")
Back to Journals » Biologics: Targets and Therapy » Volume 17
Molecular Biology Mechanisms and Emerging Therapeutics of Triple-Negative Breast Cancer
Authors Zhang Z, Zhang R, Li D
Received 12 July 2023
Accepted for publication 7 September 2023
Published 21 September 2023 Volume 2023:17 Pages 113—128
DOI https://doi.org/10.2147/BTT.S426392
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Zhiying Zhang, Rui Zhang, Donghai Li
Inner Mongolia Medical University, Department of Thyroid Breast Surgery, Affiliated Hospital of Inner Mongolia Medical University, Inner Mongolia, 010050, People’s Republic of China
Correspondence: Rui Zhang, Inner Mongolia Medical University, Department of Thyroid Breast Surgery, Affiliated Hospital of Inner Mongolia Medical University, Hohhot North Street, Inner Mongolia, 010050, People’s Republic of China, Tel +8613347158522, Email [email protected]
Abstract: Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer that is conventionally characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2), accounting for approximately 15– 20% of all breast cancers. Compared to other molecular phenotypes, TNBC is typically associated with high malignancy and poor prognosis. Cytotoxic agents have been the mainstay of treatment for the past few decades due to the lack of definitive targets and limited therapeutic interventions. However, recent developments have demonstrated that TNBC has peculiar molecular classifications and biomarkers, which provide the possibility of evolving treatment from basic cytotoxic chemotherapy to an expanding domain of targeted therapies. This review presents a framework for understanding the current clinical experience surrounding molecular biology mechanisms in TNBC (Figure 1). Including immunotherapy, polymerase (PARP) and PI3K/AKT pathway inhibitors, antibody-drug conjugates, and androgen receptor (AR) blockade. Additionally, the role of miRNA therapeutics targeting TNBC and potential strategies targeting cancer stem cells (CSCs) are discussed and highlighted. As more and more treatments arise on the horizon, we believe that patients with TNBC will have a new sense of hope.
Keywords: triple-negative breast cancer, PI3K/AKT pathway inhibitors, antibody-drug conjugates, cancer stem cells, miRNA therapeutics
Introduction
Breast cancer has emerged as the most prevalent malignancy and is one of the principal causes of cancer-related fatalities in recent years. According to the World Health Organization, it is estimated that by 2022, one out of every eight women will suffer from breast cancer.1–4 Breast cancer is a complex disease that can be classified into three major tumor subtypes - hormone receptor (HR) positive, HER2-enriched, and triple-negative, based on the expression of estrogen and progesterone receptors and HER2 amplification. With the rapid advancements in medical technology, newer insights and a better understanding of the disease have emerged. The histopathological differentiation of breast cancer demands that we consider these subtypes as distinct entities and use tumor molecular signatures in clinical decision-making to guide the appropriate treatment.5–7
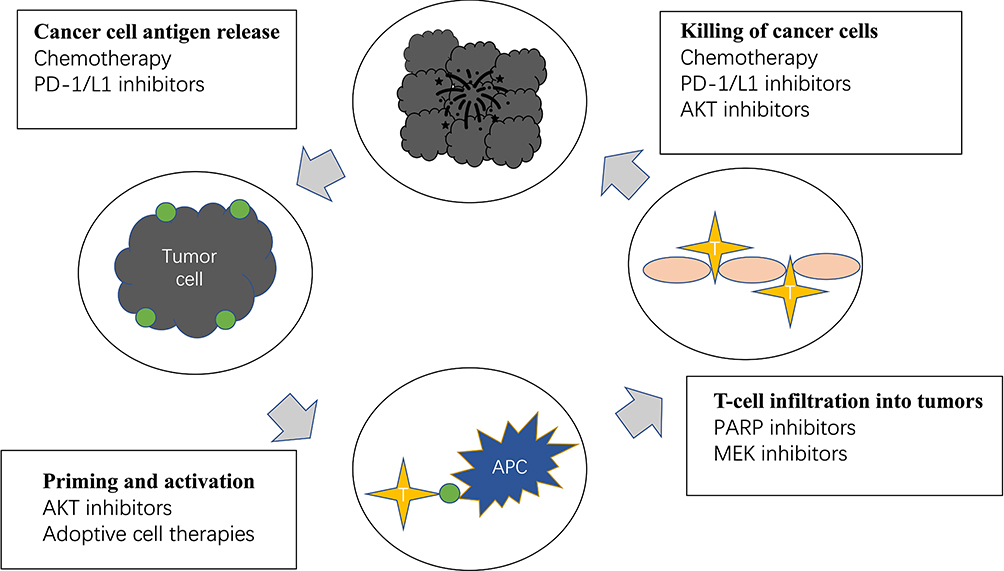 |
Figure 1 Current immunotherapy and combination therapeutic strategies for the treatment of triple-negative breast include killing of cancer cells, prevent cancer cell antigen release, promote T-cell infiltration into tumors and activate immune system. The main agents include chemotherapy, PD-1/L1 inhibitors, AKT inhibitors, PARP inhibitors and MEK inhibitors. |
Triple-negative breast cancer (TNBC) is characterized by the absence of hormone receptors and human epidermal growth factor. As the lethal subtype of breast cancer, it accounts for approximately 15% of all cases, demonstrating unique clinical and molecular characteristics.8,9 Epidemiological data indicates that TNBC is more commonly diagnosed in younger women, has a shorter time to relapse in the early stages, and carries a higher risk for hepatic, pulmonary, and central nervous system metastasis.10–12 Within five years of diagnosis, the mortality rate for TNBC patients is 40%, but for those who experience recurrence within three months, the mortality rate can reach 75%.13,14
In terms of pathology, TNBC predominantly presents with high-grade and high proliferation rates, particularly in invasive ductal carcinoma NOS, metaplastic carcinoma, apocrine carcinoma, and medullary carcinoma, illustrating its favorable response to chemotherapy with cytotoxic-based drugs. Additionally, some rare histological subtypes such as adenoid-cystic carcinoma, secretory carcinoma, adenomyoepithelioma, polymorphous carcinoma, and mucoepidermoid carcinoma are low-grade neoplasms.15,16 Therefore, conventional postoperative adjuvant chemoradiotherapy may not be adequate, and residual metastatic lesions can lead to tumor recurrence.7 Taken together these breast cancer subtypes demonstrate that the TNBC family is heterogeneous and intricated, opening new frontiers for innovative prognostic therapies for TNBC.
Currently, chemotherapy remains the primary systemic treatment option for both early and advanced stages of the disease due to its special molecular phenotype. However, the residual metastatic lesions that result from adjuvant chemoradiotherapy may eventually lead to tumor recurrence. As a result, there is an urgent to develop innovative ideas and directions for the treatment of TNBC, including novel treatment regimens and targets.
Diagnosis
As we know, it is the integral diagnosis and typing of TNBC tumors is the initial step in establishing personalized, targeted medications for each TNBC individual. At present, TNBC is predominantly diagnosed through imaging and immunohistochemistry (IHC).17 The major methods of breast imaging include mammogram, breast ultrasound, and magnetic resonance imaging (MRI). Mammography can identify breast cancer at an early stage by analyzing distribution, size, morphology, and microcalcifications. However, the positive predictive value of mammography needs to be enhanced, which can affect the judgment of doctors.18 Ultrasound is widely used in evaluating of lumps, which could detect small breast cancers that may not be visible on mammography. Currently, the BIRADS-US classification system is extensively applied in categorizing focal breast lesions.19 Additionally, the outcomes of a large clinical trial revealed that adding MRI to mammography improve the rate of cancer detection in high-risk breast cancer patients, but it also increased false-positive findings.20
IHC is typically used for staining cells with biomarkers, including hormone receptors and HER2 markers, to type breast carcinoma. The American Society of Clinical Oncology (ASCO)and the College of American Pathologists (CAP) have been provided approximately 126 latest guidelines to enhance reliability, reproducibility, and reduce false-negative results from IHC testing.21,22
Furthermore, several promising strategies have been presented for future TNBC diagnosis such as positron emission tomography (PET), nano biosensor, and circulating tumor nucleic acids (ctNAs), which could diagnosis TNBC in real-time, precisely, and minimally invasive ways.23
Molecular Characteristics of TNBC
In recent years, there have been significant developments made in illustrating the gene expression patterns and molecular subtypes of TNBC, which have potential therapeutic associations.24,25 Moreover, methylome sequencing of The Cancer Genome Atlas (TCGA) samples has identified biologically distinct subsets within TNBC that respond differently to standard chemotherapy and novel targeted agents.
Molecular Subtypes
Traditionally, breast carcinomas were divided into 18 different subtypes based on their morphological characteristics. However, since 2001, molecular characteristics have substituted morphological characteristics as the new approach to characterizing breast tumors.26 Based on gene expression, the largest overall difference is observed between hormone receptor (HR)-positive and HR-negative tumors. The subtypes are now referred to as luminal A, luminal B, basal-like, HER2-positive and normal-like breast cancer, each associated with distinct prognoses and treatment possibilities.26 Figure 2 shows the changes in breast cancer typing from 2010 to 2016.
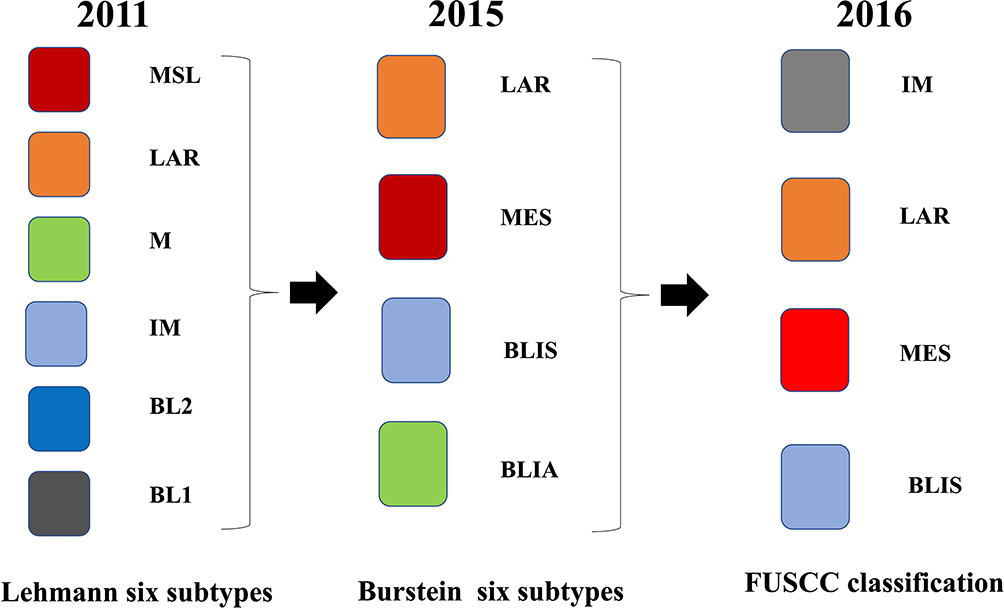 |
Figure 2 In 2011, Lehmann et al categorize TNBC into six different subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), mesenchymal stem-like (MSL), immunomodulatory (IM), and luminal androgen receptor (LAR). In 2015, Burstein identified four distinct TNBC subtypes: luminal androgen receptor (LAR), mesenchymal (MES), basal-like immunosuppressed (BLIS), and basal-like immune-activated (BLIA). In 2016, Liu and colleagues developed a novel TNBC classification system that integrates the expression profiles of both mRNAs and lncRNAs. The system divided TNBC subtypes into four subtypes: immunomodulatory (IM), luminal androgen receptor (LAR), mesenchymal-like (MES), basal-like and immune suppressed (BLIS). |
In 2011, Lehmann et al conducted a gene analysis of 21 breast cancer data sets, wherein they identified 587 TNBC cases. This helped them categorize TNBC into six different subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), mesenchymal stem-like (MSL), immunomodulatory (IM), and luminal androgen receptor (LAR).27
The BL1 subtype is typically characterized by heavy enrichments in cell division pathway components and DNA damage response pathways. There is some evidence indicating that BL1 patients may respond well to Poly (ADP-ribose) polymerase (PARP) inhibitors and genotoxic agents.28 In contrast, the BL2 subtype has high activations in growth factor signaling, glycolysis, and the expression of myoepithelial markers. Potential targeted therapeutic drugs for BL2 subtype include mTOR inhibitors and growth factor inhibitors.28
The M subtype is generally referred to as metaplastic breast cancer due to its tissue characteristics, which resemble sarcoma-like and squamous epithelial cells.28 In addition, the high expression of cell migration-related signaling pathways, extracellular matrix–receptor interaction pathways, and differentiation pathways make M-subtype patients more sensitive to mTOR inhibitors and drugs targeting epithelial–mesenchymal transition.29 Similarly, the MSL subtype also exhibits aberrant activation in cell differentiation pathways, epithelial to mesenchymal transition (EMT), transforming growth factor (TGF)-beta signaling pathway, and processes associated with growth factor signaling pathways. However, compared to the M subtype, the MSL subtype demonstrates lower levels of cell proliferation-related genes and higher levels of stemness-related genes. The potential targeted therapeutic drugs for both M subtype and MSL subtype patients include dasatinib and Abl/Src inhibitors.30
The IM subtype exhibits distinct expressions in immune cell-associated genes, cytokine signaling, immune cell signaling, antigen processing and presentation, chemokine signaling pathway, and immune signal transduction pathway. Several studies have indicated that immune checkpoint inhibitors such as PD1, PDL1, CTLA-4 could be utilized for treating patients with IM subtype breast cancer.31
On the other hand, unlike other TNBC subtypes, the LAR subtype does not express ER receptors but displays high activation in hormonal-related signaling pathways, which involve androgen and estrogen metabolism, steroid hormone biosynthesis, porphyrin, and chlorophyll metabolism. As a result, the use of anti-AR therapy may well benefit patients with LAR-subtype breast cancer.32
In 2015, Burstein and colleagues furthered the above molecular subtyping by identified and confirmed four distinct TNBC subtypes: luminal androgen receptor (LAR), mesenchymal (MES), basal-like immunosuppressed (BLIS), and basal-like immune-activated (BLIA). Through Spearman correlation analysis, these two classification systems were substantially associated with each other (P = 0.039). Compared to Perou’s “PAM50” TNBC molecular classification, this classification allows for distinct clinical prognoses and definition the clinical outcome of each subtype.33
In 2016, considering the emerging significant role of long noncoding RNAs (lncRNAs) in cellular processes, Liu and colleagues developed a novel TNBC classification system that integrates the expression profiles of both mRNAs and lncRNAs. The system divided TNBC subtypes into four subtypes: immunomodulatory (IM), luminal androgen receptor (LAR), mesenchymal-like (MES), basal-like and immune suppressed (BLIS).34
In concordance with the Lehmann/Pietenpol classification, the IM subtype predominantly exhibits immune cell process, such as cytokine-cytokine receptor interaction, T-cell receptor signaling pathway and B-cell receptor signaling pathway. The upregulated gene functions in this subtype are closely associated with immunity. In the LAR type, although immunohistochemical analysis has confirmed these tumors as TNBC, gene expression profiling has demonstrated significant elevations in androgen and estrogen metabolism, steroid hormone biosynthesis, porphyrin, and chlorophyll metabolism. This indicates that anti-androgen and anti-estrogen therapies could be effective.27,33 The MES subtype has enriched pathways, including extracellular matrix (ECM)-receptor interaction, focal adhesion, transforming growth factor (TGF)-beta signaling pathway, and low levels of genes related to cell proliferation. The MES subtype also exhibits enriched pathways in cell division and cell cycle-related pathways.
After analyzing the FUSCC classification system, it was revealed that the new subtype IM and LAR virtually identical to the Lehmann/Pietenpol IM and LAR types. Similarly, the new subtype BLIS predominantly contained the Lehmann/Pietenpol BL1 and M types. The new subtype MES contained all six of the Lehmann/Pietenpol subtypes, with the MSL and M subtypes being the most prevalent. Additionally, the novel TNBC classification system identified subtype-specific lncRNAs that could potentially serve as biomarkers and treatment targets, facilitating patient counseling and individualize the treatment of TNBC.
Molecule Mutations
Apart from discussing the distinct molecular subtypes, another important aspect of TNBC molecular characteristics is the alteration status of BRCA. The data has reveals that the prevalence of BRCA mutations of TNBC is about 10–30%. Moreover, approximately 80% of breast cancers with BRCA1 germline mutations are basal-like profile tumors.35,36 Additionally, studies have proved the association between deleterious BRCA1 mutations and the risk of developing TNBC.37 The loss or mutation of tumor suppressors TP53, PTEN, retinoblastoma gene (RB1) and the KRAS oncogene are more frequently lost or mutated in TNBC.38,39 The enhanced expression of Ki-67, vimentin, laminin and the low expression of Bcl-2 are also distinguishing features of TNBC from other breast tumors.40,41 Virtually, TNBC is not essentially a homogeneous disease entity; instead, it may be seen more as a symptom than as an individual disease that comprises several types of cancers characterized by molecular profiles.
TNBC Cytotoxic Therapy and Efficacy Evaluation
Compared to other types of breast cancer, cytotoxic chemotherapy remains the mainstay of systemic treatment for TNBC. A substantial body of literature over the past two decades has demonstrated its significant advantage in the neoadjuvant, adjuvant and metastatic setting.42 The main reason for this is the absence of high-frequency molecular alterations and the limited number of known biomarkers, rendering specific endocrine therapies and targeted therapies ineffective for TNBC. In recent years, neoadjuvant chemotherapy regimens have increasingly been used in TNBC treatment,43 with several studies highlighting its significantly higher pathological remission rates and significant enhancement of the prognosis for TNBC patients. Approximately 30–40% out of early-stage TNBC patients treated with standard neoadjuvant achieve a complete response (pCR) after treatment.44 Some existing evidence suggests that in the neoadjuvant or adjuvant setting, using dose-dense and high-dose regimens and choosing taxanes and anthracyclines are more effective in TNBC patients.45–48 The NCCN Guidelines recommended the most active agents in the first-line setting using numerous lines of chemotherapy based on taxane, anthracycline, cyclophosphamide, cisplatin, and fluorouracil for TNBC patients. Despite the combination chemotherapy regimens at the expense of increased toxicity, the outcomes of TNBC patients have been studied to have higher response rates compared with single agents.49 After considering the performance status, the risk of adverse events, prior chemotherapy regimens, disease burden, and patient preferences of chemotherapeutics, currently available chemotherapy regimens are adriamycin + cyclophosphamide + taxel (ACT), adriamycin + cyclophosphamide (AC), docetaxel + cyclophosphamide (TC), cyclophosphamide + methotrexate + fluorouracil (CMF), and cyclophosphamide + adriamycin + fluorouracil (CAF).50 Therefore, choosing appropriate chemotherapy regimens, considering the specific posology and safety profile of each agent, is necessary to ensure adequate treatment outcomes and prognosis for TNBC patients.
Taxanes
As inhibitors of microtubules that can impede tumor angiogenesis, taxanes have been extensively utilized ever since their approval by the FDA in 1984 for the treatment of progressive ovarian carcinoma. Their use as an adjuvant in the treatment of early breast cancer marked a new era in the therapy, beginning in 1989.51 The primary target of taxanes is β-tubulin of polymerized microtubules, to which they bind with high affinity to hinder microtubule depolymerization and impair mitosis.52 Taxanes function in a complicated manner, regulating various processes and pathways in cancer biology, including apoptosis, mitosis, angiogenesis and ROS production.53 Taxanes such as docetaxel and paclitaxel have exhibited a higher response rate for triple-negative breast cancers than for receptor- positive cancers.54–56 The US Oncology 9735 trial was the first randomized controlled trial to directly compare the efficacy of TC adjuvant regimens with AC adjuvant regimens in breast cancer patients. After a median of seven years of follow-up, the results revealed that four cycles of TC adjuvant regimens had overall survival rates comparable to standard AC adjuvant regimens, regardless of patient age, hormone receptor status, and lymph node status.57
Anthracycline
Anthracyclines and anthracycline antibiotics have become essential components of adjuvant treatment and are considered among the most potent drugs in breast cancer therapy. The anti-tumor effect of anthracyclines is generally attributed to intercalate with DNA, which induces DNA damage through the generation of free radicals. However, it is noteworthy that their use is limited by cardiotoxicity at high cumulative doses and hematologic malignancies.58 Based on a large number of clinical studies, the optimal dosing schedules of anthracycline adjuvant therapy for breast cancer are 60 mg/m2 of doxorubicin and 100 mg/m2 of epirubicin.59 The MINDACT trial compared an anthracycline‐based regimen to a non-anthracycline regimen. Despite both groups having comparable outcomes, the anthracycline‐taxane regimen is superior in TNBC, so we continue to recommend it as an adjuvant anthracycline in higher‐risk triple‐negative disease.60
Cyclophosphamide
As one of the most successful chemotherapy drugs, cyclophosphamide has been registered on the World Health Organizations List of Essential Medicines. It has been used to treat a range of cancers, including lymphomas, breast and ovarian cancers, both as a single agent and as adjuvant therapy.61 The primary antitumor mechanism of low dose cyclophosphamide is its ability to deplete the suppressive regulatory T cells (Tregs) population. This is a fundamental component of the tumor immune evasion arsenal that is correlated with tumor progression.62–64 Recently, docetaxel plus cyclophosphamide (TC) has been established as a standard adjuvant chemotherapy regimen for triple‐negative operable breast cancer. In addition, a prospective study by Yokoyama et al evaluating the efficacy and safety of capecitabine + epirubicin + cyclophosphamide combination therapy (CEX) as neoadjuvant chemotherapy for 18 patients with HER-2-negative breast cancer reported positive results. All patients finished four cycles of CEX, and the total pathological complete response (pCR) rate was 50%, with virtually all the pCRs observed in TNBC patients. The findings highlight the effectiveness of this regimen in treating triple-negative breast cancer.65
Capecitabine
As an orally administered prodrug of fluorouracil, capecitabine is preferentially metabolically activated preferentially at the tumor site, demonstrating its antineoplastic activity and synergizing with other cytotoxic agents, indicating potential as a component of first-line combination cytotoxic chemotherapy. Currently, in US and Europe, capecitabine combined with docetaxel is widely used for metastatic breast cancer treatment.66 Results from the CREATE-X trial indicates that the safety and effectiveness of six to eight cycles of adjuvant capecitabine following standard anthracycline and taxane neoadjuvant chemotherapy in early TNBC with a residual tumor burden.67 In addition, a meta-analysis of randomized controlled trials also suggested the possibility of improved disease-free survival and overall survival for TNBC patients following the addition of capecitabine to standard chemotherapy, though the known toxicity profile requires caution.68
Platinum Agents
Platinum-based agents can cause DNA crosslink strand breaks, leading to apoptosis specifically in cells with dysfunctional repair pathways. The BLBC subtype, accounting for most TNBC tumors, is particularly sensitive to platinum-based agents due to genomic instability, BRCA1 abnormalities, and defective DNA repair pathway.69–71 Preclinical and clinical data has indicated that BRCA1 carriers treated with cisplatin in neoadjuvant chemotherapy exhibit high pCR rates, while metastatic BRCA1 carriers also respond well.72,73 Consistent with preclinical and recent clinical data, research examining the use of platinum-based chemotherapy for TNBC has shown modest efficacy. Subsequent clinical trials have demonstrated that platinum-based monotherapy can be particularly beneficial for BRCA1/2 mutation carriers.74–76
Steven J. et al conducted a multicenter Phase II clinical trial of single-agent platinum for metastatic TNBC (TBCRC009) to evaluate its monotherapy by assessing biomarkers. The results showed that the overall response rate was 25.6%, with a response rate of 18.7% for carboplatin and 2.6% for cisplatin. Additionally, 54.5% of those who responded had BRCA1/2 mutations.74 This research results suggest that platinum-based agents have particularly effective for TNBC.
Emerging Targeted Therapies in TNBC
As contemporary technology platforms have contributed to the understanding of the molecular diversity of TNBC, we have been able to identify promising therapeutic targets for TNBC patients. Currently, several tumor-driving signaling pathways targeted therapies in TNBC, including EGFR targeted therapy, VEGF targeted therapy and PI3K/AKT/mTOR targeted therapy in TNBC are under clinical investigation.
EGFR Targeted Therapy
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase (RTK) that comprises of an extracellular ligand binding domain, a transmembrane domain, and a cytoplasmic tyrosine kinase domain. When a ligand binds to EGFR, its kinase activity is activated to participate in various downstream signaling pathways, promoting cell proliferation and motility. The EGFR gene overexpression is frequently observed in TNBC, making it a valid therapeutic target, particularly for BL2-subtype tumors.77,78 Inhibitors of EGFR including TKIs and mAbs, have already been developed and are currently available for use in the clinic, making it an attractive option for the treatment of TNBC patients.
EGFR TKIs, including gefitinib, erlotinib, afatinib, osimertinib and erlotinib, can restrain EGFR kinase activity by binding to the ATP binding site of the EGFR tyrosine kinase, and are currently approved for and highly effective against NSCLC.79,80 Several clinical trials have been conducted to investigate the toxicity and efficacy of TKIs in breast cancer. However, results have not demonstrated it to be an effective therapeutic for TNBC. For instance, a phase II clinical trial of gefitinib as a monotherapy in metastatic breast cancer concluded that gefitinib monotherapy at 500 mg daily was not exhibit efficacious to metastatic breast cancer patients.81 Another randomized phase II trial evaluated the effect of adding gefitinib to neoadjuvant EC, and showed a lack of significant difference in the outcomes of pathologic complete response (pCR) did not show a significant difference between patients treated with gefitinib and placebo. Nevertheless, the difference between TNBC and non-TNBC can be observed for gefitinib.82 Similarly, cetuximab appeared to have little activity as monotherapy, but the patient outcomes were enhanced when combined with carboplatin.83 Although the data of clinical trials of EGFR inhibitors therapies have shown mixed results in TNBC, several EGFR-TKI clinical trials are ongoing, including TKI monotherapy and TKI in combination with chemotherapy.84
VEGF Targeted Therapy
Vascular endothelial growth factor (VEGF) is the most significant angiogenic factor that promotes tumor cell proliferation and the formation of new vessels in breast cancer.85 The level of VEGF is an independent factor related to poor prognosis. Patients with TNBC show significantly higher levels of VEGF and shorter recurrence-free survival and overall survival compared to non-TNBC patients.86
Bevacizumab, a recombinant humanized monoclonal antibody against VEGF-A, was approved by the FDA in 2004 for use as part of combination therapy regimens to treat metastatic colorectal cancer. It is effectiveness and generally well-tolerated nature as a VEGF inhibitor making it promising as a single agent or as part of combination regimens for breast cancer, according to the results from clinical studies.87 However, it is important to note that the addition of bevacizumab may increase the incidence of adverse events such as hypertension, neutropenia, and mucositis. Two large, randomized studies demonstrated a significant increase in the rate of pathological complete response in HER2-negative patients when bevacizumab was added to neoadjuvant chemotherapy, and the efficacy was restricted mainly to TNBC patients.88,89 The GeparQuinto trial also investigated the effect on pCR of adding bevacizumab to standard neoadjuvant chemotherapy, and the results showed a higher pCR rate in the bevacizumab arm, particularly in the BRCAg mutations patients. However, the outcome of the BEATRICE study did not show a significant improvement in disease-free or overall survival. The heterogeneity of TN tumors could be the reason for these divergent results, and bevacizumab may have different reactions to molecular subgroups.90 In conclusion, bevacizumab shows promising potential as a positive function in metastatic TNBC. However, it still requires further follow-up to evaluate its potential effect.
PAM Targeted Therapy
The PI3K-Akt-mTOR (PAM) signaling pathway is known to play a crucial role in angiogenesis, tumor proliferation, and inhibition of apoptosis. The Cancer Genome Atlas (TCGA) has revealed significant mutation within the PI3K-Akt-mTOR (PAM) pathway in TNBC.5,91 Several preclinical studies also demonstrated that the PAM pathway was activated and upregulated in TNBC, and the M, MSL and BL subsets of TNBC showed sensitivity to PI3K/mTOR inhibitors.27,92 While these studies confirmed the PAM pathway is a promising therapeutic target, the implication of inhibitors in breast cancer are not yet well-defined.
The mTOR inhibitor, everolimus, has been approved for the treatment of metastatic breast cancer that is ER-positive and resistant to aromatase inhibitors.93 However, data on its clinical efficacy for TNBC is limited. A neoadjuvant clinical trial was conducted to assess the effects of adding everolimus to paclitaxel and anthracycline in TNBC. The results showed that there was no statistically significant increase in pCR. Further investigation is required to determine the most effective combinations and the subgroup of patients that will respond positively mTOR inhibitors.94
AKT is another critical node in the PAM signaling pathway, and some preclinical studies have demonstrated the antitumor activity of its inhibitors.95,96 Ipatasertib is a highly selective ATP-competitive AKT inhibitor. In the LOTUS phase II trial, the effectiveness of the combination of ipatasertib and paclitaxel in metastatic TNBC was evaluated. The results demonstrate the progression-free survival was longer when compared to paclitaxel alone. This is the first outcome suggesting the efficacy of AKT-targeted therapy for TNBC.97 Another AKT inhibitor, capivasertib, has also shown very promising preliminary data in a phase II study, and a larger Phase III trial is ongoing to validate its efficacy.97 More clinical trials are currently underway to further clarify the role of PAM-targeted therapy in the treatment of TNBC.
Upcoming Treatment Options
At present, there is no specific targeted agent approved by the FDA or EMA for the treatment of TNBC. However, in this article, we will discuss the most recent data on promising and novel targeted approaches for TNBC.
Immunotherapy
Immunotherapy is a promising treatment strategy for TNBC, with documented prolonged survival in other solid tumors. Immune checkpoint inhibitors (ICIs) have proven to be the most successful immunotherapeutic agents. These work by blocking immunosuppressive receptors and improving the cytotoxicity and proliferative capacity of tumor-infiltrating lymphocytes (TILs).98,99 Some of the popular monoclonal antibodies against PD-1, such as pembrolizumab, nivolumab, and against PD-L1 including atezolizumab, durvalumab, avelumab, along with ipilimumab against CTLA-4, have been observed to generate durable responses across various tumor types.100,101
PARP Inhibitors
Polyadenosine diphosphate (ADP) ribose polymerase (PARP) is a highly conserved multifunctional enzyme that plays a significant role in the mechanism of single-strand DNA break repair. Its primary function of PARP is to facilitate DNA repair, cellular proliferation, and catalyzing the transfer of ADP-ribose from NAD+ to target proteins.102 The effects of PARP inhibitors result in the catalytic inhibition of PARP1 and locking PARP1 on damaged DNA to stall the progress of replication forks. In normal cells, damaged DNA sequences can be restored back to their native form by HR proteins. However, in tumor cells lacking HR proteins, such as BRCA1, BRCA2, PALB2, or RAD51 would use error-prone DNA repair pathways, leading to the death of the cells.103 Especially in BRCA-deficient breast cancer, the repair of DNA damage exclusively depends on the action of PARP1, making the sensitivity of PARP inhibitors higher than others.104–106 This scientific evidence provides positive proof of concept for PARP inhibition in BRCA-deficient breast cancers and demonstrates a novel targeted treatment strategy for TNBC.
In 2009, Olaparib, a PARP inhibitor, was clinically validated for its synthetic lethal interaction with BRCA1/BRCA2 deficiency. Further clinical research also evaluated the efficacy, safety, and tolerability of olaparib alone in breast cancer patients and demonstrated that a dosage of 400 mg b.i.d was both safe and effective for patients with BRCA1/2 mutation.107
In a randomized phase III Olaparib trial of metastatic BRCA mutation breast cancer, 302 HER2-negative metastatic breast cancer patients with a germline BRCA mutation were randomly assigned to either receive olaparib tablets or standard single chemotherapy. The results showed that patients treated with olaparib had a higher response rate and longer median progression-free survival.108 Based on these clinical trials, the PARP inhibitor olaparib was recently approved for TNBC.
In addition to olaparib, a number of other PARP inhibitors including veliparib, talazoparib, niraparib and rucaparib are currently undergoing clinical trials, with particular focus on their potential use for metastatic breast cancer.109–111 One such trial, the I-SPY 2 trial, evaluated the effectiveness of veliparib and carboplatin when added to standard chemotherapy regimens for patients with TNBC. While the addition of veliparib did increase pCR rates from 26% to 52%, its precise impact on patient outcomes remains unclear.112 Another PARP inhibitor that is being evaluated for breast cancer is talazoparib. In the phase III EMBRACA study, talazoparib was compared as a monotherapy against capecitabine or eribulin chemotherapy regimens for patients with BRCA mutation breast cancer. The results of this study showed that median progression-free survival was significantly improved with talazoparib treatment, and interim analysis of overall survival also showed a positive trend. As a result, talazoparib has also received FDA approval for use in the treatment of BRCA mutation breast cancer.113
There are numerous ongoing clinical trials exploring the potential of PARP inhibitors for early-stage TNBC patients. One such trial is the phase III OlympiA trial, which aims to assess the effectiveness of olaparib treatment for high-risk TNBC patients with germline BRCA1/2 mutations who have undergone standard neoadjuvant chemotherapy and local treatment (NCT02032823). Another phase III clinical trial is currently underway to establish the safety and efficacy of adding olaparib to neoadjuvant platinum-based chemotherapy for TNBC patients (NCT03150576). Beyond olaparib, a Phase I trial is ongoing to evaluate the effectiveness of neoadjuvant veliparib combined with radiation therapy for residual breast cancer following neoadjuvant chemotherapy (NCT01618357).
Anti-Androgen Therapy
Androgen receptor (AR) belongs to the nuclear steroid hormone receptor family, playing a critical role in signaling pathways and regulating gene expression as transcription factors. Emerging evidence indicated that AR signaling is a critical determinant of normal and malignant breast tissue, performing a pivotal function in breast carcinoma progression.114,115 In breast cancer, AR is the most widely expressed nuclear hormone receptor, and its expression is always associated with a favorable prognosis. A systematic review examined 7693 patients across 19 different studies, finding that AR is expressed in 60.5% of patients, who exhibited better overall survival (OS) and disease-free survival (DFS) than AR-negative breast cancer patients.116 The LAR subtype of TNBC is enriched in hormonally regulated pathways, predominantly dependent on AR signaling. However, the mechanism of AR in TNBC still ambiguous. Existing evidence suggests that AR is activated in extracellular signal-regulated kinase (ERK) and HER2 signaling through PI3K, Src, and RAS in TNBC.117 Moreover, AR overexpressed cell lines were found to frequently carry PIK3CA-activating mutations. These cells are sensitive to PI3K inhibitors and AR inhibitors, highlighting a correlation between AR and the PI3K pathway.118–120
This evidence leads to new treatment strategies and potential clinical implications for TNBC, and ongoing clinical trials for AR-targeted therapies. The oral nonsteroidal AR antagonist, bicalutamide, has been approved by The US Food and Drug Administration for the treatment of metastatic prostate cancer. Evidence of using bicalutamide’s effectiveness in the treating TNBC has been proven in phase II clinical trial where patients with metastatic AR-positive TNBC were given bicalutamide. Among patients with AR-positive tumors, the clinical benefit rate was 19%, and the median progression-free survival was 12 weeks.121 These findings form the basis of anti-androgen therapy in AR-positive TNBC. Subsequently, another phase II clinical trial with enzalutamide, a more potent AR inhibitor, was conducted. It was reported that 35% of patients showed clinical benefits in advanced-stage AR-positive TNBC.122
Emerging Treatment Strategies
Antibody-Drug Conjugates
In recent years, antibody-drug conjugates (ADCs) have emerged as viable antitumor agents that can deliver cytotoxic payloads on monoclonal antibodies through specific conjugations. ADCs have unique tumor specificity and potency that traditional drugs cannot match, making them a useful tool in personalized cancer medicine.123–125 The clinical use of ADCs is poised to become a significant modality in the therapy of tumors in the near future, with novel ADCs entering clinical studies for the treatment of TNBC patients.
The Human trophoblast cell-surface antigen 2 (Trop-2) has exhibited high potential as a therapeutic target due to its overexpressed in metastatic TNBC.126 Sacituzumab-govitecan, an ADC targeting Trop-2, incorporates a humanized monoclonal antibody (hRS7) to deliver the active metabolite of irinotecan SN-38 (govitecan).127 The randomized phase III ASCENT (NCT01631552) trial reported a response rate of 33.3% with sacituzumab govitecan, with longer progression-free survival than standard chemotherapy. This led to the approval of the new ADC for refractory metastatic TNBC.128
Another novel HER2-targeted ADC, trastuzumab deruxtecan (T-DXd), was designed to deliver topoisomerase I inhibitor to HER2-expressing cancer cells. The phase I study aims to evaluate its antitumor activity and safety in patients with advanced breast cancer expressing low levels of HER2.129 The confirmed objective response rate is 37.0%, and the median duration of response of 10.4 months. Moreover, a phase III study compares the efficacy and safety of T-DXd with capecitabine chemotherapy regimen (NCT03734029). T-DXd has demonstrated promising clinical antitumor activity in HER2-negative patients with a manageable safety profile and may provide a novel targeted treatment option for TNBC.
Cancer Stem-Cell Population Inhibitors
It is now evident that certain tumors can be initiated and sustained cancer stem cells, which possess the ability to renew and differentiate themselves to drive tumorigenesis.130 Breast cancer stem cells, for example, can repopulate a heterogeneous tumor from a single cell131 and can be identified through the measurement of aldehyde dehydrogenase activity,132,133 assessment of integrin receptors expression, or a decrease in the ability to exclude the ATP-binding cassette (ABC) transporter.134 These stem cells exhibit slower rates of proliferation and higher levels of resistance to chemotherapy, which contributes to resistance and accelerated recurrence after traditional chemotherapy therapies.131 Therefore, exploring the novel target therapies for breast cancer stem cells in combination with chemotherapy may be a potential modality for treating TNBC. Although inhibitors of breast cancer stem cells can be established clearly, the pathways that contribute to the maintenance of breast cancer stem cells, such as the mitogen-activated protein kinase (MAPK), JAK/STAT, Wnt, TGF-β, Hedgehog, and Notch1 pathways, have been identified in independent studies.135–139 Apart from that, other agents that target various aspects of CSCs, including specific markers, signaling pathways, dormancy, proliferation, metabolism, autophagy, and drug resistance, have shown promising preclinical results and are now entering clinical trials.140
MiRNA Targets and Therapies
The dysregulation of miRNAs is known to play significant role in silencing gene expression, suppressing or activating multiple genes in TNBC. This dysregulation can occur through various mechanisms such as and amplification, deletion, epigenetic, transcriptional dysregulation or defective miRNA biogenesis.141 These oncogenic miRNAs can act as tumor suppressor genes or oncogenes, ultimately promoting tumor invasion, metastasis, and drug resistance.142 For instance, miR-155, miR-320a, and miR-205 expression have been linked to lymph node metastasis,143 while miR-206 affects the migration and invasion of TNBC cells, and miR-340 inhibits invasion and metastasis by Rho Kinase 1 (ROCK1).144,145 Moreover, miRNA-222, miR-5195-3p and miRNA-449 have been found to mediate multi-drug resistance to chemotherapeutic agents via various mechanism in TNBC cells.146–148 Beyond this, miRNA regulators also contribute to pathobiology of TNBC by influencing the tumor microenvironment, including inflammation, target expression, and secretion from fibroblasts.149 For instance, in PTEN-deleted fibroblasts, miR-320 mediates E26 oncogene homolog 2 (ETS2) target to active oncogenic secretome, promoting angiogenesis and invasion in the TNBC microenvironment, thereby enhancing TNBC cell migration and invasion.150 Additionally, miR-9, a pro-metastatic miRNA, is up-regulated in TNBC tumor cells to transfer to fibroblasts.151 While most of the literature on using miRNAs to target breast cancer is still in the preclinical stage, miRNA therapies are showing great potential for treating TNBC.152
Summary and Outlook
TNBC is a unique disease entity characterized by molecular and immunological heterogeneity, which manifests a variety of clinical subtypes. While chemotherapy remains the principal treatment for TNBC, massively parallel sequencing technologies have contributed to our novel understanding of this subtype. These molecular advances have enabled us to exploit promising therapeutic targets for TNBC by targeting actionable genomic alterations and molecular pathways, numerous experimental therapies are ongoing. To update the latest developments. This has resulted in numerous ongoing experimental therapies. To provide the latest developments in TNBC treatment, this article comprehensively reviews the molecular classification of TNBC and targeted therapy developments in signaling pathways, targeted therapy, immunotherapy, PARP inhibitors, antibody-drug conjugates, and androgen receptor blockade. Treatment options for TNBC have evolved from basic cytotoxic chemotherapy to an expanding domain of targeted therapies for this heterogeneous disease. Ongoing efforts of researchers have opened up new avenues of hope for TNBC patients.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Nature Science Foundation of Inner Mongolia Autonomous Region (Grant numbers 2022MS08010) and Postgraduate Talent Excellence Program (YKDD2023ZY001).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Siegel RL, Ward EM, et al. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
2. Saraiva DP, Guadalupe Cabral M, Jacinto A, et al. How many diseases is triple negative breast cancer: the protagonism of the immune microenvironment. ESMO Open. 2017;2(4):e000208. doi:10.1136/esmoopen-2017-000208
3. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
4. Bilani N, Zabor EC, Elson L, et al. Breast Cancer in the United States: a Cross-Sectional Overview. J Cancer Epidemiol. 2020;2020:6387378. doi:10.1155/2020/6387378
5. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
6. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi:10.1038/nature12634
7. Chaudhary LN, Wilkinson KH, Kong A. Triple-Negative Breast Cancer: who Should Receive Neoadjuvant Chemotherapy? Surg Oncol Clin N Am. 2018;27(1):141–153. doi:10.1016/j.soc.2017.08.004
8. Bauer KR, Brown M, Cress RD, et al. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–1728. doi:10.1002/cncr.22618
9. Kohler BA, Sherman RL, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, 1975–2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015;107(6):djv048. doi:10.1093/jnci/djv048
10. Sharma P. Biology and Management of Patients With Triple-Negative Breast Cancer. Oncologist. 2016;21(9):1050–1062. doi:10.1634/theoncologist.2016-0067
11. Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. doi:10.1158/1078-0432.CCR-06-3045
12. Lin NU, Claus E, Sohl J, et al. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645. doi:10.1002/cncr.23930
13. Zhang L, Fang C, Xu X, et al. Androgen receptor, EGFR, and BRCA1 as biomarkers in triple-negative breast cancer: a meta-analysis. Biomed Res Int. 2015;2015:357485. doi:10.1155/2015/357485
14. Gluz O, Liedtke C, Gottschalk N, et al. Triple-negative breast cancer--current status and future directions. Ann Oncol. 2009;20(12):1913–1927. doi:10.1093/annonc/mdp492
15. Bando Y, Kobayashi T, Miyakami Y, et al. Triple-negative breast cancer and basal-like subtype: pathology and targeted therapy. J Med Invest. 2021;68(3.4):213–219. doi:10.2152/jmi.68.213
16. Livasy CA, Karaca G, Nanda R, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19(2):264–271. doi:10.1038/modpathol.3800528
17. Penault-Llorca F, Viale G. Pathological and molecular diagnosis of triple-negative breast cancer: a clinical perspective. Ann Oncol. 2012;23(Suppl 6):vi19–22. doi:10.1093/annonc/mds190
18. Gosling S, Scott R, Greenwood C, et al. Calcification Microstructure Reflects Breast Tissue Microenvironment. J Mammary Gland Biol Neoplasia. 2019;24(4):333–342. doi:10.1007/s10911-019-09441-3
19. Gokhale S. Ultrasound characterization of breast masses. Indian J Radiol Imaging. 2009;19(3):242–247. doi:10.4103/0971-3026.54878
20. Berg WA, Zhang Z, Lehrer D, et al. Detection of breast cancer with addition of annual screening ultrasound or a single screening MRI to mammography in women with elevated breast cancer risk. JAMA. 2012;307(13):1394–1404. doi:10.1001/jama.2012.388
21. Wolff AC. Guideline Summary: American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Human Epidermal Growth Factor Receptor HER2 Testing in Breast Cancer. J Oncol Pract. 2007;3(1):48–50. doi:10.1200/JOP.0718501
22. Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med. 2010;134(7):e48–72. doi:10.5858/134.7.e48
23. Dass SA, Tan KL, Selva Rajan R, et al. Triple Negative Breast Cancer: a Review of Present and Future Diagnostic Modalities. Medicina. 2021;57(1).
24. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi:10.1038/35021093
25. Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi:10.1038/nature10933
26. Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi:10.1073/pnas.191367098
27. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi:10.1172/JCI45014
28. Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232(2):142–150. doi:10.1002/path.4280
29. Gibson GR, Qian D, Ku JK, et al. Metaplastic breast cancer: clinical features and outcomes. Am Surg. 2005;71(9):725–730. doi:10.1177/000313480507100906
30. Yin L, Duan -J-J, Bian X-W, et al. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020;22(1):61. doi:10.1186/s13058-020-01296-5
31. Bertucci F, Finetti P, Cervera N, et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006;66(9):4636–4644. doi:10.1158/0008-5472.CAN-06-0031
32. Hayes MJ, Thomas D, Emmons A, et al. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res. 2008;14(13):4038–4044. doi:10.1158/1078-0432.CCR-07-4379
33. Burstein MD, Tsimelzon A, Poage GM, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–1698. doi:10.1158/1078-0432.CCR-14-0432
34. Curigliano G, Romieu G, Campone M, et al. A phase I/II trial of the safety and clinical activity of a HER2-protein based immunotherapeutic for treating women with HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2016;156(2):301–310. doi:10.1007/s10549-016-3750-y
35. Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi:10.1186/bcr2635
36. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and Pathologic Characteristics of Patients With BRCA -Positive and BRCA -Negative Breast Cancer. J Clin Oncol. 2008;26(26):4282–4288. doi:10.1200/JCO.2008.16.6231
37. de Ruijter TC, Veeck J, de Hoon JPJ, et al. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 2011;137(2):183–192. doi:10.1007/s00432-010-0957-x
38. Engebraaten O, Vollan HKM, Borresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183(4):1064–1074. doi:10.1016/j.ajpath.2013.05.033
39. Hu X, Stern HM, Ge L, et al. Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol Cancer Res. 2009;7(4):511–522. doi:10.1158/1541-7786.MCR-08-0107
40. Rodriguez-Pinilla SM, Sarrio D, Honrado E, et al. Vimentin and laminin expression is associated with basal-like phenotype in both sporadic and BRCA1-associated breast carcinomas. J Clin Pathol. 2007;60(9):1006–1012. doi:10.1136/jcp.2006.042143
41. Jaafar R, Mnich K, Dolan S, et al. RIP2 enhances cell survival by activation of NF-kB in triple negative breast cancer cells. Biochem Biophys Res Commun. 2018;497(1):115–121. doi:10.1016/j.bbrc.2018.02.034
42. Breast Cancer Trialists’ Collaborative Group (EBCTCG) E; Early Breast Cancer Trialists’ Collaborative, G. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet. 2012;379(9814):432–444. doi:10.1016/S0140-6736(11)61625-5
43. Rastogi P, Anderson SJ, Bear HD, et al. Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J Clin Oncol. 2008;26(5):778–785. doi:10.1200/JCO.2007.15.0235
44. von Minckwitz G, Untch M, Blohmer J-U, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012;30(15):1796–1804. doi:10.1200/JCO.2011.38.8595
45. Citron ML, Berry DA, Cirrincione C, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21(8):1431–1439. doi:10.1200/JCO.2003.09.081
46. Gluz O, Nitz UA, Harbeck N, et al. Triple-negative high-risk breast cancer derives particular benefit from dose intensification of adjuvant chemotherapy: results of WSG AM-01 trial. Ann Oncol. 2008;19(5):861–870. doi:10.1093/annonc/mdm551
47. Henderson IC, Berry DA, Demetri GD, et al. Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 2003;21(6):976–983. doi:10.1200/JCO.2003.02.063
48. Hayes DF, Thor AD, Dressler LG, et al. HER2 and response to paclitaxel in node-positive breast cancer. N Engl J Med. 2007;357(15):1496–1506. doi:10.1056/NEJMoa071167
49. Conlin AK, Seidman AD. Taxanes in breast cancer: an update. Curr Oncol Rep. 2007;9(1):22–30. doi:10.1007/BF02951422
50. Telli ML, Gradishar WJ, Ward JH. NCCN Guidelines Updates: breast Cancer. J Natl Compr Canc Netw. 2019;17(5.5):552–555. doi:10.6004/jnccn.2019.5006
51. Ring AE, Ellis PA. Taxanes in the treatment of early breast cancer. Cancer Treat Rev. 2005;31(8):618–627. doi:10.1016/j.ctrv.2005.09.005
52. Alushin GM, Lander G, Kellogg E, et al. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157(5):1117–1129. doi:10.1016/j.cell.2014.03.053
53. Mosca L, Ilari A, Fazi F, et al. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist Updat. 2021;54:100742. doi:10.1016/j.drup.2020.100742
54. Martin M, Rodríguez-Lescure Á, Ruiz A, et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res Treat. 2010;123(1):149–157. doi:10.1007/s10549-009-0663-z
55. Sparano JA, Wang M, Martino S, et al. Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med. 2008;358(16):1663–1671. doi:10.1056/NEJMoa0707056
56. Juul N, Szallasi Z, Eklund AC, et al. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: a retrospective analysis of five clinical trials. Lancet Oncol. 2010;11(4):358–365. doi:10.1016/S1470-2045(10)70018-8
57. Jones S, Holmes FA, O’Shaughnessy J, et al. Docetaxel With Cyclophosphamide Is Associated With an Overall Survival Benefit Compared With Doxorubicin and Cyclophosphamide: 7-Year Follow-Up of US Oncology Research Trial 9735. J Clin Oncol. 2009;27(8):1177–1183. doi:10.1200/JCO.2008.18.4028
58. Kwok JC, Richardson DR. Examination of the mechanism(s) involved in doxorubicin-mediated iron accumulation in ferritin: studies using metabolic inhibitors, protein synthesis inhibitors, and lysosomotropic agents. Mol Pharmacol. 2004;65(1):181–195. doi:10.1124/mol.65.1.181
59. Trudeau M, Charbonneau F, Gelmon K, et al. Selection of adjuvant chemotherapy for treatment of node-positive breast cancer. Lancet Oncol. 2005;6(11):886–898. doi:10.1016/S1470-2045(05)70424-1
60. Shah AN, Gradishar WJ. Adjuvant Anthracyclines in Breast Cancer: what Is Their Role? Oncologist. 2018;23(10):1153–1161. doi:10.1634/theoncologist.2017-0672
61. Madondo MT, Quinn M, Plebanski M. Low dose cyclophosphamide: mechanisms of T cell modulation. Cancer Treat Rev. 2016;42:3–9. doi:10.1016/j.ctrv.2015.11.005
62. Needham DJ, Lee JM, Beilharz MW. Intra-tumoural regulatory T cells: a potential new target in cancer immunotherapy. Biochem Biophys Res Commun. 2006;343(3):684–691. doi:10.1016/j.bbrc.2006.03.018
63. Sugiyama D, Nishikawa H, Maeda Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3 + CD4 + regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A. 2013;110(44):17945–17950. doi:10.1073/pnas.1316796110
64. Awwad M, North RJ. Cyclophosphamide (Cy)-facilitated adoptive immunotherapy of a Cy-resistant tumour. Evidence that Cy permits the expression of adoptive T-cell mediated immunity by removing suppressor T cells rather than by reducing tumour burden. Immunology. 1988;65(1):87–92.
65. Yokoyama T, Makino H, Seki N, et al. Capecitabine + Epirubicin + Cyclophosphamide Combination Therapy (CEX Therapy) as Neoadjuvant Chemotherapy for HER-2-Negative Breast Cancer: a Retrospective, Single-Center Study. J Nippon Med Sch. 2020;87(2):73–79. doi:10.1272/jnms.JNMS.2020_87-204
66. Wagstaff AJ, Ibbotson T, Goa KL. Capecitabine: a review of its pharmacology and therapeutic efficacy in the management of advanced breast cancer. Drugs. 2003;63(2):217–236. doi:10.2165/00003495-200363020-00009
67. Masuda N, Lee S-J, Ohtani S, et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N Engl J Med. 2017;376(22):2147–2159. doi:10.1056/NEJMoa1612645
68. Natori A, Ethier J-L, Amir E, et al. Capecitabine in early breast cancer: a meta-analysis of randomised controlled trials. Eur J Cancer. 2017;77:40–47. doi:10.1016/j.ejca.2017.02.024
69. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–871. doi:10.1111/j.1349-7006.2004.tb02195.x
70. Kennedy RD, Quinn JE, Mullan PB, et al. The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst. 2004;96(22):1659–1668. doi:10.1093/jnci/djh312
71. Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25(43):5846–5853. doi:10.1038/sj.onc.1209876
72. Byrski T, Gronwald J, Huzarski T, et al. Pathologic Complete Response Rates in Young Women With BRCA1 -Positive Breast Cancers After Neoadjuvant Chemotherapy. J Clin Oncol. 2010;28(3):375–379. doi:10.1200/JCO.2008.20.7019
73. Silver DP, Richardson AL, Eklund AC, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28(7):1145–1153. doi:10.1200/JCO.2009.22.4725
74. Isakoff SJ, Mayer EL, He L, et al. TBCRC009: a Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33(17):1902–1909. doi:10.1200/JCO.2014.57.6660
75. Denkert C, Liedtke C, Tutt A, et al. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389(10087):2430–2442. doi:10.1016/S0140-6736(16)32454-0
76. Byrski T, Dent R, Blecharz P, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012;14(4):R110. doi:10.1186/bcr3231
77. Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–118. doi:10.1111/j.1365-2559.2007.02889.x
78. Sobande F, Dušek L, Matějková A, et al. EGFR in triple negative breast carcinoma: significance of protein expression and high gene copy number. Cesk Patol. 2015;51(2):80–86.
79. Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66(16):8163–8171. doi:10.1158/0008-5472.CAN-06-0453
80. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi:10.1056/NEJMoa040938
81. von Minckwitz G, Jonat W, Fasching P, et al. A multicentre phase II study on gefitinib in taxane- and anthracycline-pretreated metastatic breast cancer. Breast Cancer Res Treat. 2005;89(2):165–172. doi:10.1007/s10549-004-1720-2
82. Bernsdorf M, Ingvar C, Jörgensen L, et al. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res Treat. 2011;126(2):463–470. doi:10.1007/s10549-011-1352-2
83. Schuler M, Awada A, Harter P, et al. A phase II trial to assess efficacy and safety of Afatinib in extensively pretreated patients with HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2012;134(3):1149–1159. doi:10.1007/s10549-012-2126-1
84. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6(8):1609–1623.
85. Ribatti D, Nico B, Ruggieri S, et al. Angiogenesis and Antiangiogenesis in Triple-Negative Breast cancer. Transl Oncol. 2016;9(5):453–457. doi:10.1016/j.tranon.2016.07.002
86. Linderholm BK, Hellborg H, Johansson U, et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20(10):1639–1646. doi:10.1093/annonc/mdp062
87. Shih T, Lindley C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28(11):1779–1802. doi:10.1016/j.clinthera.2006.11.015
88. von Minckwitz G, Eidtmann H, Rezai M, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012;366(4):299–309. doi:10.1056/NEJMoa1111065
89. Bear HD, Tang G, Rastogi P, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med. 2012;366(4):310–320. doi:10.1056/NEJMoa1111097
90. Cameron D, Brown J, Dent R, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, Phase 3 trial. Lancet Oncol. 2013;14(10):933–942. doi:10.1016/S1470-2045(13)70335-8
91. Marotti JD, de Abreu FB, Wells WA, et al. Triple-Negative Breast Cancer: next-Generation Sequencing for Target Identification. Am J Pathol. 2017;187(10):2133–2138. doi:10.1016/j.ajpath.2017.05.018
92. Marty B, Maire V, Gravier E, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008;10(6):R101. doi:10.1186/bcr2204
93. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. doi:10.1056/NEJMoa1109653
94. Gonzalez-Angulo AM, Green MC, Murray JL, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancer. Ann Oncol. 2014;25(6):1122–1127. doi:10.1093/annonc/mdu124
95. Sangai T, Akcakanat A, Chen H, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18(20):5816–5828. doi:10.1158/1078-0432.CCR-12-1141
96. Hudis C. A Phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15(6):R110. doi:10.1186/bcr3577
97. Kim SB, Dent R, Im SA, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, Phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372. doi:10.1016/S1470-2045(17)30450-3
98. Rosenberg JE. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi:10.1016/S0140-6736(16)00561-4
99. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060
100. Erber R, Hartmann A. Understanding PD-L1 Testing in Breast Cancer: a Practical Approach. Breast Care. 2020;15(5):481–490. doi:10.1159/000510812
101. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690
102. Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279(53):55117–55126. doi:10.1074/jbc.M404524200
103. Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30(9):1437–1447. doi:10.1093/annonc/mdz192
104. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
105. Turner NC, Lord CJ, Iorns E, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27(9):1368–1377. doi:10.1038/emboj.2008.61
106. Tutt ANJ, Garber JE, Kaufman B, et al. Adjuvant Olaparib for Patients with BRCA1 - or BRCA2 -Mutated Breast Cancer. N Engl J Med. 2021;384(25):2394–2405. doi:10.1056/NEJMoa2105215
107. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi:10.1016/S0140-6736(10)60892-6
108. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377(6):523–533. doi:10.1056/NEJMoa1706450
109. Isakoff SJ, Puhalla S, Domchek SM, et al. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in BRCA1 / 2 metastatic breast cancer: design and rationale. Future Oncol. 2017;13(4):307–320. doi:10.2217/fon-2016-0412
110. Somlo G, Frankel PH, Arun BK, et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1 - or BRCA2 -Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin Cancer Res. 2017;23(15):4066–4076. doi:10.1158/1078-0432.CCR-16-2714
111. Anampa J, Chen A, Wright J, et al. Phase I Trial of Veliparib, a Poly ADP Ribose Polymerase Inhibitor, Plus Metronomic Cyclophosphamide in Metastatic HER2-negative Breast Cancer. Clin Breast Cancer. 2018;18(1):e135–e142. doi:10.1016/j.clbc.2017.08.013
112. Wulfkuhle JD. Prediction of complete pathologic response to veliparib/carboplatin plus standard neoadjuvant therapy in HER2 negative breast cancer: exploratory protein pathway marker results from the I-SPY 2 trial. Cancer Res. 2016;76.
113. Lee KH, Sohn J, Goodwin A, et al. Talazoparib versus Chemotherapy in Patients with HER2-Negative Advanced Breast Cancer and a Germline BRCA1/2 Mutation Enrolled in Asian Countries: exploratory Subgroup Analysis of the Phase III EMBRACA Trial. Cancer Res Treatment. 2021;53(4):1084–1095. doi:10.4143/crt.2020.1381
114. Peters AA, Buchanan G, Ricciardelli C, et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009;69(15):6131–6140. doi:10.1158/0008-5472.CAN-09-0452
115. Wong YC, Xie B. The role of androgens in mammary carcinogenesis. Ital J Anat Embryol. 2001;106(2 Suppl 1):111–125.
116. Vera-Badillo FE, Templeton AJ, de Gouveia P, et al. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106(1):djt319. doi:10.1093/jnci/djt319
117. Narayanan R, Dalton JT. Androgen Receptor: a Complex Therapeutic Target for Breast Cancer. Cancers. 2016;8(12):108. doi:10.3390/cancers8120108
118. Adamczyk A, Niemiec J, Janecka A, et al. Prognostic value of PIK3CA mutation status, PTEN and androgen receptor expression for metastasis-free survival in HER2-positive breast cancer patients treated with trastuzumab in adjuvant setting. Pol J Pathol. 2015;66(2):133–141. doi:10.5114/pjp.2015.53009
119. Lehmann BD, Bauer JA, Schafer JM, et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014;16(4):406. doi:10.1186/s13058-014-0406-x
120. Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15(7):2472–2478. doi:10.1158/1078-0432.CCR-08-1763
121. Gucalp A, Tolaney S, Isakoff SJ, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19(19):5505–5512. doi:10.1158/1078-0432.CCR-12-3327
122. Traina TA, Miller K, Yardley DA, et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR plus triplenegative breast cancer (TNBC). J Clin Oncol. 2015;33(15):1003. doi:10.1200/jco.2015.33.15_suppl.1003
123. Thomas A, Teicher BA, Hassan R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016;17(6):e254–e262. doi:10.1016/S1470-2045(16)30030-4
124. Ponziani S, Di Vittorio G, Pitari G, et al. Antibody-Drug Conjugates: the New Frontier of Chemotherapy. Int J Mol Sci. 2020;21(15):5510. doi:10.3390/ijms21155510
125. Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2021;384(16):1529–1541. doi:10.1056/NEJMoa2028485
126. Goldenberg DM, Cardillo TM, Govindan SV, et al. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget. 2015;6(26):22496–22512. doi:10.18632/oncotarget.4318
127. Bardia A, Mayer IA, Diamond JR, et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2017;35(19):2141–2148. doi:10.1200/JCO.2016.70.8297
128. Bardia A, Mayer IA, Vahdat LT, et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2019;380(8):741–751. doi:10.1056/NEJMoa1814213
129. Modi S, Park H, Murthy RK, et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: results From a Phase Ib Study. J Clin Oncol. 2020;38(17):1887–1896. doi:10.1200/JCO.19.02318
130. Passegue E. Cancer biology: a game of subversion. Nature. 2006;442(7104):754–755. doi:10.1038/442754a
131. Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825. doi:10.1073/pnas.0905718106
132. Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. doi:10.1158/1078-0432.CCR-09-1630
133. Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69(8):3382–3389. doi:10.1158/0008-5472.CAN-08-4418
134. Pontier SM, Muller WJ. Integrins in mammary-stem-cell biology and breast-cancer progression--a role in cancer stem cells? J Cell Sci. 2009;122(Pt 2):207–214. doi:10.1242/jcs.040394
135. Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi:10.1172/JCI44745
136. DiMeo TA, Anderson K, Phadke P, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69(13):5364–5373. doi:10.1158/0008-5472.CAN-08-4135
137. Bhola NE, Balko JM, Dugger TC, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. doi:10.1172/JCI65416
138. Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. doi:10.1158/0008-5472.CAN-06-0054
139. Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709–718. doi:10.1158/0008-5472.CAN-09-1681
140. Lee KL, Kuo Y-C, Ho Y-S, et al. Triple-Negative Breast Cancer: current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers. 2019;11(9):1334. doi:10.3390/cancers11091334
141. Ding L, Gu H, Xiong X, et al. MicroRNAs Involved in Carcinogenesis, Prognosis, Therapeutic Resistance and Applications in Human Triple-Negative Breast Cancer. Cells. 2019;8(12):1492. doi:10.3390/cells8121492
142. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20(8):460–469. doi:10.1016/j.molmed.2014.06.005
143. Lukianova NY, Borikun TV, Chekhun VF. Tumor microenvironment-derived miRNAs as prognostic markers of breast cancer. Exp Oncol. 2019;41(3):242–247. doi:10.32471/exp-oncology.2312-8852.vol-41-no-3.13615
144. Fan C, Liu N, Zheng D, et al. MicroRNA-206 inhibits metastasis of triple-negative breast cancer by targeting transmembrane 4 L6 family member 1. Cancer Manag Res. 2019;11:6755–6764. doi:10.2147/CMAR.S199027
145. Shi P, Chen C, Li X, et al. MicroRNA124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol Med Rep. 2019;19(5):3667–3675. doi:10.3892/mmr.2019.10044
146. Najminejad H, Kalantar SM, Abdollahpour‐Alitappeh M, et al. Emerging roles of exosomal miRNAs in breast cancer drug resistance. IUBMB Life. 2019;71(11):1672–1684. doi:10.1002/iub.2116
147. Liu M, Gong C, Xu R, et al. MicroRNA-5195-3p enhances the chemosensitivity of triple-negative breast cancer to paclitaxel by downregulating EIF4A2. Cell Mol Biol Lett. 2019;24(1):47. doi:10.1186/s11658-019-0168-7
148. Tormo E, Ballester S, Adam-Artigues A, et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci Rep. 2019;9(1):5316. doi:10.1038/s41598-019-41472-y
149. Rupaimoole R, Calin GA, Lopez-Berestein G, et al. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016;6(3):235–246. doi:10.1158/2159-8290.CD-15-0893
150. Bronisz A, Godlewski J, Wallace JA, et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2011;14(2):159–167. doi:10.1038/ncb2396
151. Baroni S, Romero-Cordoba S, Plantamura I, et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016;7(7):e2312. doi:10.1038/cddis.2016.224
152. Qattan A. Novel miRNA Targets and Therapies in the Triple-Negative Breast Cancer Microenvironment: an Emerging Hope for a Challenging Disease. Int J Mol Sci. 2020;21(23):8905. doi:10.3390/ijms21238905
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.