")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 9
Molecular docking as a popular tool in drug design, an in silico travel
Authors de Ruyck J, Brysbaert G, Blossey R, Lensink M
Received 29 January 2016
Accepted for publication 12 March 2016
Published 24 June 2016 Volume 2016:9 Pages 1—11
DOI https://doi.org/10.2147/AABC.S105289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Jerome de Ruyck, Guillaume Brysbaert, Ralf Blossey, Marc F Lensink
University Lille, CNRS UMR8576 UGSF, Lille, France
Abstract: New molecular modeling approaches, driven by rapidly improving computational platforms, have allowed many success stories for the use of computer-assisted drug design in the discovery of new mechanism- or structure-based drugs. In this overview, we highlight three aspects of the use of molecular docking. First, we discuss the combination of molecular and quantum mechanics to investigate an unusual enzymatic mechanism of a flavoprotein. Second, we present recent advances in anti-infectious agents' synthesis driven by structural insights. At the end, we focus on larger biological complexes made by protein–protein interactions and discuss their relevance in drug design. This review provides information on how these large systems, even in the presence of the solvent, can be investigated with the outlook of drug discovery.
Keywords: structure-based drug design, protein–protein docking, quaternary structure prediction, residue interaction networks, RINs, water position
Introduction
Sequencing of the human genome has led to an increase in the number of new therapeutic targets for pharmaceutical research. In addition, high-throughput crystallography and nuclear magnetic resonance methods have been further developed and contributed to the acquisition of the atomic structures of proteins and protein–ligand complexes of an increasing level of detail.1 When the three-dimensional structure of the target, even from experiments or computing, exists, a frequently used technique to design inhibitor molecules is structure-based drug design (SBDD), which is depicted in Figure 1.
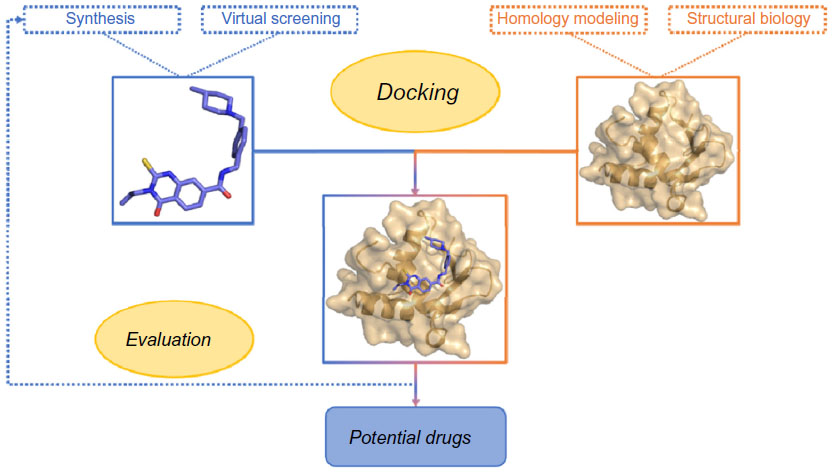 | Figure 1 Summary of a classical SBDD approach. |
The most popular method in SBDD is molecular docking. Initially, docking – a term which was coined in the late 1970s – meant the refinement of a model of a complex structure by optimization of the separation between the partners, but with fixed relative orientations. Later, this relative orientation was allowed to vary, but the internal geometry of each of the partners was held fixed. This type of modeling is often being referred to as rigid docking.2,3 Currently, thanks to further increases in computational resources, it has become possible to model changes in internal geometry of the interacting partners that may occur when a complex is formed. This type of modeling is also known as flexible docking.
Moreover, computational modeling of the quaternary structure of complexes, formed by two or more molecular interaction partners, is nowadays also feasible. Examples are protein–protein complexes and complexes between proteins and nucleic acids.4
In this review, we focus on modern usages of molecular docking. The first sections are dedicated to the design of new drug candidates starting from known crystal structures of crucial proteins. We then turn to protein–protein docking, including a discussion of the importance of water molecules in the docking procedure – how they are managed and, in the end, how they can influence binding probes. A list of the modeling programs discussed in this review is presented in Table 1.
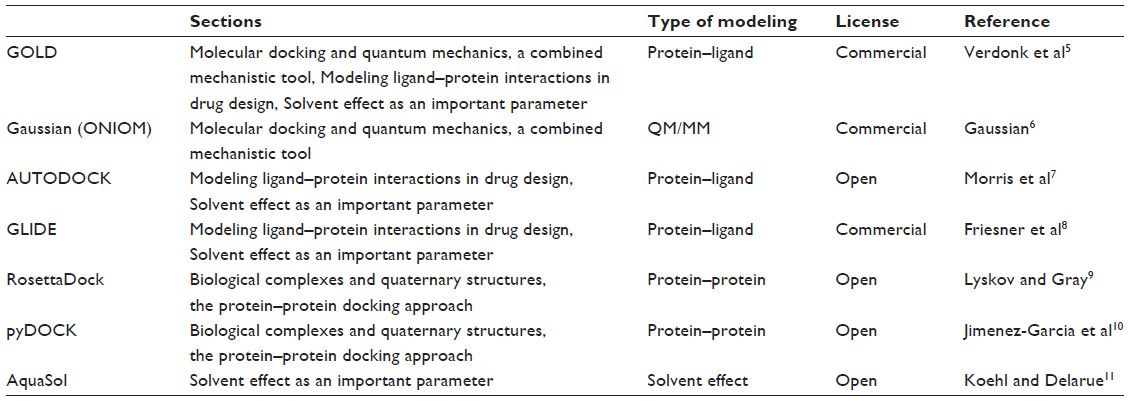 | Table 1 Summary of the modeling programs listed in this review |
Molecular docking and quantum mechanics, a combined mechanistic tool
A detailed understanding of the mechanisms of enzymes at atomic and electronic detail is of crucial importance in biomedical research.12,13 This would require solving the quantum mechanics (QM) of molecules, and thus, the computational costs of ab initio QM methods have limited their application. It is, for example, tenuous to elucidate a complete enzymatic mechanism, and therefore, methods have been devised to approximate the treatment. Several groups used combined approaches where calculations typically use a molecular mechanics (MM) force field for the system as a whole and apply an ab initio (QM) treatment to the site of interest. Using this QM/MM method, they were able to tackle different aspects of the biological systems studied such as electronic properties,14,15 interaction sites,16 or even conformational changes occurring in the protein active sites.17 Nowadays, a more advanced application of this approach is the ONIOM method.18 ONIOM stands for “our own N-layered integrated molecular orbital and molecular mechanics”. Originally developed by Morokuma in Dapprich et al19 and Svensson et al20 this computational technique models large molecules by defining more than two layers within the structure that are treated at different accuracy levels. By this way, the ONIOM method can treat relatively large molecules and can be applied in many areas of research and specifically organic and enzymatic reaction mechanisms.21 The modeling process involves two major steps: building the model and then mapping the enzymatic chemical process.
In order to highlight the importance of the very first step, we now describe the use of molecular docking to obtain a good starting point needed to elucidate the mechanistic pathway of an unusual flavoenzyme, isopentenyl diphosphate isomerase (IDI). Usually, flavoproteins are listed as redox catalysts, but in this specific case, there is no redox exchange observed.22 This enzyme catalyzes the isomerization of isopentenyl pyrophosphate into dimethylallyl pyrophosphate,23–25 which is the primary building block for all isoprenoid compounds. These are vital for every (micro-) organism, as these molecules are involved in every single cellular mechanism.26,27 Two types of IDIs have been reported. On the one hand, type I IDI (IDI-1) was discovered some decades ago. It has been extensively studied, and the mechanism was well established.28–30 On the other hand, type 2 IDI (IDI-2) was discovered more recently and is not so well characterized.31 This is a flavoprotein that requires reduced flavin mononucleotide cofactor and a divalent cation,32–34 but the mechanism is still vague and difficult to approach by experimental methods. As a presumption, it could be a protonation from N5 of reduced flavin mononucleotide leading to a carbocation, subsequently followed by a deprotonation step (Figure 2A).35,36 Thus, it is a critical enzyme for several classes of pathogenic microorganisms, and it is totally absent from human. Considering that novel chemotherapeutic strategies are urgently needed,37 new mechanism-based inhibitors of IDI-2 were sought. Recently, a structure-based approach was initiated for inhibitor development, since a high-resolution structure had been published.38 The goal was to investigate the putative mechanism by using QM/MM techniques. In this context, molecular docking provided the starting and ending points of the reaction path. As the apoprotein had been crystallized, the strategy was to dock both the substrate, isopentenyl pyrophosphate, and the product, dimethylallyl pyrophosphate, to the structure (Figure 2B). Then, protonation states were carefully inspected, and the energy of both the structures was minimized. Several other research groups are now using these methods to address the enzymatic mechanisms of a wide range of potential drug targets and to develop new mechanism-based inhibitors.39–44
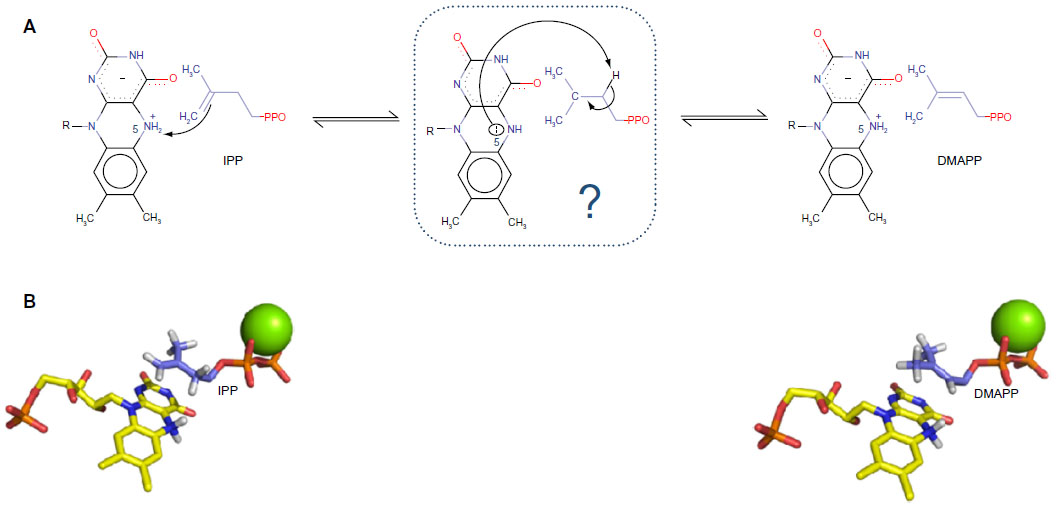 | Figure 2 (A) Proposed mechanistic scheme of the enzymatic reaction. N5 of the cofactor protonates the alkene moiety of IPP. The intermediate is still unclear, but the same N5 could, subsequently, act as a basis in order to yield DMAPP. (B) Docked structures of IPP (starting point) and DMAPP (ending point) in IDI-2 active site. The reduced FMN cofactor is represented in yellow and the divalent cation in green. |
Modeling ligand–protein interactions in drug design
Ligand binding is the key step in enzymatic reactions and, thus, for their inhibition. Therefore, a detailed understanding of interactions between small molecules and proteins may form the basis for a rational drug design strategy.45–48 This approach was widely considered in order to design molecules addressing a broad range of major pathologies such as cancers49,50 or cardiovascular diseases.51–53
Another example, which is emphasized here, is the successful use of docking to design lead compounds as new anti-infectious agents against Mycobacterium tuberculosis or Plasmodium falciparum. These two pathogens are the key actors in the development of tuberculosis and malaria, respectively, which are the two major causes of mortality in developing countries.54 In order to target this scourge, several research teams have studied, for a long time now, the nonmevalonate isoprenoid biosynthesis pathway (2-methyl-d-erythritol-4-phosphate [MEP] pathway). Indeed, these parasites rely on this cascade to produce their own isoprenoid compounds, critical for their survival.55–57 The second step of the pathway is the reduction of 1-deoxy-d-xylulose-5-phosphate to MEP catalyzed by 1-deoxy-d-xylulose-5-phosphate reductoisomerase (DXR).58 In addition, humans and animals do not rely on the MEP pathway, making DXR an attractive target in the search for novel families of drugs. Currently, several inhibitors of DXR have been synthesized and evaluated.59
The purpose of this subsection is to present the usage of structural data in order to improve the efficiency of a new family of drugs. In the absence of crystallographic structures of DXR from P. falciparum (pf-DXR) or M. tuberculosis, molecular modeling, based on the structure of DXR from Escherichia coli,60 allowed several research groups to further elucidate the structure and function of the enzyme and also facilitated structure-based inhibitor design. Consequently, models of DXR from the pathogens were built61 and used to develop efficient screening methods in order to identify potential lead compounds.62 Later, these models were validated by X-ray crystallography.63,64
Thereafter, on the basis of previous quantitative structure–activity relationship65 and crystallographic studies,66 several novel pyridine-containing fosmidomycin derivatives were designed and synthesized. They were found to be highly potent inhibitors of pf-DXR, having Ki values in the nanomolar range. Thus, these molecules were more active than fosmidomycin, the reference in the field of DXR inhibition67 (Figure 3). Recently, structure-guided design68 and virtual screening69 were successfully applied in order to identify and evaluate new molecules with a potent inhibitory effect on P. falciparum.
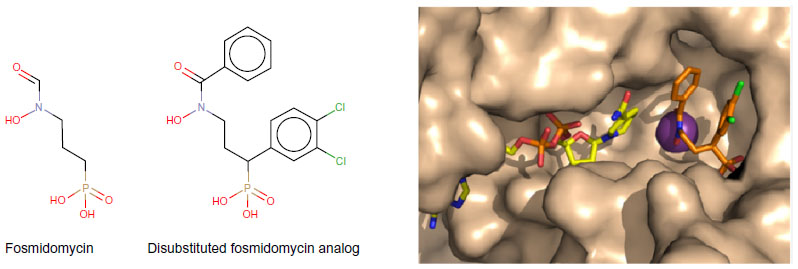 | Figure 3 Representation of a fosmidomycin analog more potent than fosmidomycin in terms of inhibiting Plasmodium falciparum. |
In light of these promising results, one can conclude that considerable progress has been made in the past, and the goal of obtaining clinically effective antimalarial drugs seems reachable.70–77
Biological complexes and quaternary structures, the protein–protein docking approach
Protein–protein interactions (PPIs) play a central role in all biological processes. These processes result from the physical interaction of several protein molecules, thus forming the macromolecular assemblies that effectuate cellular function. Many large-scale studies focusing on PPI have emerged in recent years, using graphs with nodes and edges to represent the protein components interacting with each other.78,79 Such binary representations capture a wealth of information but are inherently abstract and incomplete, since they contain no information as to time, place, or specificity. Such detailed information is indispensable for the guidance of mutagenesis studies or the design of inhibitor molecules.
Protein–protein docking actually predates protein–ligand (small molecule) docking, as the concept of protein docking introduced by Wodak and Janin80 was later extended to the interaction between macromolecules and small ligands.81 The treatment of flexibility in the binding process is considerably easier with small molecules, even though a considerable computational cost is involved, and small molecule docking has become one of the most active research areas in computational drug discovery. Most if not all of present-day protein–protein docking algorithms have been developed in light of the critical assessment of prediction of interactions (CAPRI) experiment, which is a community-wide collaboration that has accelerated the development of computational protein docking methods.82 CAPRI, which is modeled after critical assessment of protein structure prediction,83 organizes blind prediction trials; participants model their complex, and the models are assessed through comparison with an unknown crystal structure,82 made available to the assessors on a confidential basis and prior to publication.
The early years have been essential for the development of docking algorithms,84,85 with the incorporation of more elaborate scoring functions owing to efficient implementations of fast Fourier transform algorithms in docking86–88 as one of the key advancements. This also spawned the CAPRI scoring experiment, which was designed to help developers test scoring functions independently from docking calculations.89 In the scoring experiment, participants are given access to an enriched ensemble of docking models, contributed on a voluntary basis by participants in the docking experiment. The scorers select models from this ensemble and make a docking submission that is assessed using the standard CAPRI evaluation criteria. Although scorers are generally apt at discriminating near-native solutions from a selection of incorrect decoys, a correct ranking of these remains problematic.90 Benchmark data sets dedicated to scoring protein complexes have been developed recently that should facilitate the development of scoring functions.91,92
During the last years, development was shifted to more realistic docking scenarios. CAPRI no longer offers so-called “bound” targets, where the structure of one or both of the partners is supplied in their bound conformation. Dockers nowadays routinely use unbound structures, that is, the structures of the binding partners as they occur in solution, and some degree of conformational change needs to be taken into account.93 Moreover, often only the sequence of the interacting partners is provided, and a step of homology modeling is required prior to docking. Although these matters significantly complicate the docking procedure, they represent the realistic scenarios that computational biologists nowadays are presented with.
The PPI interfaces have received increased attention during the past years.94,95 A prediction of the residues involved in the interaction interface may be used to guide the docking itself.96–97 Subsequent optimization of the interaction interface requires modification of side chain orientation; protein docking algorithms increasingly include flexibility treatments in their docking procedures, and more recent implementations favor the simultaneous docking of ensembles of unbound conformers.98,100 Together, the reliable prediction of interface residues and the incorporation of global and local flexibility in the docking algorithms provide invaluable information to inform mutagenesis studies and to steer drug design applications.101–104
The scoring functions of docking programs have been successfully improved with additional descriptors based on residue interaction networks (RINs).105,106 RINs consist of networks generated from three-dimensional structures, where nodes correspond to residues and edges to detected interactions. RINs are small-world networks, and their topological analyses have been used in particular to study protein–protein interfaces107 and protein–ligand binding108–111 and to optimize scoring functions for the evaluation of docking poses.105,106 Using different approaches, it has been demonstrated that combining the network measures such as closeness centrality, betweenness centrality, degree, or clustering coefficient with energy terms can improve the ranking in scoring functions. Chang et al used two different types of RINs, a hydrophobic one and a hydrophilic one, for each complex, and then calculated a network-based score considering the average degrees and clustering coefficients. They developed a scoring method that enhanced discrimination of the scoring method of RosettaDock112 by >10% on a subset of protein–protein docking benchmark 2.0.113 Pons et al generated the RIN of each protein individually before docking and calculated four measures of the network, including closeness centrality. By integrating a score based on the closeness values into the pyDock scoring function,114 they improved by as much as 36% the top ten success rate on the protein–protein docking benchmark 4.0.115 Furthermore, residue centrality analyses as performed by del Sol et al,116 which are based on the average shortest path length, can also be used on docked poses to evaluate the central residues located in the interface. These residues could subsequently be targeted for mutagenesis experiments or drug design.117
The docking predictions can be used in combination with homology-based methodologies and integrated into PPI networks to enhance these with structural information.118,119 The Interactome3D120 web service incorporates structural data into PPI networks to improve them with interface information. These structural data either come from experiments or are modeled through a comparative modeling pipeline. Mosca et al120 illustrate the value of this tool by showing it allowed them to suggest a potential mechanism of action common to several disease-causing mutations. Indeed, they observed the mutations on structures of the complement cascade pathway involving, in particular, the complement component 3 (C3) and the component factor H (CFH) interaction. Several disease-causing mutations were located at the interface of proteins, and these key elements could be targeted by drugs in order to stabilize the C3–CFH interface. Thus, with this type of network, it is possible to contextualize mutations related to different diseases involved in a pathway and draw potential links between them. It can help to better define the target to aim for and, hence, improve the drug design.120 Docking predictions could then be additionally integrated to these networks. Furthermore, it was predicted that on average a drug binds to six different targets, including both the primary target and additional “off-targets”.121 Following this idea, reverse docking can be performed, which consists in the screening of one single molecule against multiple receptors instead of screening multiple small molecules against several receptors.122,123 Homology modeling may be useful to enrich the screening, when experimental structures are not available. The building of structural PPI networks may then be used in drug design to predict the targets the drug may bind to, with their related potential adverse drug reactions.122 They can help to identify which proteins would be affected by a drug designed to disrupt a particular interface because they highlight the domains that are involved in PPI. These structural PPI networks can also be exploited for drug repositioning, considering the use of known approved drugs or the reconsideration of late-stage failures.
Solvent effect as an important parameter
Proteins in solution are surrounded by water molecules. Water molecules around proteins organize in hydration shells that show correlated fluctuations.124 They are responsible for electrostatic screening125 and make important contributions to enzyme substrate recognition and catalysis and to molecular recognition in general.126,127
Considerable effort has been devoted to the modeling of water molecules in protein–ligand docking procedures, where the importance of water-mediated contacts has long been recognized. Well-known docking packages such as GOLD, AUTODOCK, or GLIDE can incorporate water molecules explicitly to predict protein–ligand docking poses.128–130 But very few methods exist that allow the prediction of hydration water positions at protein–protein interfaces.
The important contribution of water in the binding105 between proteins is readily realized when considering the high-affinity barnase–barstar complex.131 The extracellular ribonuclease barnase is always expressed with its inhibitor barstar in order to prevent the bacterium from degrading its own RNA. The complex is noted for its extremely tight binding, with a kon rate of 108 M−1 s−1 and an affinity kd ≈10−14 M. The complex has been extensively studied both experimentally and computationally, explaining in detail its binding energetics.132 The binding interface, which is mainly composed of polar and charged residues, contains as many as 51 associated water molecules, of which no less than 18 are fully buried.133 Water plays a key role in the binding process; it was shown that interfacial layers of water molecules exhibit anisotropic behavior and form a collaborative network that facilitates the binding of the interfaces.134
Interfacial water molecules play a critical role also in both the stability and the specificity of colicin DNase–immunity protein complexes.135 The complex between endonuclease colicin E2 and its Im2 immunity protein (E2/Im2) was presented as CAPRI target T47 with an addition to the docking experiment: groups submitting standard docking predictions were invited to also predict the positions of water molecules in the interface of the complex, using the method of their choice.136 These were then compared to the water positions in the crystal structure, a high-resolution (1.72 Å) structure determined at cryogenic temperatures (100 K).137 The docking itself presented little challenge, as both cognate (PDB 1emv; E9/Im9, PDB 7cei; E7/Im7) and noncognate (PDB 2wpt; E9/Im2; CAPRI T41) templates were available, but the prediction of interfacial water molecules proved to be much more difficult: only four of the 88 high-quality (root mean square with target <1.0 Å) models submitted, that is, <5%, were found to have a water-mediated contact recall fraction >50%. A water-mediated contact is defined as a receptor–ligand contact where either ligand and receptor molecules have one or more heavy atoms within a 3.5 Å distance of the same water molecule. These results attest the relative immaturity of protein interface water prediction and show that further work is needed to attain a performance that is of practical use for drug design applications. Nevertheless, some promising observations could be made, namely, that three highly conserved water molecules, which are believed to be part of the protein–protein interface hotspot, were among the best predicted water positions and that another water molecule, involved in the specificity for the family of complexes, was also relatively well predicted.136
Hydrophilic association characterizes most nonobligate protein complexes. Also in transient protein–protein interactions, which lie at the basis of most cellular processes, water plays an essential, mediating role.138 Although larger in size, protein–protein interfaces constitute weaker binding sites with respect to small molecules. Successful well-known drugs such as aspirin and ibuprofen transiently bind such protein–protein interfaces and do not shut down, but rather modulate overstimulated signal transduction pathways. PPIs are increasingly targeted in drug design, which is now entering the systems biology era.139 The successful development of drugs targeting such protein–protein interfaces indubitably benefits from a reliable prediction of interfacial water molecules.140
For the association of large assemblies, continuum approaches may prove useful for the prediction of water molecule positions at interfaces and, in particular, for the energetic characterization of (large) complexes. Recently, Smaoui et al141 modeled the formation of amyloid fibrils, protein aggregates that cause brain tissue damage, and compared the findings with experiment. They employed molecular dynamics simulations and, for the calculation of solvation-free energies, a continuum description using an extension of the standard Poisson–Boltzmann equation. This extension, the Poisson–Boltzmann–Langevin equation, considers the water molecules as point dipoles.142 A solver for the Poisson–Boltzmann–Langevin equation had previously been developed by Koehl and Delarue.11
Conclusion
High-throughput X-ray crystallography of a target alone or in complex with small molecules has significantly grown these last years. With the development of increasingly more sophisticated computational tools, SBDD is becoming a key step in the development of target-based therapies. These integrative approaches, which are primarily driven by increasingly powerful computational platforms, have allowed many success stories of the use of computer-assisted drug design in the discovery of new drugs. In addition, molecular docking approaches are being used to reach other goals such as the elucidation of noncanonical enzymatic mechanisms or the depiction of the quaternary structure of biological protein complexes. Such analyses, which are used in close coupling with traditional medicinal chemistry techniques, are increasingly relevant with drug design entering the systems biology era.
Acknowledgments
We acknowledge the support from the research federation FRABio (CNRS FR3688), “Structural and Functional Biochemistry of Biomolecular Assemblies”. JdR acknowledges funding from the Nord-Pas-de-Calais Regional Council and M de Wergifosse for fruitful discussion about ONIOM method. RB and MFL acknowledge financial support from the French Agence Nationale de Recherche, project “Fluctuations in Structured Coulomb Fluids”, grant number ANR-12-BSV5-0009-01. MFL is grateful to the French Agence Nationale de Recherche for his grant number ANR-13-BSV8-0002-0.
Disclosure
The authors report no conflicts of interest in this work.
References
Gore M, Desai NS. Computer-aided drug designing. Methods Mol Biol. 2014;1168:313–321. | |
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery:methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. | |
Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking:a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. | |
Zacharias M. Protein-Protein Complexes:Analysis, Modeling and Drug Design. London, Imperial College Press. 2010. | |
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52(4):609–623. | |
Gaussian, Inc. Gaussian 09 [Computer Program]. Wallingford, CT:Gaussian, Inc.; 2009. | |
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4:automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | |
Friesner RA, Banks JL, Murphy RB, et al. Glide:a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. | |
Lyskov S, Gray JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36(Web Server issue):W233–W238. | |
Jimenez-Garcia B, Pons C, Fernandez-Recio J. pyDockWEB:a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics. 2013;29(13):1698–1699. | |
Koehl P, Delarue M. AQUASOL:an efficient solver for the dipolar Poisson-Boltzmann-Langevin equation. J Chem Phys. 2010;132(6):064101. | |
Naray-Szabo G, Olah J, Kramos B. Quantum mechanical modeling:a tool for the understanding of enzyme reactions. Biomolecules. 2013;3(3):662–702. | |
Polyak I, Reetz MT, Thiel W. Quantum mechanical/molecular mechanical study on the mechanism of the enzymatic Baeyer-Villiger reaction. J Am Chem Soc. 2012;134(5):2732–2741. | |
de Wergifosse M, de Ruyck J, Champagne B. How the second-order nonlinear optical response of the collagen triple helix appears:a theoretical investigation. J Phys Chem C. 2014;118(16):8595–8602. | |
de Ruyck J, Famerée M, Wouters J, Perpète EA, Preat J, Jacquemin D. Towards the understanding of the absorption spectra of NAD(P)H/NAD(P)+ as a common indicator of dehydrogenase enzymatic activity. Chem Phys Lett. 2007;450:119–122. | |
Gresh N, Audiffren N, Piquemal JP, de Ruyck J, Ledecq M, Wouters J. Analysis of the interactions taking place in the recognition site of a bimetallic Mg(II)-Zn(II) enzyme, isopentenyl diphosphate isomerase. A parallel quantum-chemical and polarizable molecular mechanics study. J Phys Chem B. 2010;114(14):4884–4895. | |
Goldwaser E, de Courcy B, Demange L, et al. Conformational analysis of a polyconjugated protein-binding ligand by joint quantum chemistry and polarizable molecular mechanics. Addressing the issues of anisotropy, conjugation, polarization, and multipole transferability. J Mol Model. 2014;20(11):2472. | |
Vreven T, Byun KS, Komaromi I, et al. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J Chem Theory Comput. 2006;2:815–826. | |
Dapprich S, Komaromi I, Byun KS, Morokuma K, Frish MJ. A new ONIOM implementation in Gaussian 98. Part 1. The calculation of energies, gradients and vibrational frequencies and electric field derivatives. J Mol Struct (Theochem). 1999;462:1. | |
Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. ONIOM:a multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels–Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J Phys Chem. 1996;100(50):19357–19363. | |
Chung LW, Sameera WM, Ramozzi R, et al. The ONIOM method and its applications. Chem Rev. 2015;115(12):5678–5796. | |
Unno H, Yamashita S, Ikeda Y, et al. New role of flavin as a general acid-base catalyst with no redox function in type 2 isopentenyl-diphosphate isomerase. J Biol Chem. 2009;284(14):9160–9167. | |
Sobrado P. Noncanonical reactions of flavoenzymes. Int J Mol Sci. 2012;13(11):14219–14242. | |
Berthelot K, Estevez Y, Deffieux A, Peruch F. Isopentenyl diphosphate isomerase:a checkpoint to isoprenoid biosynthesis. Biochimie. 2012;94(8):1621–1634. | |
de Ruyck J, Wouters J, Poulter CD. Inhibition studies on enzymes involved in isoprenoid biosynthesis:focus on two potential drug targets:DXR and IDI-2 enzymes. Curr Enzym Inhib. 2011;7(2):79–95. | |
Hahn FM, Xuan JW, Chambers AF, Poulter CD. Human isopentenyl diphosphate:dimethylallyl diphosphate isomerase:overproduction, purification, and characterization. Arch Biochem Biophys. 1996;332(1):30–34. | |
Schwender J, Seemann M, Lichtenthaler HK, Rohmer M. Biosynthesis of isoprenoids (carotenoids, sterols, prenyl side-chains of chlorophylls and plastoquinone) via a novel pyruvate/glyceraldehyde 3-phosphate non-mevalonate pathway in the green alga Scenedesmus obliquus. Biochem J. 1996;316(pt 1):73–80. | |
de Ruyck J, Durisotti V, Oudjama Y, Wouters J. Structural role for Tyr-104 in Escherichia coli isopentenyl-diphosphate isomerase:site-directed mutagenesis, enzymology, and protein crystallography. J Biol Chem. 2006;281(26):17864–17869. | |
Durbecq V, Sainz G, Oudjama Y, et al. Crystal structure of isopentenyl diphosphate:dimethylallyl diphosphate isomerase. EMBO J. 2001;20(7):1530–1537. | |
Lee S, Poulter CD. Escherichia coli type I isopentenyl diphosphate isomerase:structural and catalytic roles for divalent metals. J Am Chem Soc. 2006;128(35):11545–11550. | |
Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. An unusual isopentenyl diphosphate isomerase found in the mevalonate pathway gene cluster from Streptomyces sp. strain CL190. Proc Natl Acad Sci U S A. 2001;98(3):932–937. | |
Dutoit R, de Ruyck J, Durisotti V, Legrain C, Jacobs E, Wouters J. Overexpression, physicochemical characterization, and modeling of a hyperthermophilic pyrococcus furiosus type 2 IPP isomerase. Proteins. 2008;71(4):1699–1707. | |
de Ruyck J, Rothman SC, Poulter CD, Wouters J. Structure of Thermus thermophilus type 2 isopentenyl diphosphate isomerase inferred from crystallography and molecular dynamics. Biochem Biophys Res Commun. 2005;338(3):1515–1518. | |
Rothman SC, Helm TR, Poulter CD. Kinetic and spectroscopic characterization of type II isopentenyl diphosphate isomerase from Thermus thermophilus:evidence for formation of substrate-induced flavin species. Biochemistry. 2007;46(18):5437–5445. | |
Nagai T, Unno H, Janczak MW, Yoshimura T, Poulter CD, Hemmi H. Covalent modification of reduced flavin mononucleotide in type-2 isopentenyl diphosphate isomerase by active-site-directed inhibitors. Proc Natl Acad Sci U S A. 2011;108(51):20461–20466. | |
Heaps NA, Poulter CD. Type-2 isopentenyl diphosphate isomerase:evidence for a stepwise mechanism. J Am Chem Soc. 2011;133(47):19017–19019. | |
Shenoy ES, Paras ML, Noubary F, Walensky RP, Hooper DC. Natural history of colonization with methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE):a systematic review. BMC Infect Dis. 2014;14:177. | |
de Ruyck J, Janczak MW, Neti SS, et al. Determination of kinetics and the crystal structure of a novel type 2 isopentenyl diphosphate:dimethylallyl diphosphate isomerase from Streptococcus pneumoniae. Chembiochem. 2014;15(10):1452–1458. | |
Singh T, Adekoya OA, Jayaram B. Understanding the binding of inhibitors of matrix metalloproteinases by molecular docking, quantum mechanical calculations, molecular dynamics simulations, and a MMGBSA/MMBappl study. Mol Biosyst. 2015;11(4):1041–1051. | |
Nitoker N, Major DT. Understanding the reaction mechanism and intermediate stabilization in mammalian serine racemase using multiscale quantum-classical simulations. Biochemistry. 2015;54(2):516–527. | |
Selvaraj C, Singh P, Singh SK. Molecular insights on analogs of HIV PR inhibitors toward HTLV-1 PR through QM/MM interactions and molecular dynamics studies:comparative structure analysis of wild and mutant HTLV-1 PR. J Mol Recognit. 2014;27(12):696–706. | |
Kumar RP, Roopa L, Nongthomba U, Sudheer Mohammed MM, Kulkarni N. Docking, molecular dynamics and QM/MM studies to delineate the mode of binding of CucurbitacinE to F-actin. J Mol Graph Model. 2016;63:29–37. | |
Samanta PN, Das KK. Prediction of binding modes and affinities of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamide inhibitors to the carbonic anhydrase receptor by docking and ONIOM calculations. J Mol Graph Model. 2016;63:38–48. | |
Qiu L, Lin J, Bertaccini EJ. Insights into the nature of anesthetic-protein interactions:an ONIOM study. J Phys Chem B. 2015;119(40):12771–12782. | |
Jazayeri A, Dias JM, Marshall FH. From G protein-coupled receptor structure resolution to rational drug design. J Biol Chem. 2015;290(32):19489–19495. | |
Singh R, Singh S, Nath Pandey P. In-silico analysis of Sirt2 from Schistosoma monsoni:structures, conformations and interactions with inhibitors. J Biomol Struct Dyn. 2016. 34(5):1042-51. doi:10.1080/07391102.2015.1065205. | |
Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66(1):334–395. | |
Alvarez Dorta D, Sivignon A, Chalopin T, et al. The Antiadhesive Strategy in Crohn’s Disease:Orally Active Mannosides to Decolonize Pathogenic Escherichia coli from the Gut. Chembiochem. 2016;17(10):936–952. | |
Amin KM, Anwar MM, Kamel MM, Kassem EM, Syam YM, Elseginy SA. Synthesis, cytotoxic evaluation and molecular docking study of novel quinazoline derivatives as PARP-1 inhibitors. Acta Pol Pharm. 2013;70(5):833–849. | |
Sabbah DA, Saada M, Khalaf RA, et al. Molecular modeling based approach, synthesis, and cytotoxic activity of novel benzoin derivatives targeting phosphoinostide 3-kinase (PI3Kalpha). Bioorg Med Chem Lett. 2015;25(16):3120–3124. | |
Frederick R, Robert S, Charlier C, et al. 3,6-disubstituted coumarins as mechanism-based inhibitors of thrombin and factor Xa. J Med Chem. 2005;48(24):7592–7603. | |
Dong MH, Chen HF, Ren YJ, Shao FM. Molecular modeling studies, synthesis and biological evaluation of dabigatran analogues as thrombin inhibitors. Bioorg Med Chem. 2016;24(2):73–84. | |
Mena-Ulecia K, Tiznado W, Caballero J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS One. 2015;10(11):e0142774. | |
Vitoria M, Granich R, Gilks CF, et al. The global fight against HIV/AIDS, tuberculosis, and malaria:current status and future perspectives. Am J Clin Pathol. 2009;131(6):844–848. | |
Jomaa H, Wiesner J, Sanderbrand S, et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285(5433):1573–1576. | |
Obiol-Pardo C, Rubio-Martinez J, Imperial S. The methylerythritol phosphate (MEP) pathway for isoprenoid biosynthesis as a target for the development of new drugs against tuberculosis. Curr Med Chem. 2011;18(9):1325–1338. | |
Guggisberg AM, Amthor RE, Odom AR. Isoprenoid biosynthesis in Plasmodium falciparum. Eukaryot Cell. 2014;13(11):1348–1359. | |
Takahashi S, Kuzuyama T, Watanabe H, Seto H. A 1-deoxy-D-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-D-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis. Proc Natl Acad Sci U S A. 1998;95(17):9879–9884. | |
Jackson ER, Dowd CS. Inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr):a review of the synthesis and biological evaluation of recent inhibitors. Curr Top Med Chem. 2012;12(7):706–728. | |
Reuter K, Sanderbrand S, Jomaa H, et al. Crystal structure of 1-deoxy-D-xylulose-5-phosphate reductoisomerase, a crucial enzyme in the non-mevalonate pathway of isoprenoid biosynthesis. J Biol Chem. 2002;277(7):5378–5384. | |
Singh N, Avery MA, McCurdy CR. Toward Mycobacterium tuberculosis DXR inhibitor design:homology modeling and molecular dynamics simulations. J Comput Aided Mol Des. 2007;21(9):511–522. | |
Goble JL, Adendorff MR, de Beer TA, Stephens LL, Blatch GL. The malarial drug target Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase (PfDXR):development of a 3-D model for identification of novel, structural and functional features and for inhibitor screening. Protein Pept Lett. 2010;17(1):109–120. | |
Andaloussi M, Henriksson LM, Wieckowska A, et al. Design, synthesis, and X-ray crystallographic studies of alpha-aryl substituted fosmidomycin analogues as inhibitors of Mycobacterium tuberculosis 1-deoxy-D-xylulose 5-phosphate reductoisomerase. J Med Chem. 2011;54(14):4964–4976. | |
Umeda T, Tanaka N, Kusakabe Y, Nakanishi M, Kitade Y, Nakamura KT. Crystallization and preliminary X-ray crystallographic study of 1-deoxy-D-xylulose 5-phosphate reductoisomerase from Plasmodium falciparum. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(pt 3):330–332. | |
Silber K, Heidler P, Kurz T, Klebe G. AFMoC enhances predictivity of 3D QSAR:a case study with DOXP-reductoisomerase. J Med Chem. 2005;48(10):3547–3563. | |
Henriksson LM, Unge T, Carlsson J, Aqvist J, Mowbray SL, Jones TA. Structures of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase provide new insights into catalysis. J Biol Chem. 2007;282(27):19905–19916. | |
Xue J, Diao J, Cai G, et al. Antimalarial and structural studies of pyridine-containing inhibitors of 1-deoxyxylulose-5-phosphate reductoisomerase. ACS Med Chem Lett. 2013;4(2):278–282. | |
Cobb RE, Bae B, Li Z, DeSieno MA, Nair SK, Zhao H. Structure-guided design and biosynthesis of a novel FR-900098 analogue as a potent Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr) inhibitor. Chem Commun (Camb). 2015;51(13):2526–2528. | |
Chaudhary KK, Prasad CV. Virtual screening of compounds to 1-deoxy-dxylulose 5-phosphate reductoisomerase (DXR) from Plasmodium falciparum. Bioinformation. 2014;10(6):358–364. | |
Verbrugghen T, Vandurm P, Pouyez J, Maes L, Wouters J, Van Calenbergh S. Alpha-heteroatom derivatized analogues of 3-(acetylhydroxyamino)propyl phosphonic acid (FR900098) as antimalarials. J Med Chem. 2013;56(1):376–380. | |
Jansson AM, Wieckowska A, Bjorkelid C, et al. DXR inhibition by potent mono- and disubstituted fosmidomycin analogues. J Med Chem. 2013;56(15):6190–6199. | |
Masini T, Hirsch AK. Development of inhibitors of the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J Med Chem. 2014;57(23):9740–9763. | |
Chofor R, Sooriyaarachchi S, Risseeuw MD, et al. Synthesis and bioactivity of beta-substituted fosmidomycin analogues targeting 1-deoxy-D-xylulose-5-phosphate reductoisomerase. J Med Chem. 2015;58(7):2988–3001. | |
Hasan MA, Mazumder MH, Chowdhury AS, Datta A, Khan MA. Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment. Source Code Biol Med. 2015;10:7. | |
Mendoza-Martinez C, Correa-Basurto J, Nieto-Meneses R, et al. Design, synthesis and biological evaluation of quinazoline derivatives as anti-trypanosomatid and anti-plasmodial agents. Eur J Med Chem. 2015;96:296–307. | |
Mbengue A, Bhattacharjee S, Pandharkar T, et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 2015;520(7549):683–687. | |
Shah P, Tiwari S, Siddiqi MI. Recent progress in the identification and development of anti-malarial agents using virtual screening based approaches. Comb Chem High Throughput Screen. 2015;18(3):257–268. | |
Guruharsha KG, Rual JF, Zhai B, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147(3):690–703. | |
Havugimana PC, Hart GT, Nepusz T, et al. A census of human soluble protein complexes. Cell. 2012;150(5):1068–1081. | |
Wodak SJ, Janin J. Computer analysis of protein-protein interaction. J Mol Biol. 1978;124(2):323–342. | |
Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161(2):269–288. | |
Janin J, Henrick K, Moult J, et al. CAPRI:a critical assessment of predicted interactions. Proteins. 2003;52(1):2–9. | |
Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP) – round x. Proteins. 2014;82(suppl 2):1–6. | |
Mendez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein-protein interactions:current status of docking methods. Proteins. 2003;52(1):51–67. | |
Mendez R, Leplae R, Lensink MF, Wodak SJ. Assessment of CAPRI predictions in rounds 3-5 shows progress in docking procedures. Proteins. 2005;60(2):150–169. | |
Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition:determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89(6):2195–2199. | |
Ritchie DW, Kemp GJ. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39(2):178–194. | |
Ritchie DW, Kozakov D, Vajda S. Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics. 2008;24(17):1865–1873. | |
Lensink MF, Mendez R, Wodak SJ. Docking and scoring protein complexes:CAPRI 3rd Edition. Proteins. 2007;69(4):704–718. | |
Lensink MF, Wodak SJ. Docking and scoring protein interactions:CAPRI 2009. Proteins. 2010;78(15):3073–3084. | |
Kastritis PL, Bonvin AM. Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J Proteome Res. 2010;9(5):2216–2225. | |
Lensink MF, Wodak SJ. Score_set:a CAPRI benchmark for scoring protein complexes. Proteins. 2014;82(11):3163–3169. | |
Lensink MF, Mendez R. Recognition-induced conformational changes in protein-protein docking. Curr Pharm Biotechnol. 2008;9(2):77–86. | |
Lensink MF, Wodak SJ. Blind predictions of protein interfaces by docking calculations in CAPRI. Proteins. 2010;78(15):3085–3095. | |
Fleishman SJ, Whitehead TA, Strauch EM, et al. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J Mol Biol. 2011;414(2):289–302. | |
Dominguez C, Boelens R, Bonvin AM. HADDOCK:a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–1737. | |
May A, Zacharias M. Protein-protein docking in CAPRI using ATTRACT to account for global and local flexibility. Proteins. 2007;69(4):774–780. | |
Mustard D, Ritchie DW. Docking essential dynamics eigenstructures. Proteins. 2005;60(2):269–274. | |
Krol M, Chaleil RA, Tournier AL, Bates PA. Implicit flexibility in protein docking:cross-docking and local refinement. Proteins. 2007;69(4):750–757. | |
Chaudhury S, Gray JJ. Conformer selection and induced fit in flexible backbone protein-protein docking using computational and NMR ensembles. J Mol Biol. 2008;381(4):1068–1087. | |
Sable R, Jois S. Surfing the protein-protein interaction surface using docking methods:application to the design of PPI inhibitors. Molecules. 2015;20(6):11569–11603. | |
Bier D, Thiel P, Briels J, Ottmann C. Stabilization of protein-protein interactions in chemical biology and drug discovery. Prog Biophys Mol Biol. 2015;119(1):10–19. | |
Persico M, Di Dato A, Orteca N, et al. From protein communication to drug discovery. Curr Top Med Chem. 2015;15(20):2019–2031. | |
Kuenemann MA, Sperandio O, Labbe CM, Lagorce D, Miteva MA, Villoutreix BO. In silico design of low molecular weight protein-protein interaction inhibitors:overall concept and recent advances. Prog Biophys Mol Biol. 2015;119(1):20–32. | |
Pons C, Glaser F, Fernandez-Recio J. Prediction of protein-binding areas by small-world residue networks and application to docking. BMC Bioinformatics. 2011;12:378. | |
Chang S, Jiao X, Li CH, Gong XQ, Chen WZ, Wang CX. Amino acid network and its scoring application in protein-protein docking. Biophys Chem. 2008;134(3):111–118. | |
del Sol A, O’Meara P. Small-world network approach to identify key residues in protein-protein interaction. Proteins. 2005;58(3):672–682. | |
Amitai G, Shemesh A, Sitbon E, et al. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344(4):1135–1146. | |
Hu Z, Bowen D, Southerland WM, et al. Ligand binding and circular permutation modify residue interaction network in DHFR. PLoS Comput Biol. 2007;3(6):e117. | |
Liu R, Hu J. Computational prediction of heme-binding residues by exploiting residue interaction network. PLoS One. 2011;6(10):e25560. | |
Xue W, Ban Y, Liu H, Yao X. Computational study on the drug resistance mechanism against HCV NS3/4A protease inhibitors vaniprevir and MK-5172 by the combination use of molecular dynamics simulation, residue interaction network, and substrate envelope analysis. J Chem Inf Model. 2014;54(2):621–633. | |
Gray JJ, Moughon S, Wang C, et al. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331(1):281–299. | |
Mintseris J, Wiehe K, Pierce B, et al. Protein-protein docking benchmark 2.0:an update. Proteins. 2005;60(2):214–216. | |
Cheng TM, Blundell TL, Fernandez-Recio J. pyDock:electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins. 2007;68(2):503–515. | |
Hwang H, Vreven T, Janin J, Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78(15):3111–3114. | |
del Sol A, Fujihashi H, Amoros D, Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol Syst Biol. 2006;2(2006):0019. | |
Villoutreix BO, Kuenemann MA, Poyet JL, et al. Drug-like protein- protein interaction modulators:challenges and opportunities for drug discovery and chemical biology. Mol Inform. 2014;33(6–7):414–437. | |
Hammad N, Jingdong J. Structure-based protein-protein interaction networks and drug design. Quantitative Biology. 2013;1(Issue 3):183–191. | |
Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol. 2012;30(2):159–164. | |
Mosca R, Ceol A, Aloy P. Interactome3D:adding structural details to protein networks. Nat Methods. 2013;10(1):47–53. | |
Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. The topology of drug-target interaction networks:implicit dependence on drug properties and target families. Mol Biosyst. 2009;5(9):1051–1057. | |
Xie L, Xie L, Bourne PE. Structure-based systems biology for analyzing off-target binding. Curr Opin Struct Biol. 2011;21(2):189–199. | |
Yang L, Chen J, He L. Harvesting candidate genes responsible for serious adverse drug reactions from a chemical-protein interactome. PLoS Comput Biol. 2009;5(7):e1000441. | |
Zhang L, Yang Y, Kao YT, Wang L, Zhong D. Protein hydration dynamics and molecular mechanism of coupled water-protein fluctuations. J Am Chem Soc. 2009;131(30):10677–10691. | |
Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44(4):400–417. | |
Ben-Naim A. Molecular recognition – viewed through the eyes of the solvent. Biophys Chem. 2002;10(1–102):309–319. | |
Bienstock RJ. Solvation methods for protein-ligand docking. Methods Mol Biol. 2015;1289:3–12. | |
Verdonk ML, Chessari G, Cole JC, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48(20):6504–6515. | |
Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures:incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46(1):34–40. | |
Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide:docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. | |
Guillet V, Lapthorn A, Hartley RW, Mauguen Y. Recognition between a bacterial ribonuclease, barnase, and its natural inhibitor, barstar. Structure. 1993;1(3):165–176. | |
Wang T, Tomic S, Gabdoulline RR, Wade RC. How optimal are the binding energetics of barnase and barstar? Biophys J. 2004;87(3):1618–1630. | |
Buckle AM, Schreiber G, Fersht AR. Protein-protein recognition:crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry. 1994;33(30):8878–8889. | |
Ahmad M, Gu W, Geyer T, Helms V. Adhesive water networks facilitate binding of protein interfaces. Nat Commun. 2011;2:261. | |
Meenan NA, Sharma A, Fleishman SJ, et al. The structural and energetic basis for high selectivity in a high-affinity protein-protein interaction. Proc Natl Acad Sci U S A. 2010;107(22):10080–10085. | |
Lensink MF, Moal IH, Bates PA, et al. Blind prediction of interfacial water positions in CAPRI. Proteins. 2014;82(4):620–632. | |
Wojdyla JA, Fleishman SJ, Baker D, Kleanthous C. Structure of the ultra-high-affinity colicin E2 DNase – Im2 complex. J Mol Biol. 2012;417(1–2):79–94. | |
Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions:structural, functional, and network properties. Structure. 2010;18(10):1233–1243. | |
Keskin O, Gursoy A, Ma B, Nussinov R. Towards drugs targeting multiple proteins in a systems biology approach. Curr Top Med Chem. 2007;7(10):943–951. | |
Ahmed MH, Spyrakis F, Cozzini P, et al. Bound water at protein-protein interfaces:partners, roles and hydrophobic bubbles as a conserved motif. PLoS One. 2011;6(9):e24712. | |
Smaoui MR, Poitevin F, Delarue M, Koehl P, Orland H, Waldispuhl J. Computational assembly of polymorphic amyloid fibrils reveals stable aggregates. Biophys J. 2013;104(3):683–693. | |
Azuara C, Orland H, Bon M, Koehl P, Delarue M. Incorporating dipolar solvents with variable density in Poisson-Boltzmann electrostatics. Biophys J. 2008;95(12):5587–5605. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.