Back to Journals » Cancer Management and Research » Volume 10
Molecular characterization and biomarker identification in colorectal cancer: Toward realization of the precision medicine dream
Authors Goel G ![]()
Received 11 June 2018
Accepted for publication 31 August 2018
Published 19 November 2018 Volume 2018:10 Pages 5895—5908
DOI https://doi.org/10.2147/CMAR.S162967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Gaurav Goel
Division of Hematology-Oncology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Abstract: Colorectal cancer (CRC) is a major public health problem, both in the USA and globally. Over the past 20 years, significant advances have been made in the treatment of patients with metastatic CRC (mCRC). Recent efforts in the field of biomarkers have focused on the development of molecular diagnostics to define the subset of patients with mCRC that is likely to derive most benefit from anti-EGFR therapy. Herein, we review the recent advancements in molecular stratification of CRC and the role of current as well as emerging biomarkers in this disease. It is now clear that the presence of activating mutations in the KRAS and NRAS genes serves as reliable predictive markers for resistance to anti-EGFR therapy in mCRC. It is also clear that further improvements in the survival of mCRC patients will probably be made possible only with identification of new predictive molecular biomarkers and their evaluation using rational and innovative clinical trials. The recent advances in DNA sequencing technology and “omics”-based approaches have provided promising new strategies for the development of novel molecular biomarkers in this disease.
Keywords: colorectal cancer, biomarkers, circulating tumor DNA, next-generation sequencing, personalized medicine, omics
Introduction
Colorectal cancer (CRC) is a major public health problem, both in the USA and globally. In the USA, it is the second leading cause of cancer-related mortality, and in 2018, it is estimated that nearly 50,630 deaths will be attributed to this disease.1 When metastatic disease is diagnosed, CRC is usually associated with poor prognosis with 5-year survival rates less than 15%.1 With the advances in the treatment of metastatic CRC (mCRC) over the past 20 years, the median overall survival (OS) has been steadily increasing, and it has now reached beyond 40 months in a select group of patients.2–4 This improvement in OS has been made possible with the identification of new agents and incorporation of targeted biologic agents into the cytotoxic chemotherapy regimens used for treatment of mCRC patients.5,6 Although the progress made thus far is encouraging, the existing treatment paradigm usually employs a “one-size-fits-all” approach, which translates into demonstrable clinical benefit from any given chemotherapeutic regimen in only a small subset of treated patients.
It is now being increasingly realized that CRC is not a single disease entity, but a heterogeneous group of tumors, both at the intertumoral and intratumoral levels. Given the complex molecular makeup of these tumors, significant efforts have been focused in recent years on identifying potential molecular markers that would provide further information regarding tumor response to anticancer therapies, which in turn would help personalize treatment for mCRC. “Predictive” biomarkers identify patients who are most likely to benefit from a specific treatment, and they can, therefore, play a critical role in guiding treatment decisions. Research in the identification of predictive biomarkers for mCRC has been directed toward the targets that are downstream in the EGFR signal transduction cascade, including mutations in the KRAS, NRAS, BRAF, and PIK3CA genes, and PTEN protein expression. “Prognostic” biomarkers, on the other hand, provide information about the tumor’s aggressiveness, and they help distinguish patients based on their expected disease outcome, independent of the treatment received. It is believed that molecular stratification of CRC can identify biomarkers to provide such additional prognostic and predictive information, over and beyond the conventional TNM staging system. Such validated biomarkers will also help optimize efficacy, minimize toxicity, and reduce treatment costs.
This article will review the recent advancements in molecular stratification of CRC and the role of current as well as emerging biomarkers in this disease.
Subtypes of colorectal tumors
CRC subtyping based on traditional oncogenic pathways
CRC has been theorized as a stepwise model of genetic and epigenetic events by Fearon and Vogelstein, nearly three decades ago.7 It usually begins with the inactivation of APC tumor suppressor gene, resulting in increased Wnt pathway signaling.8 Thereafter, depending on the mechanism of underlying genomic alterations, CRC develops through one of the three distinct molecular pathways – chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP) pathways. Of these, CIN pathway is the most commonly occurring (85%), and it involves widespread loss of heterozygosity and gross chromosomal abnormalities leading to hypermutability. MSI and CIMP pathways result in development of colorectal tumors due to a defective DNA mismatch repair (MMR) system. MMR system is a multiprotein system responsible for proofreading and eliminating any nucleotide mismatches that escape the checking system of DNA polymerase during DNA replication process. While the MSI pathway is characterized by the presence of genetic or sporadic mutations in the MMR genes, the CIMP pathway involves epigenetic silencing of the MMR gene MLH1 by causing hypermethylation of the CpG dinucleotide sequences located in the gene’s promoter region.
Transcriptome-based CRC subtypes
The international Colorectal Cancer Subtyping Consortium studied six independent transcriptome-based classification systems of CRC, and the resulting collection of 27 subtypes were coalesced into four distinct Consensus Molecular Subtype (CMS) groups.9 Transcriptional molecular signatures refine disease subclassification and help to identify subtypes with distinct biologic behavior. CMS1 group (MSI immune subtype, 14%) consists of hypermutated MSI tumors with strong immune activation. These tumors have overexpression of proteins involved in DNA damage repair consistent with defective DNA mismatch repair. BRAF mutations occur frequently in these tumors, and an increased expression of genes associated with diffuse immune infiltrate is noted. CMS2 (canonical subtype, 37%) group is characterized by epithelial differentiation and chromosomally unstable tumors. The tumors in this group display high somatic copy number alterations, and upregulation of WNT and MYC downstream targets. CMS3 (metabolic subtype, 13%) tumors show epithelial differentiation and marked metabolic dysregulation. Nearly 30% of these tumors are hypermutated as well. There is a higher prevalence of CIMP and KRAS activating mutations in CMS3. CMS4 (mesenchymal subtype, 23%) consists of tumors with prominent TGF-β activation, stromal invasion, and angiogenesis.
Established biomarkers in current clinical practice
RAS (KRAS and NRAS) mutations
The RAS gene family is composed of three well-known genes: KRAS, NRAS, and HRAS.10 KRAS mutations are estimated to be present in ~30%–45% of CRC patients and include single-point mutations in exons 2, 3, and 4.11 Another 5% of CRC tumors harbor activating mutations in NRAS, in the same exons as KRAS. These genes encode GTP/GDP binding protein, which is a key player in the MAP kinase signal transduction pathway.
The role of RAS mutations as a negative predictive marker for anti-EGFR therapy in the treatment of mCRC has been confirmed in multiple clinical trials and is now well established.12–18 Several systematic reviews and meta-analyses have also confirmed that among the anti-EGFR monoclonal antibody-treated mCRC patients, KRAS wild-type patients perform better than KRAS-mutant population; and that among the KRAS wild-type patients, treatment with anti-EGFR monoclonal antibody-containing regimen is associated with better response than chemotherapy alone.19–22 Studies have also clearly established that in contrast to the KRAS wild-type tumors, the addition of anti-EGFR therapy to chemotherapy does not improve the outcomes in KRAS-mutant population and is even potentially harmful.16,17
It is, however, worth noting that in these earlier studies, only mutations in exon 2 of the KRAS gene were assessed. More recent data suggest that in addition to mutations in KRAS exon 2, other RAS mutations in KRAS exons 3 and 4, and NRAS exons 2, 3, and 4 are also associated with resistance to anti-EGFR therapy in mCRC.23–25 The most convincing evidence establishing the predictive value of “expanded” RAS testing is provided by the meta-analysis of nine randomized clinical trials consisting of nearly 6,000 patients.24 In this meta-analysis, the tumors with new RAS mutations (KRAS exons 3 and 4, NRAS exons 2, 3, and 4) were compared with tumors without any RAS mutation, with respect to anti-EGFR treatment-related progression-free survival (PFS) and OS benefit. Patients with tumors that were KRAS exon 2 wild-type, but harbored RAS mutations in KRAS exons 3 and 4, or NRAS exons 2, 3, and 4 had significantly inferior survival outcomes compared to those without any RAS mutation.
Taken together, it is now well established that presence of any activating mutation in KRAS or NRAS predicts for resistance to anti-EGFR therapy in mCRC. A recent guideline on molecular testing for CRC issued jointly by the American Society for Clinical Pathology (ASCP), College of American Pathologists (CAP), Association of Molecular Pathology (AMP), and American Society of Clinical Oncology (ASCO), recommends expanded RAS testing, which includes mutational analysis of KRAS and NRAS codons 12 and 13 of exon 2, codons 59 and 61 of exon 3, and codons 117 and 146 of exon 4 for mCRC patients being considered for anti-EGFR therapy.26 The mCRC patients whose tumors harbor these mutations should not receive anti-EGFR therapy as they will not derive clinical benefit from such treatment. Perhaps, a greater concern is that such patients are likely to experience an even worse clinical outcome. It is also worth noting that the absence of RAS mutation is not the most accurate predictor of tumor response to anti-EGFR monoclonal antibody, as only 40%–50% of patients with KRAS/NRAS wild-type disease will ultimately respond to such therapy.
In contrast to the predictive value, the role of KRAS mutation in CRC as an “independent” prognostic marker has yielded inconsistent results. In one of the early efforts, several researchers with information about the KRAS tumor genotype and outcome of CRC patients came together to form a collaborative database.27 The data on 3,439 patients recruited from 42 centers in 21 countries were entered into a multivariate analysis, and showed that KRAS mutation in codon 12 was associated with an increased risk of tumor recurrence and worse survival. In another retrospective analysis of tumor samples obtained from CRC patients treated with chemotherapy (without anti-EGFR monoclonal antibody) in the MRC FOCUS trial, the presence of mutation in either KRAS (codons 12, 13, and 61) or BRAF was associated with significantly decreased survival when compared to the wild-type tumors.28 In contrast, the CO.17 study13 and the study by Kim et al29 did not demonstrate any significant survival difference between the wild-type KRAS and mutant-KRAS subgroups. In a systematic review with meta-analysis also, KRAS mutation status did not correlate with prognosis in CRC patients.30 Due to the inconsistencies in data, KRAS mutational status cannot be used reliably as an independent prognostic marker for mCRC patients at this time.
Table 1 provides a summary of established and emerging biomarkers in CRC and their potential role in clinical practice.
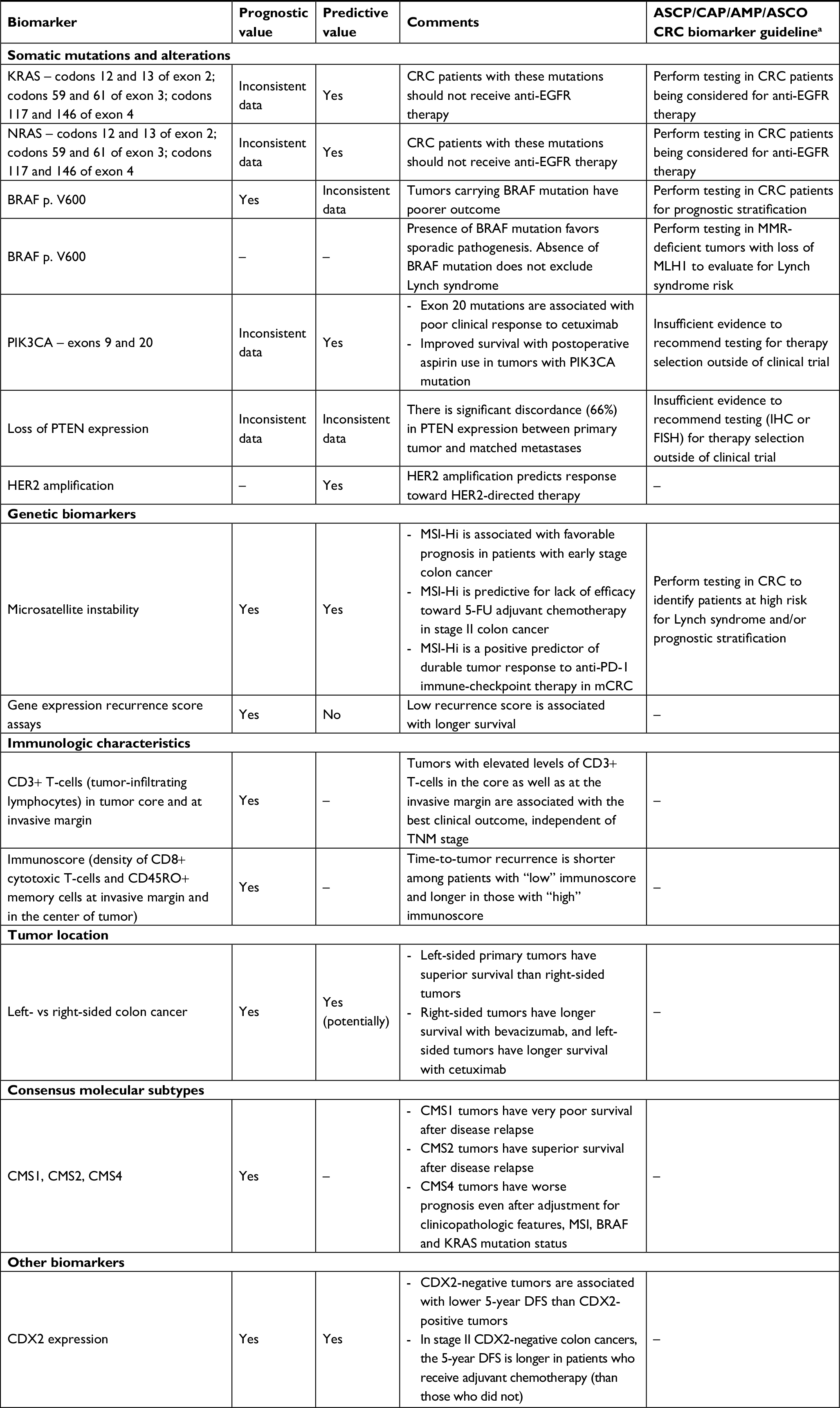 | Table 1 Summary of established and emerging biomarkers in the management of colorectal cancer Note: aSepulveda et al.26 Abbreviations: 5-FU, 5-fluorouracil; AMP, Association of Molecular Pathology; ASCO, American Society of Clinical Oncology; ASCP, American Society for Clinical Pathology; CAP, College of American Pathologists; CRC, colorectal cancer; DFS, disease-free survival; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; MMR, DNA mismatch repair; MSI-Hi, microsatellite instability-high; PD-1, programmed death-1. |
BRAF mutations
BRAF is a member of the RAF proto-oncogene family, and a key player of the RAS/RAF/MAP kinase pathway. Activating point mutations in BRAF are present in ~5%–10% of CRC.31 This mutation results in constitutive activation of BRAF kinase, which then leads to activation of the downstream RAS/RAF/MAP kinase pathway.
Several independent studies have consistently demonstrated that the presence of BRAFV600E mutations is associated with poor prognosis in patients with mCRC.23,31–35 As the population of BRAF-mutant mCRC patients is relatively small due to the low mutation prevalence, several meta-analyses and systematic reviews have been performed to provide more conclusive evidence on the prognostic and predictive value of BRAF mutation as it relates to anti-EGFR therapy.36–41 In one of the largest meta-analyses of 21 clinical trials that included 5,229 mCRC patients treated with anti-EGFR monoclonal antibody, 343 patients displayed BRAF mutations out of 4,616 (7.4%) patients with known BRAF status.38 Patients with BRAF wild-type showed an improved PFS (HR, 0.38; 95% CI 0.29–0.51) and an improved OS (HR, 0.35; 95% CI 0.29–0.42), compared to the BRAF-mutant group. These results indicate that the presence of BRAF mutation is associated with poor prognosis in mCRC patients.
In mCRC, the presence of BRAF mutation has been suggested as a negative predictor of response toward anti-EGFR therapy, based on the results from the retrospective study by Di Nicolantonio et al32 and the PICCOLO trial.42 On the other hand, in a meta-analysis of eight randomized clinical trials that included RAS wild-type mCRC patients treated with anti-EGFR therapy, there was lack of statistically significant difference in OS (P=0.43) between the BRAF-mutant and BRAF wild-type subgroups.40 In another meta-analysis, the addition of anti-EGFR therapy in the BRAF-mutant advanced CRC patients did not significantly improve the survival compared to the control regimens.41
The available clinical evidence suggests that the presence of BRAF mutations has a clear prognostic value in mCRC as it confers a significantly worse clinical outcome. Recent guidelines recommend that BRAFV600E mutational analysis should be performed in all mCRC patients for prognostic stratification.26 However, the available data on BRAF mutations as a predictive marker for anti-EGFR therapy are limited due to being largely retrospective in nature. The relatively low prevalence of BRAF mutation in colon cancer poses the greatest challenge in addressing this question prospectively. Therefore, at the present time, there is insufficient evidence to recommend BRAF mutation testing on colorectal tumors to predict response to anti-EGFR therapy.26
MSI and tumor mutational burden
The loss of MMR proteins leads to MMR deficiency, and these tumors are also referred to as microsatellite instability-high (MSI-Hi) tumors. In fact, MSI is considered as the molecular fingerprint of a deficient MMR system. The Cancer Genome Atlas (TCGA) project reported that ~16% of all CRCs are hypermutated or MSI-Hi.43 The majority of MSI-Hi CRCs (11%–13%) are caused by somatic changes, either resulting from epigenetic silencing of the MLH1 gene in the majority of cases or due to somatic mutation in any of the four most common MMR genes (MLH1, MSH2, MSH6, and PMS2) or POLE gene.43–45 The remaining 3%–5% MSI-Hi CRCs are caused by germline mutation in the MMR genes, and these patients are referred to as having the hereditary nonpolyposis CRC (Lynch syndrome).44–47
MSI status has been evaluated as a prognostic and predictive biomarker in CRC. MSI-Hi CRCs are more often located in the proximal colon, are poorly differentiated, and have mucinous or medullary histology. These tumors are also considered highly immunogenic.48 It is now understood that the length variations in the microsatellites of coding sequence in MSI-Hi tumors lead to frame-shift mutations. The high mutational burden associated with these tumors results in formation of tumor-specific neoantigens and subsequent T-cell infiltration, which suggests presence of a robust antitumoral immune response.48 This explains the observation that MSI-Hi is associated with favorable prognosis in patients with early stage colon cancer.49 It is now recommended that MMR status should be checked in patients with CRC for prognostic stratification.26
MSI status also has predictive value, both in early stage and advanced CRC.50 The presence of MSI-Hi has been shown to be predictive for lack of efficacy toward 5-FU adjuvant chemotherapy in early-stage colon cancer.51 Therefore, adjuvant 5-FU should be avoided for stage II MSI-Hi colon cancer. In patients with advanced CRC, the data from early clinical trials suggest that the presence of MSI-Hi status is a positive predictor of durable tumor response to anti-programmed death-1 (PD-1) immune-checkpoint therapy.52–55 A Phase II study evaluated the clinical efficacy of anti-PD-1 agent pembrolizumab in patients with chemorefractory mCRC, with and without MMR deficiency.54 In this study, the MSI-Hi CRCs were found to have encouraging responses to pembrolizumab. The overall response rate (ORR) was 50% for MSI-Hi CRC and 0% for microsatellite stable (MSS) CRC.55 The median OS was not reached in the MSI-Hi CRC cohort, as opposed to 6 months in the MSS CRC cohort (HR, 0.247; P=0.001). Based on these encouraging results, a phase III study (KEYNOTE-177, NCT02563002) of pembrolizumab in MSI-Hi advanced CRC is ongoing to confirm these early observations.
Similarly, in another phase II study (CheckMate-142), nivolumab monotherapy was shown to provide clinical benefit (ORR, 31%; 12-month PFS, 50%; 12-month OS, 73%) in previously treated patients with MSI-Hi mCRC patients.56 Nivolumab was also combined with the CTLA-4 inhibitor ipilimumab to synergistically promote T-cell antitumor activity and further improve the outcomes among MSI-Hi mCRC patients.57 In the nivolumab plus ipilimumab cohort of the CheckMate-142 study, patients received nivolumab plus ipilimumab every 3 weeks for four doses, followed by nivolumab once every 2 weeks. Among the 119 patients treated with this combination, ORR was 55% at a median follow-up of 13.4 months. The 12-month PFS and OS rates were 71% and 85%, respectively. Based on these encouraging results, the Food and Drug Administration granted accelerated approval to the combination of nivolumab and ipilimumab in MSI-Hi mCRC patients that have progressed beyond first-line chemotherapy.
Recent evidence suggests that the presence of a high tumor mutational load might be a more important and accurate predictive marker of response to immunotherapy than the tumor’s MSI status itself. This is illustrated by the fact that PD-1 immunotherapy has shown efficacy in colorectal tumors carrying the POLE mutation.58 This type of tumor is classified as an MSS tumor but is characterized by the presence of very high mutational burden and tumor-infiltrating lymphocytes in its microenvironment.
Gene expression profiling-based recurrence score assays
Multigene expression signatures have been developed for use as biomarkers in early stage colon cancer, such as stage II disease where the role of adjuvant chemotherapy is still unclear. Examples of gene expression signatures that can guide CRC prognosis include Oncotype DX and ColoPrint assays. The Oncotype DX is a 12-gene RT-PCR-based recurrence score assay that was shown to have prognostic value in patients with stage II colon cancer, independent of the traditional clinicopathologic factors.59,60 ColoPrint is an 18-gene microarray-based assay that differentiates stage II tumors into low and high risk of recurrence.61 Stage II colon cancer patients who were identified as having low-risk disease based on the ColoPrint assay had significantly improved disease-free survival (DFS) than those in the high-risk group (87.6% vs 67.2%; HR, 2.5; P=0.005). Although these recurrence score assays have a definite prognostic value in early stage colon cancer, their ability to predict benefit from adjuvant chemotherapy has not been established yet.62
Emerging biomarkers
PIK3CA mutations
PIK3CA gene encodes for the p110α catalytic subunit of the PI3K protein, which is a component of the PI3K/PTEN/AKT signaling pathway. PIK3CA mutations occur in 10%–18% of CRCs, mainly in exons 9 and 20.31,43,63,64 When mutated, PIK3CA induces phosphorylation of AKT, which promotes cell growth and suppresses apoptosis in CRCs.
The impact of PIK3CA mutations on the outcome of mCRC has been evaluated in retrospective studies. In the study by De Roock et al, it was suggested that not all PIK3CA mutations behave similarly in terms of response to anti-EGFR therapy.31 PIK3CA exon 9 mutations had no effect, whereas exon 20 mutations were associated with poor clinical response to cetuximab compared to wild-type PIK3CA tumors (OS, 34 weeks vs 51 weeks [HR, 3.29; P=0.0057]). Several meta-analyses have also been performed to study the association between PIK3CA mutational status and clinical response to anti-EGFR therapy in mCRC patients.39,65,66 In one such systematic review with meta-analysis, Yang et al evaluated the predictive value of specific exon mutations of PIK3CA (exons 9 and 20) in response to anti-EGFR therapy in KRAS wild-type mCRC patients.39 PIK3CA exon 20 mutations (but not PIK3CA exon 9 mutations) were associated with shorter PFS and OS. In another meta-analysis by Wu et al, the presence of a PIK3CA mutation was associated with poorer PFS (HR, 1.53; P<0.001) and OS (HR, 1.28; P=0.015) in the unselected mCRC patients treated with anti-EGFR therapy.66 The negative predictive value of PIK3CA mutation toward anti-EGFR therapy was even stronger when the analysis was restricted to patients with only KRAS wild-type disease (HR for PFS was 2.44; P=0.004).
Based on these results, the presence of PIK3CA mutations may be considered a negative predictive biomarker for anti-EGFR therapy in patients with wild-type KRAS mCRC, and this is largely driven by PIK3CA mutations in exon 20. However, as the available clinical data are largely retrospective in nature, the recent CRC biomarker guideline recommends against routine PIK3CA mutational analysis for selection of therapy outside of a clinical trial.26
Loss of PTEN expression
PTEN acts as a tumor suppressor gene, where its normal function is to inhibit PI3K-initiated signaling. Accordingly, loss of PTEN expression results in activation of the PI3K/AKT pathway. PTEN loss is present in 20%–40% of the unselected CRC cases when evaluated by immunohistochemistry (IHC). The available data surrounding the role of PTEN expression as a biomarker in CRC are inconsistent and discordant.
Data from a few retrospective studies suggest that PTEN expression in CRC might have a prognostic value, such that low PTEN expression is associated with aggressive tumor behavior, liver metastases, lymphatic invasion, venous invasion, and poor survival.67–70 On the other hand, there are several studies that refute the prognostic influence of PTEN in CRC.71,72
Similarly, PTEN expression has also been evaluated for its predictive value toward anti-EGFR therapy in mCRC but has shown inconsistent results. In a few studies, the loss of PTEN expression was associated with resistance to anti-EGFR therapy.73–76 The largest of these is the retrospective study by Loupakis et al,75 which evaluated the effect of PTEN expression loss on the clinical outcome of mCRC patients treated with cetuximab-based therapy. Patients with PTEN loss had significantly shorter PFS than those with PTEN-positive tumors (3.3 months vs 4.7 months; HR, 0.49; P<0.005). In contrast, several other published reports suggest that PTEN loss is not predictive of response to anti-EGFR therapy.33,77–79
Due to these conflicting results, the precise role of PTEN status as a prognostic and predictive biomarker of CRC remains unclear. In addition to the inconsistency in data, there are other potential issues that limit the use of PTEN loss as a biomarker. PTEN loss is usually measured by IHC or fluorescence in situ hybridization (deletion), and at present, there is no international standard for IHC assessment of PTEN loss. As a result, there can be significant interobserver variation among the different clinical trials.80,81 Moreover, there appears to be a significant discordance (66%) in PTEN expression between the primary tumor and the matched metastases.82
Therefore, PTEN loss cannot be viewed as a reliable predictive marker for anti-EGFR therapy at this time. The recent CRC biomarker guideline also does not recommend routine analysis of PTEN for the purposes of therapy selection.26
HER2 amplification
HER2 (also known as ERBB2) is a proto-oncogene that belongs to the human EGFR family. It is amplified in ~5% of patients with KRAS exon 2 wild-type mCRC and has a high concordance between the primary tumor and the metastases.83 It is also mutually exclusive with alterations to KRAS, NRAS, and BRAF in CRC.43
HER2 amplification has been studied as a biomarker to predict response toward HER2-directed therapy in mCRC.84 The phase II HERACLES trial evaluated the activity of dual HER2 inhibition using trastuzumab plus lapatinib in chemorefractory mCRC patients with HER2-positive, KRAS exon 2 (codons 12 and 13) wild-type disease.84 Of the 27 patients enrolled in the study, eight patients showed a response (ORR 30%). Moreover, the responses were noted to be durable with a median duration of 38 weeks, and the treatment combination was well tolerated. These results show that HER2 amplification is probably a clinically relevant genetic alteration that has a potential role as a predictive biomarker in CRC. However, the extremely low prevalence of this genetic alteration in colorectal tumors presents a significant challenge in validating these results in a confirmatory phase III clinical trial setting.
Immunologic characteristics
Galon and his colleagues85 were the first to demonstrate the importance of immune signatures as a prognostic marker in colon cancer, beyond the conventional TNM staging. The distribution of total T-cells (CD3+), cytotoxic T-effector cells (CD8+), and memory T-cells (CD45RO+) was evaluated in the tumor core and at the invasive margin. The investigators demonstrated that tumors with elevated levels of CD3+ T-cells in the core as well as at the invasive margin were associated with the best clinical outcome, independent of the TNM stage. In a subsequent study, the prognostic role of tumor-infiltrating lymphocytes was also demonstrated in mCRC.86 In this study, high tumor-infiltrating lymphocyte density in the metastatic sites conferred a greater response to chemotherapy and was associated with a longer PFS.
Recently, an “Immunoscore” methodology has been defined with an intent to quantify the immune infiltrate inside the tumor.87 This scoring system is based on the density of CD8+ cytotoxic T-cells and CD45RO+ memory cells at the invasive margin and in the center of the tumor. In a worldwide consortium-based analysis of tissue samples from stage I–III colon cancer patients, this standardized immunoscore assay was recently validated as a prognostic biomarker in early stage colon cancer.88,89 In the training set consisting of 700 patients, the patients with “high” Immunoscore had the lowest risk of cancer recurrence at 5 years (HR for “high” vs “low” immunoscore, 0.20; P<0.0001).89 These findings were confirmed independently in the internal and external validation sets, consisting of 636 and 1,345 patients, respectively. Moreover, the prognostic value of immunoscore assay was independent of the known existing prognostic factors including T stage, N stage, and MSI status.
CDX2 expression
CDX2 is an intestine-specific transcription factor and is a major regulator of intestinal development and oncogenesis.90 In fact, it is a highly sensitive and specific marker of adenocarcinomas of intestinal origin. It is known that colon cancers that lack CDX2 expression are associated with advanced stage, poor differentiation, vascular invasion, BRAF mutation, and CIMP pathway.91
CDX2 expression has been recently evaluated for its role as a prognostic and predictive biomarker in early stage colon cancer.92 In this study’s validation data set, CDX2-negative colon cancers were associated with a lower 5-year DFS than the CDX2-positive tumors (12.1% vs 87.9%; HR, 2.42; P=0.003). These results were independent of the known prognostic factors including tumor stage and pathologic grade. Absence of CDX2 expression was also shown to be a potential predictive marker of response to adjuvant chemotherapy in colon cancer. Among the stage II CDX2-negative colon cancers, the 5-year DFS was found to be longer in patients who received adjuvant chemotherapy than those who did not (91% vs 56%; P=0.006). Although encouraging, these results are derived from retrospective patient cohorts and pooled data sets, and, therefore, require validation using prospective randomized trials before CDX2 expression can be incorporated as a biomarker in routine clinical practice.
Tumor location
It is now well established that there are distinct differences in the clinical and molecular characteristics of the right- vs left-sided colon tumors. The large genome-scale analysis of CRC samples performed by TCGA revealed that significant biologic differences exist between right-sided tumors and tumors originating from the other sites, such that the right-sided cancers were more frequently hypermethylated and hypermutated.43
The location of colorectal primary tumor has prognostic, and potentially predictive value in CRC.93–95 In the study by Loupakis et al, the left-sided tumors were found to have better prognosis than the right-sided tumors in three independent cohorts of mCRC patients derived from the PROVETTA, AVF2107g, and NO16966 trials.93 The favorable outcome of left-sided tumors was independent of the known prognostic variables. In another study, Venook et al95 studied the effect of primary tumor location on survival in mCRC patients from the CALGB/SWOG 80405 clinical trial. In the KRAS wild-type mCRC cohort, patients with left-sided primary tumor had superior survival than those with right-sided primary tumor. Post hoc subgroup analyses showed that among patients with right-sided tumors, treatment with bevacizumab was associated with longer survival than that seen with cetuximab. Conversely, among patients with left-sided tumors, treatment with cetuximab was associated with longer OS than with bevacizumab. These results suggest that in addition to the known prognostic role, the location of colorectal primary tumor might also have a predictive value toward anti-EGFR and anti-VEGF therapies.
Consensus molecular subtypes of CRC
The recently defined four transcriptome-based molecular subtypes of CRC also demonstrate clinical and prognostic associations.9 For example, the CMS4 tumors are associated with worse OS and DFS even after adjustment for clinicopathologic features, MSI, BRAF, and KRAS mutation status. The CMS1 lesions are frequently right sided with high histopathologic grade, and these patients have very poor survival after disease relapse. Conversely, CMS2 tumors are predominantly left sided and have superior survival after relapse. Due to the biologic differences between the CMS subtypes, response to therapies is also expected to differ for each subtype. Future studies that prospectively compare the efficacy of therapies in different colorectal CMS subtypes will help establish its role as a predictive biomarker to guide therapeutic decisions.
MicroRNAs (miRNAs)
miRNAs are short (18–22 nucleotides), noncoding single-stranded RNA sequences involved in the posttranscriptional regulation of gene expression.96 They cause either degradation or inhibition of translation by binding to the 3′-untranslated region of targeted mRNA. miRNAs are believed to play a significant role in colorectal tumorigenesis, and several candidate miRNAs are being evaluated for having either predictive or prognostic role in the treatment of CRC.97–102 More recently, circulating exosomal miR-27a and miR-130a were demonstrated as novel diagnostic and prognostic biomarkers of CRC.103
Novel molecular approaches in CRC management
Next-generation sequencing
Next-generation sequencing is non-Sanger-based, high-throughput DNA sequencing technology, where an extremely large number of DNA strands can be sequenced simultaneously in parallel, yielding substantially more throughput of several hundred billion bases in a single run than by conventional sequencing methods. Next-generation sequencing of entire CRC genome has shown that each tumor harbors around 75 mutations.43 Whole-genome sequencing and whole-exome sequencing are two of the more recent advances in next-generation sequencing technology.104,105 Next-generation sequencing has utility in target identification for matched therapies. Specific examples include MSI-Hi for immune checkpoint inhibitor therapy, HER2 amplification for HER2-targeted therapy.
“Omics”-based approaches
Large-scale “omics” techniques have the ability to identify tumor biology with an extraordinarily high definition that enables understanding of complex pathologies and discriminate tumors with distinct molecular phenotypes. This growing knowledge has the potential to identify biomarkers leading to the development of pathway-directed targeted therapies and help individualize anticancer treatments to improve clinical outcomes.
Transcriptomics is the study of global mRNA expression of a particular tissue.106 Recent transcriptomic and immune-based characterization of CRC has further improved our understanding of the colorectal tumor biology, and the precision medicine paradigm in this disease has now evolved and shifted from clonal perspective for targeted therapies, to a clonal–stromal–immune perspective, which involves multimolecular, multidrug approach.107 Epigenomics is the study of epigenetic modifications that are now widely known to play an important role in tumorigenesis and are prevalent in CRC.108 These alterations include DNA promoter region methylation and histone modifications, and several biomarkers of these epigenetic changes have been described.108 Proteomics refers to large-scale study of proteins to comprehensively map biologic processes such as molecular mechanisms of carcinogenesis.109,110 It involves identification and quantification of proteins, as well as the study of their location, modifications, interactions, and functions. Proteomics has identified five different CRC subtypes in TCGA cohort, two of which overlap with the MSI/CIMP transcriptomic subtype.43,109,110 Omics strategy is now being explored to guide therapies in these distinct CRC subtypes. Finally, genomics, based on the advances in the next-generation sequencing technology, is providing a great opportunity for biomarker development and realization of precision medicine in CRC.
Circulating tumor DNA
Circulating cell-free DNA is referred to the DNA fragments found in cell-free components of peripheral blood, and it is largely derived from the normal tissue. A small fraction of circulating cell-free DNA in patients with malignancy is also derived from the tumor cells and is referred to as circulating tumor DNA (ctDNA). CtDNA is present in a majority of patients with mCRC, and molecular analysis of ctDNA from a patient’s peripheral blood is a considerably less invasive means of identifying specific genetic mutations in tumors.
CtDNA has high accuracy for the detection of KRAS and other mutations in CRC patients.111,112 Moreover, it does not have the limitation of spatial selection bias owing to tumor heterogeneity that is frequently associated with biopsy of the tumor tissue sample. CtDNA has also made it significantly easier to study the clonal evolution of CRC and detection of emerging KRAS-mutated clones during anti-EGFR therapy. For example, in the studies by Diaz et al113 and Misale et al,114 the emergence of KRAS mutations was detectable in ctDNA about 6–10 months prior to radiographic evidence of disease progression while on anti-EGFR therapy.
CtDNA has an immense potential to be a noninvasive molecular biomarker. Given its ease for sample collection, it can be used to estimate tumor burden, predict radiologic tumor progression, detect minimal residual disease, monitor both tumor progression and treatment response, and study acquired resistance mechanisms.104,115,116 However, further development of ctDNA-based biomarkers would require standardization of the assay platform and prospective studies to validate ctDNA-based biomarkers in patient care.115,117
Circulating tumor cells
Tumor cells detected in the peripheral blood are referred to as circulating tumor cells (CTCs). CtDNA and CTCs are considered distinct entities, as ctDNA is often present in patients without detectable CTCs.111 Several studies have suggested a potential prognostic role of CTCs in CRC.118–120 In a prospective multicenter study, Cohen et al118 examined CTCs from 430 patients with mCRC at baseline and after starting first-, second-, or third-line chemotherapy. Patients were stratified into unfavorable and favorable prognostic groups based on CTC levels of ≥3 or <3 CTCs/7.5 mL, respectively. Patients in the baseline unfavorable CTCs group had a shorter median PFS (4.5 months vs 7.9 months; P=0.0002) and OS (9.4 months vs 18.5 months; P<0.0001) compared to those in the favorable CTCs group. Patients whose CTC level converted from the unfavorable to the favorable group after the initial 3–5 weeks of treatment had a significantly improved PFS (6.2 months vs 1.6 months; P=0.02) and OS (11.0 months vs 3.7 months; P=0.0002).
Precision medicine – goals and challenges
The evolving paradigm
Precision medicine paradigm in CRC originally started with “one gene, one drug” approach, which was a considerable advancement over nonselective patient treatment based on the results of conventional population-level clinical trials. However, it was soon realized that such strategy is unlikely to result in durable remission given the complex molecular makeup of CRC with significant cross-talk between signaling pathways. As an example, there is virtually no clinical activity of BRAF inhibitors in BRAF-mutant mCRC, which is in strike contrast to their activity in metastatic melanoma.121 This lack of response to BRAF inhibitors in CRC is due to the rapid activation of EGFR/PI3K pathway via feedback loop, resulting in the escape from downstream inhibition of targets because of the cross-talk between signaling pathways in colon cancer cells.122,123 Consequently, a “multi-gene, multi-drug” approach was evaluated and was the next big step to optimize outcomes. The encouraging activity observed with combined inhibition of BRAF, MEK, and EGFR in early trials of BRAF-mutant CRC is a perfect example of this concept.124
Impact of tumor heterogeneity
CRC is a very heterogeneous disease, and tumor heterogeneity exists at several different levels. First, the genetic makeup of a given tumor type can vary significantly between one patient and another. Next, within an individual patient, there exists a high degree of genetic heterogeneity between the primary tumor and the metastasis as well as between the different metastatic sites (intertumoral heterogeneity).125–127 Heterogeneity can also exist in different areas within a single tumor (intratumoral heterogeneity), in different anatomical regions of the same patient (spatial tumor heterogeneity), and at different time points within the same tumor (temporal tumor heterogeneity).
Intratumoral heterogeneity relates to genetic heterogeneity, functional heterogeneity, and nongenetic (epigenetic) heterogeneity.128 Genetic heterogeneity can be attributed to varying number of cancer clones depending upon the mutation rates and differences in the ability of these clones to metastasize or/and their responsiveness to therapies. Genetic heterogeneity also arises due to clonal selection from treatment pressures. For example, mutations in the driver oncogenes KRAS, NRAS, and BRAF are found to emerge in patients who were initially diagnosed with wild-type disease and were treated with anti-EGFR antibody, but subsequently developed resistance to such therapy.129 Even though nondominant, these dynamic clonal and subclonal alterations are responsible for sustaining CRC progression.
These different levels of tumor heterogeneity can affect the signaling of multiple key oncogenic pathways resulting in extensive phenotypic variation, such that each tumor clone even within the same tumor displays its own genetic, epigenetic, transcriptomic, and proteomic profile.126,130,131 Consequently, intratumoral heterogeneity poses an enormous challenge to the practice of personalized medicine because any given therapeutic agent targeted at a specific molecular alteration is likely to be successful in only a minority of patients. The four CMS groups are currently the best representation of CRC heterogeneity at the gene-expression level.107
Future directions and conclusion
Significant advances have been made in the development of molecular biomarkers for the treatment of mCRC. Unlike BRAF, mutations in RAS genes (KRAS and NRAS) do not have a definite prognostic significance in mCRC. However, the presence of activating mutations in these RAS genes is predictive for lack of response to anti-EGFR therapy. There is now a well-established body of evidence documenting that patients with RAS-mutated mCRC should not receive anti-EGFR therapy. However, it is worthwhile to note that the mere presence of wild-type RAS in the tumor does not guarantee a response to anti-EGFR therapy. This suggests that further refinement in the biomarker selection criteria beyond our current understanding is warranted.
CRC is a highly heterogeneous disease, and, therefore, molecular characterization of these tumors is necessary to determine molecularly homogeneous subtypes, understand tumor biology, and identify the most relevant driver events. Recent advances in DNA sequencing technology and “omics”-based approaches have provided promising new strategies for the development of molecular biomarkers in this disease. We have now identified and characterized genomic, transcriptomic, and immune-based subtypes of CRC. Our future success will depend on the ability to guide therapeutic decisions based on distinct biologic subtypes identified using such gene-expression signatures. Given the significant heterogeneity associated with colorectal tumors, the clonal perspective for targeted therapies is met with only limited success. Instead, a “multimolecular, multidrug” approach is now advocated that integrates knowledge from transcriptomic and immune subtyping of CRC to guide novel therapeutic strategies and precision medicine.107 For example, combination of PD-L1 blockade plus immune stimulation with MEK inhibitor in MSS colon cancer has shown early signs of clinical efficacy.132
To conclude, the goal of precision medicine is to personalize the therapeutic strategy in each given individual depending upon the clinicopathologic features, genomic profile, epigenetic changes, and other molecular characteristics of the tumor. Future clinical trials should, therefore, focus on testing therapies in molecularly defined subgroups of CRC to optimize efficacy, minimize toxicity, and reduce cost. Of course, such initiatives will require intimate collaborations between institutions in multiple countries across continents to ensure rapid clinical trial accrual and efficacy evaluation.
Disclosure
The author reports no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics. CA: Cancer J Clin. 2018;68(1):7–30. | ||
Lenz HJ, Niedzwiecki D, Innocenti F. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (MFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients with expanded RAS analyses untreated metastatic adenocarcinoma of the colon or rectum (mCRC). Paper presented at: ESMO Congress, Madrid, Spain; 2014. | ||
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. | ||
Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32(21):2240–2247. | ||
Goel G, Chauhan A, Hosein PJ. Ramucirumab: a novel anti-angiogenic agent in the treatment of metastatic colorectal cancer. Curr Colorectal Cancer Rep. 2016;12(5):232–240. | ||
Goel G. Evolution of regorafenib from bench to bedside in colorectal cancer: is it an attractive option or merely a “me too” drug? Cancer Manag Res. 2018;10:425–437. | ||
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. | ||
Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61(3):818–822. | ||
Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. | ||
Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. | ||
Wong R, Cunningham D. Using predictive biomarkers to select patients with advanced colorectal cancer for treatment with epidermal growth factor receptor antibodies. J Clin Oncol. 2008;26(35):5668–5670. | ||
Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. | ||
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. | ||
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. | ||
van Cutsem E, Köhne C-H, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med Overseas Ed. 2009;360(14):1408–1417. | ||
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27(5):663–671. | ||
Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–4705. | ||
Peeters M, Price TJ, Cervantes A, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28(31):4706–4713. | ||
Adelstein B-A, Dobbins TA, Harris CA, Marschner IC, Ward RL. A systematic review and meta-analysis of KRAS status as the determinant of response to anti-EGFR antibodies and the impact of partner chemotherapy in metastatic colorectal cancer. Eur J Cancer. 2011;47(9):1343–1354. | ||
Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: anti-epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154(1):37–49. | ||
Loupakis F, Cremolini C, Salvatore L, et al. Clinical impact of anti-epidermal growth factor receptor monoclonal antibodies in first-line treatment of metastatic colorectal cancer: meta-analytical estimation and implications for therapeutic strategies. Cancer. 2012;118(6):1523–1532. | ||
Vale CL, Tierney JF, Fisher D, et al. Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat Rev. 2012;38(6):618–625. | ||
Douillard J-Y, Oliner KS, Siena S, et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med Overseas Ed. 2013;369(11):1023–1034. | ||
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, Mckinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015; 26(1):13–21. | ||
van Cutsem E, Lenz HJ, Köhne CH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33(7):692–700. | ||
Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35(13):1453–1486. | ||
Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85(5):692–696. | ||
Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27(35):5931–5937. | ||
Kim ST, Park KH, Kim JS, Shin SW, Kim YH. Impact of KRAS mutation status on outcomes in metastatic colon cancer patients without anti-epidermal growth factor receptor therapy. Cancer Res Treat. 2013;45(1):55–62. | ||
Ren J, Li G, Ge J, Li X, Zhao Y. Is K-ras gene mutation a prognostic factor for colorectal cancer: a systematic review and meta-analysis. Dis Colon Rectum. 2012;55(8):913–923. | ||
de Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. | ||
di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. | ||
Tol J, Dijkstra JR, Klomp M, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46(11):1997–2009. | ||
van Cutsem E, Köhne CH, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–2019. | ||
Bokemeyer C, van Cutsem E, Rougier P, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–1475. | ||
Lin JS, Webber EM, Senger CA, Holmes RS, Whitlock EP. Systematic review of pharmacogenetic testing for predicting clinical benefit to anti-EGFR therapy in metastatic colorectal cancer. Am J Cancer Res. 2011;1(5):650–662. | ||
Xu Q, Xu AT, Zhu MM, Tong JL, Xu XT, Ran ZH. Predictive and prognostic roles of BRAF mutation in patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: a meta-analysis. J Dig Dis. 2013;14(8):409–416. | ||
Yuan ZX, Wang XY, Qin QY, et al. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PLoS One. 2013;8(6):e65995. | ||
Yang ZY, Wu XY, Huang YF, et al. Promising biomarkers for predicting the outcomes of patients with KRAS wild-type metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: a systematic review with meta-analysis. Int J Cancer. 2013;133(8):1914–1925. | ||
Rowland A, Dias MM, Wiese MD, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112(12):1888–1894. | ||
Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51(5):587–594. | ||
Seymour MT, Brown SR, Middleton G, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14(8):749–759. | ||
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. | ||
Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med Overseas Ed. 1998;338(21):1481–1487. | ||
Evaluation of Genomic Applications in Prevention Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. | ||
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. | ||
Sinicrope FA, Foster NR, Thibodeau SN, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011;103(11):863–875. | ||
Lanza G, Gafà R, Maestri I, Santini A, Matteuzzi M, Cavazzini L. Immunohistochemical pattern of MLH1/MSH2 expression is related to clinical and pathological features in colorectal adenocarcinomas with microsatellite instability. Mod Pathol. 2002;15(7):741–749. | ||
Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609–618. | ||
Myint ZW, Goel G. Role of modern immunotherapy in gastrointestinal malignancies: a review of current clinical progress. J Hematol Oncol. 2017;10(1):86. | ||
Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219–3226. | ||
Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19(2):462–468. | ||
Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51. | ||
Dt L, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. | ||
Dt L, Uram JN, Wang H. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J Clin Oncol. 2016;34:103. | ||
Overman MJ, Mcdermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. | ||
Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair–deficient/microsatellite instability–high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779. | ||
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Cancer Netw. 2017;15(2):142–147. | ||
Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29(35):4611–4619. | ||
Venook AP, Niedzwiecki D, Lopatin M, et al. Biologic determinants of tumor recurrence in stage II colon cancer: validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J Clin Oncol. 2013;31(14):1775–1781. | ||
Salazar R, Roepman P, Capella G, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29(1):17–24. | ||
Goel G. Evolving role of gene expression signatures as biomarkers in early-stage colon cancer. J Gastrointest Cancer. 2014;45(4):399–404. | ||
Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41(11):1649–1654. | ||
Barault L, Veyrie N, Jooste V, et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer. 2008;122(10):2255–2259. | ||
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23(6):1518–1525. | ||
Wu S, Gan Y, Wang X, Liu J, Li M, Tang Y. PIK3CA mutation is associated with poor survival among patients with metastatic colorectal cancer following anti-EGFR monoclonal antibody therapy: a meta-analysis. J Cancer Res Clin Oncol. 2013;139(5):891–900. | ||
Colakoglu T, Yildirim S, Kayaselcuk F, et al. Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: is PTEN loss predictor of local recurrence? Am J Surg. 2008;195(6):719–725. | ||
Sawai H, Yasuda A, Ochi N, et al. Loss of PTEN expression is associated with colorectal cancer liver metastasis and poor patient survival. BMC Gastroenterol. 2008;8(1):56. | ||
Xh L, Zheng HC, Takahashi H, Masuda S, Yang XH, Takano Y. PTEN expression and mutation in colorectal carcinomas. Oncol Rep. 2009;22(4):757–764. | ||
Lin MS, Huang JX, Chen WC, et al. Expression of PPARγ and PTEN in human colorectal cancer: an immunohistochemical study using tissue microarray methodology. Oncol Lett. 2011;2(6):1219–1224. | ||
Price TJ, Hardingham JE, Lee CK, et al. Prognostic impact and the relevance of PTEN copy number alterations in patients with advanced colorectal cancer (CRC) receiving bevacizumab. Cancer Med. 2013;2(3):277–285. | ||
Eklöf V, Wikberg ML, Edin S, et al. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer. 2013;108(10):2153–2163. | ||
Frattini M, Saletti P, Romagnani E, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97(8):1139–1145. | ||
Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20(1):84–90. | ||
Loupakis F, Pollina L, Stasi I, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–2629. | ||
Negri FV, Bozzetti C, Lagrasta CA, et al. PTEN status in advanced colorectal cancer treated with cetuximab. Br J Cancer. 2010;102(1):162–164. | ||
Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–5930. | ||
Ulivi P, Capelli L, Valgiusti M, et al. Predictive role of multiple gene alterations in response to cetuximab in metastatic colorectal cancer: a single center study. J Transl Med. 2012;10(1):87. | ||
Karapetis CS, Jonker D, Daneshmand M, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20(3):744–753. | ||
Molinari F, Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol. 2013;3:326. | ||
Sridharan M, Hubbard JM, Grothey A. Colorectal cancer: how emerging molecular understanding affects treatment decisions. Oncology. 2014;28(2):110–118. | ||
Cejas P, Lopez-Gomez M, Aguayo C, et al. Analysis of the concordance in the EGFR pathway status between primary tumors and related metastases of colorectal cancer patients: implications for cancer therapy. Curr Cancer Drug Targets. 2012;12(2):124–131. | ||
Valtorta E, Martino C, Sartore-Bianchi A, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol. 2015;28(11):1481–1491. | ||
Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–746. | ||
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. | ||
Halama N, Michel S, Kloor M, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011;71(17):5670–5677. | ||
Galon J, Pagès F, Marincola FM, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. 2012;10(1):205. | ||
Galon J, Mlecnik B, Marliot F, et al. Validation of the Immunoscore (IM) as a prognostic marker in stage I/II/III colon cancer: results of a worldwide consortium-based analysis of 1,336 patients. J Clin Oncol. 2016;34(Suppl); abstr 3500. | ||
Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–2139. | ||
Werling RW, Yaziji H, Bacchi CE, Gown AM. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27(3):303–310. | ||
Baba Y, Nosho K, Shima K, et al. Relationship of CDX2 loss with molecular features and prognosis in colorectal cancer. Clin Cancer Res. 2009;15(14):4665–4673. | ||
Dalerba P, Sahoo D, Paik S, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med Overseas Ed. 2016;374(3):211–222. | ||
Loupakis F, Yang D, Yau L, et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst. 2015;107(3):dju427. | ||
Tejpar S, Stintzing S, Ciardiello F, et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: retrospective analyses of the CRYSTAL and FIRE-3 trials. JAMA Oncol. 2016;3(2):194–201. | ||
Venook AP, Niedzwiecki D, Innocenti F, et al. Impact of primary (1°) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol. 2016;34(suppl):abstr 3504. | ||
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. | ||
Schetter AJ, Okayama H, Harris CC. The role of microRNAs in colorectal cancer. Cancer J. 2012;18(3):244–252. | ||
Lou X, Qi X, Zhang Y, Long H, Yang J. Decreased expression of microRNA-625 is associated with tumor metastasis and poor prognosis in patients with colorectal cancer. J Surg Oncol. 2013;108(4):230–235. | ||
Yang I-P, Tsai H-L, Huang C-W, et al. The functional significance of microRNA-29c in patients with colorectal cancer: a potential circulating biomarker for predicting early relapse. PLoS One. 2013;8(6):e66842. | ||
Zhang YU, Wang X, Xu B, et al. Epigenetic silencing of miR-126 contributes to tumor invasion and angiogenesis in colorectal cancer. Oncol Rep. 2013;30(4):1976–1984. | ||
Dong Y, Zhao J, Wu CW, Cw W, et al. Tumor suppressor functions of miR-133a in colorectal cancer. Mol Cancer Res. 2013;11(9):1051–1060. | ||
Karaayvaz M, Zhai H, Ju J. miR-129 promotes apoptosis and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cell Death Dis. 2013;4:e659. | ||
Liu X, Pan B, Sun L, et al. Circulating exosomal miR-27a and miR-130a act as novel diagnostic and prognostic biomarkers of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2018;27(7):746–754. | ||
Wang L, Wheeler DA. Genomic sequencing for cancer diagnosis and therapy. Annu Rev Med. 2014;65:33–48. | ||
Wang X, Kuang YY, Hu XT, Xt H. Advances in epigenetic biomarker research in colorectal cancer. World J Gastroenterol. 2014;20(15):4276–4287. | ||
Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33(3):285–289. | ||
Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17(2):79–92. | ||
van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. J Clin Oncol. 2011;29(10):1382–1391. | ||
Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. | ||
Uhlen M, Fagerberg L, Hallstrom BM, et al. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. | ||
Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra224. | ||
Thierry AR, Mouliere F, El Messaoudi S, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20(4):430–435. | ||
Diaz LA, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. | ||
Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. | ||
Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. | ||
Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–586. | ||
Sausen M, Parpart S, Diaz LA. Circulating tumor DNA moves further into the spotlight. Genome Med. 2014;6(5):35. | ||
Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213–3221. | ||
Romiti A, Raffa S, di Rocco R, et al. Circulating tumor cells count predicts survival in colorectal cancer patients. J Gastrointestin Liver Dis. 2014;23(3):279–284. | ||
Huang X, Gao P, Song Y, et al. Relationship between circulating tumor cells and tumor response in colorectal cancer patients treated with chemotherapy: a meta-analysis. BMC Cancer. 2014;14:976. | ||
Kopetz S, Desai J, Chan E, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(15_Suppl):3534. | ||
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. | ||
Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–235. | ||
Triple therapy improves colorectal cancer response. Cancer Discov. 2016;6(12):OF6. | ||
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. | ||
Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–364. | ||
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. | ||
Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14(4):235–246. | ||
Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21(9):2157–2166. | ||
Burrell RA, Mcgranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. | ||
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. | ||
Bendell JC, Kim TW, Goh BC, et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J Clin Oncol. 2016;34(Suppl):abstr 3502. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.