledgments
We acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, India for financial support via the networking project BSC0121 at the CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow. We are thankful to the Director of CSIR-CIMAP, Lucknow for rendering essential research facilities and support. Author DKY is thankful to the Indian Council of Medical Research (ICMR), New Delhi for the award of Senior Research Fellowship (SRF) and Research Associate (RA). This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), which was funded by the Ministry of Education, Science and Technology (No. 2012R1A6A3A04038302 and 2017R1C1B2003380).
Disclosure
The authors report no conflicts of interest in this work.
References
Qurishi Y, Hamid A, Majeed R, et al. Interaction of natural products with cell survival and signaling pathways in the biochemical elucidation of drug targets in cancer. Future Oncol. 2011;7(8):1007–1021. | ||
Ali R, Mirza Z, Ashraf GM, et al. New anticancer agents: recent developments in tumor therapy. Anticancer Res. 2012;32(7):2999–3005. | ||
Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazón J. Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules. 2009;14(7):2373–2393. | ||
Vanden Berghe W, Sabbe L, Kaileh M, Haegeman G, Heyninck K. Molecular insight in the multifunctional activities of Withaferin A. Biochem Pharmacol. 2012;84(10):1282–1291. | ||
Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): a review. Altern Med Rev. 2000;5(4):334–346. | ||
Misra L, Mishra P, Pandey A, Sangwan RS, Sangwan NS, Tuli R. Withanolides from Withania somnifera roots. Phytochemistry. 2008;69(4):1000–1004. | ||
Chaurasiya ND, Uniyal GC, Lal P, et al. Analysis of withanolides in root and leaf of Withania somnifera by HPLC with photodiode array and evaporative light scattering detection. Phytochem Anal. 2008;19(2):148–154. | ||
Widodo N, Kaur K, Shrestha BG, et al. Selective killing of cancer cells by leaf extract of Ashwagandha: identification of a tumor-inhibitory factor and the first molecular insights to its effect. Clin Cancer Res. 2007;13(7):2298–2306. | ||
Padmavathi B, Rath PC, Rao AR, Singh RP. Roots of Withania somnifera inhibit forestomach and skin carcinogenesis in mice. Evid Based Complement Alternat Med. 2005;2(1):99–105. | ||
Devi PU, Sharada AC, Solomon FE. Antitumor and radiosensitizing effects of Withania somnifera (Ashwagandha) on a transplantable mouse tumor, Sarcoma-180. Indian J Exp Biol. 1993;31(7):607–611. | ||
Kumar P, Kushwaha RA. Medicinal evaluation of Withania somnifera (L.) Dunal (Ashwagandha). Asian J Chem. 2006;18(2):1401–1404. | ||
Sharada AC, Solomon FE, Devi PU. Toxicity of Withania somnifera root extract in rats and mice. Int J Pharm. 1993;31(3):205–212. | ||
Kupchan SM, Anderson WK, Bollinger P, et al. Tumor inhibitors. XXXIX. Active principles of Acnistus arborescens. Isolation and structural and spectral studies of withaferin A and withacnistin. J Org Chem. 1969;34(12):3858–3866. | ||
Shohat B, Gitter S, Abraham A, Lavie D. Antitumor activity of withaferin A (NSC-101088). Cancer Chemother Rep. 1967;51:271–276. | ||
Jayaprakasam B, Zhang Y, Seeram NP, Nair MG. Growth inhibition of human tumor cell lines by withanolides from Withania somnifera leaves. Life Sci. 2003;74(1):125–132. | ||
al-Hindawi MK, al-Khafaji SH, Abdul-Nabi MH. Anti-granuloma activity of Iraqi Withania somnifera. J Ethnopharmacol. 1992;37(2):113–116. | ||
Shohat B, Kirson I, Lavie D. Immunosuppressive activity of two plant steroidal lactones withaferin A and withanolide E. Biomedicine. 1978;28(1):18–24. | ||
Wang HC, Tsai YL, Wu YC, et al. Withanolides-induced breast cancer cell death is correlated with their ability to inhibit heat protein 90. PLoS One. 2012;7(5):e37764. | ||
Hahm ER, Lee J, Kim SH, et al. Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J Natl Cancer Inst. 2013;105(15):1111–1122. | ||
Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One. 2011;6(8):e23354. | ||
Stan SD, Hahm ER, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008;68(18):7661–7669. | ||
Lee J, Hahm ER, Singh SV. Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis. 2010;31(11):1991–1998. | ||
Lee J, Sehrawat A, Singh SV. Withaferin A causes activation of Notch2 and Notch4 in human breast cancer cells. Breast Cancer Res Treat. 2012;136(1):45–56. | ||
Dwivedi GR, Maurya A, Yadav DK, Khan F, Darokar MP, Srivastava SK. Drug resistance reversal potential of ursolic acid derivatives against nalidixic acid- and multidrug-resistant Escherichia coli. Chem Biol Drug Des. 2015;86(3):272–283. | ||
Gaur R, Yadav DK, Kumar S, Darokar MP, Khan F, Bhakuni RS. Molecular modeling based synthesis and evaluation of in vitro anticancer activity of indolyl chalcones. Curr Top Med Chem. 2015;15(11):1003–1012. | ||
Yadav DK, Rai R, Kumar N, et al. New arylated benzo[h]quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Sci Rep. 2016;6:38128. | ||
Attri P, Kumar N, Park JH, et al. Influence of reactive species on the modification of biomolecules generated from the soft plasma. Sci Rep. 2015;5:8221. | ||
Machin RP, Veleiro AS, Nicotra VE, Oberti JC, M Padrón J. Antiproliferative activity of withanolides against human breast cancer cell lines. J Nat Prod. 2010;73(5):966–968. | ||
Yadav DK, Dhawan S, Chauhan A, et al. QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Curr Drug Targets. 2014;15(8):753–761. | ||
Yadav DK, Kalani K, Singh AK, Khan F, Srivastava SK, Pant AB. Design, synthesis and in vitro evaluation of 18β-glycyrrhetinic acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Curr Med Chem. 2014;21(9):1160–1170. | ||
Yadav DK, Ahmad I, Shukla A, Khan F, Negi AS, Gupta A. QSAR and docking studies on chalcone derivatives for antitubercular activity against M. tuberculosis H37Rv. J Chemom. 2014;28:499–507. | ||
Antony ML, Lee J, Hahm ER, et al. Growth arrest by the antitumor steroidal lactone withaferin A in human breast cancer cells is associated with down-regulation and covalent binding at cysteine 303 of β-tubulin. J Biol Chem. 2014;289(3):1852–1865. | ||
Yadav DK, Khan F. QSAR, docking and ADMET studies of camptothecin derivatives as inhibitors of DNA topoisomerase-I. J Chemom. 2013;27:21–33. | ||
Yadav DK, Mudgal V, Agrawal J, et al. Molecular docking and ADME studies of natural compounds of Agarwood oil for topical anti-inflammatory activity. Curr Comput Aided Drug Des. 2013;9(3):360–370. | ||
Yadav DK, Kalani K, Khan F, Srivastava SK. QSAR and docking based semi-synthesis and in vitro evaluation of 18β-glycyrrhetinic acid derivatives against human lung cancer cell line A-549. Med Chem. 2013;9(8):1073–1084. | ||
QikProp. QikProp version 3.2. New York, NY: Schrödinger, LLC; 2013. | ||
Yadav DK, Khan F, Negi AS. Pharmacophore modeling, molecular docking, QSAR, and in silico ADMET studies of gallic acid derivatives for immunomodulatory activity. J Mol Model. 2012;18(6):2513–2525. | ||
Yadav DK, Meena A, Srivastava A, Chanda D, Khan F, Chattopadhyay SK. Development of QSAR model for immunomodulatory activity of natural coumarinolignoids. Drug Des Devel Ther. 2010;4:173–186. | ||
Kumar S, Tiwari M. Grid potential analysis, virtual screening studies and ADME/T profiling on N-arylsulfonylindoles as anti-HIV-1 agents. J Chemom. 2013;27(6):143–154. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.