")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 10
Molecular and Clinical Features of Hepatocellular Carcinoma in Patients with HBV-HDV Infection
Authors Costante F, Stella L, Santopaolo F, Gasbarrini A, Pompili M, Asselah T, Ponziani FR
Received 4 January 2023
Accepted for publication 6 April 2023
Published 25 April 2023 Volume 2023:10 Pages 713—724
DOI https://doi.org/10.2147/JHC.S384751
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Manal Hassan
Federico Costante,1,* Leonardo Stella,1,* Francesco Santopaolo,1,2 Antonio Gasbarrini,1,2 Maurizio Pompili,1,2 Tarik Asselah,3,* Francesca Romana Ponziani1,2
1Internal Medicine and Gastroenterology - Hepatology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, 00168, Italy; 2Department of Translational Medicine and Surgery, Catholic University, Rome, 00168, Italy; 3Service d’Hépatologie, Hôpital Beaujon UMR 1149 Inserm - Université de Paris, Clichy, France
*These authors contributed equally to this work
Correspondence: Francesca Romana Ponziani; Federico Costante, Internal Medicine and Gastroenterology - Hepatology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Largo Francesco Vito 1, Rome, 00168, Italy, Tel +390630156264, Email [email protected]; [email protected]
Abstract: Hepatitis D virus (HDV) infection affects more than 10 million people worldwide, with an estimated prevalence of nearly 4.5% among HBsAg-positive individuals. Epidemiological studies have shown a significant increase in the prevalence of hepatocellular carcinoma (HCC) in patients with chronic HDV infection compared to those with chronic hepatitis B virus (HBV) mono-infection. Despite the clinical findings, data on molecular oncogenic mechanisms are limited and fragmentary. Moreover, the role of HDV in promoting the development of HCC has so far been controversial, because it is difficult to weigh the respective contributions of the two viruses. In this review, we focused on the direct oncogenic action of HDV, its role in modifying the tumor microenvironment, and the genetic signature of HDV-related HCC, comparing these features with HBV-related HCC.
Keywords: hepatitis B virus, hepatitis delta virus, chronic hepatitis B, CHB, chronic delta hepatitis, CDH, dual infection, HCC
Introduction
Hepatocellular carcinoma (HCC) is the most frequent histological subtype of primary liver cancer, accounting for nearly 75% of all liver malignancies.1 With 800,000 new cases annually, a cumulative incidence of 16/100,000 cases and 745,000 deaths per year, HCC ranks as the sixth most frequent cancer in the world, and represents one of the leading causes of cancer-related mortality in men. Moreover, the incidence is expected to increase after 2025, with more than 1,000,000 cases yearly.2 Asia and Africa have the highest incidence rates worldwide;1 however, as recent epidemiological data show, the mortality rate from HCC is rising in North America and Europe, where HCC is reported to be the cause of death in 54–70% of individuals with compensated cirrhosis from various etiologies.3
In most cases, HCC is preceded by advanced fibrosis/cirrhosis, resulting from chronic liver inflammation.4 Globally, the underlying aetiology of liver disease is related to chronic viral hepatitis sustained by hepatitis B virus (HBV) or hepatitis C virus (HCV) infections in about 80% of patients.5 On the other hand, the relationship with hepatitis delta virus (HDV) remains unclear.6 Further independent risk factors for HCC are alcohol abuse, diabetes mellitus, overweight and obesity, metabolic dysfunction associated fatty liver disease (MAFLD), aflatoxin B1 exposure, hemochromatosis and other more rare conditions.7 Given its rising prevalence and the steady decline in viral hepatitis-related HCC due to immunization and efficient antiviral therapies, MAFLD is expected to overtake viral infections as the primary cause of HCC in the next ten years.7,8 However, HCC still remains a fearsome consequence of HBV infection, especially when associated with delta virus (HDV) infection.
In this paper, we will discuss the pathophysiological background and characteristics of HCC arising in this specific clinical setting.
HBV: Microbiological Characteristics and Natural History
The hepatitis B virion consists of an inner nucleocapsid core that contains the viral DNA and an outside lipoprotein envelope. Viral genome encodes seven proteins: envelope antigens (Small-protein or S or HBsAg, Middle-protein or M, Large-protein or L), a non-structural antigen (HBeAg), the core antigen (HBcAg), HBV polymerase and X protein (HBx). After endocytosis in the hepatocyte, HBV-DNA is incorporated into the genome of the host but it is also transformed into a double-stranded circular DNA structure (the “covalently closed circular DNA”), which serves both as a transcriptional template for viral RNA and as a reservoir for the reactivation of viral replication.
When the host fails to resolve acute HBV infection it becomes chronic, and up to 40% of untreated patients progresses to cirrhosis (Figure 1).9–11 Roughly one-third of patients with untreated HBV-related liver cirrhosis develops HCC.11 Although nearly 80% of HCC develop in a cirrhotic liver,12 and a small percentage in the absence of cirrhosis.13 Overall, at least one-third of HCC-related death is caused by chronic HBV infection.14
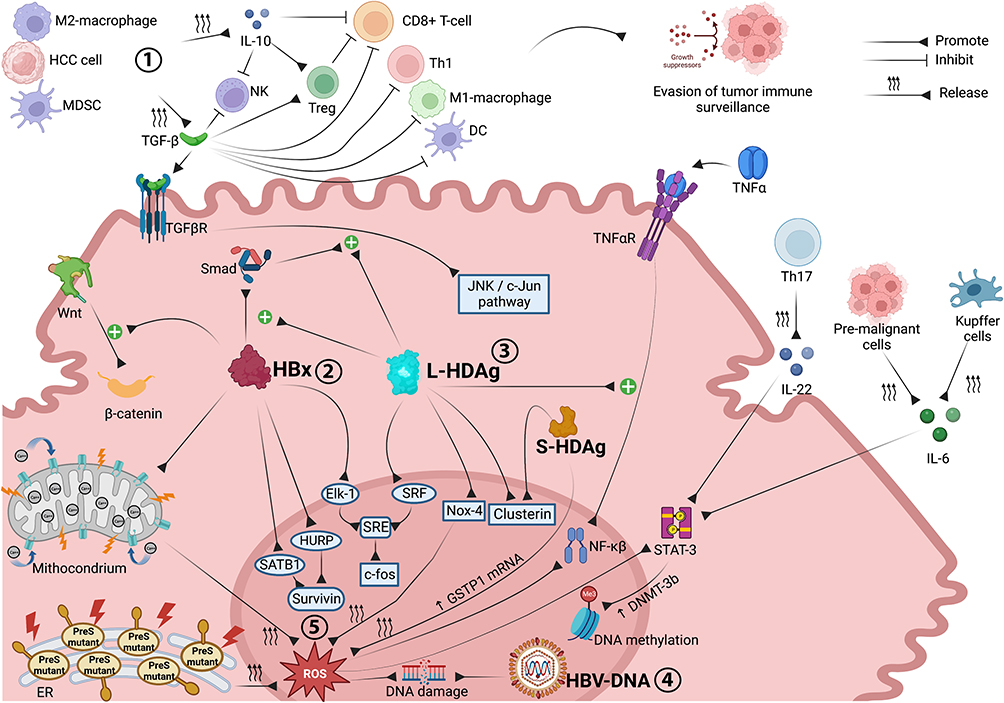 |
Figure 1 Molecular interactions and oncogenic mechanisms occurring in the HCC microenvironment associated with chronic HBV-HDV infection. Both immune cell impairment and stimulation of pro-oncogenic intracellular signaling pathways are involved in CHB- and CDH-driven hepatocarcinogenesis. Through the release of TGF-β and IL-10, M2 macrophages, MDSCs and tumor cells inhibit the antitumor functions of immune effector cells, promoting tumor immune escape. HBx upregulates Wnt/β-catenin signaling and promotes survivin protein synthesis, supporting tumor cell proliferation and preventing apoptosis. Synergistically, L-HDAg and HBx trigger the JNK/c-Jun and SRE/c-fos cascades, both of which are involved in cancer cell growth, proliferation and survival. The integration of HBV-DNA into the host genome and the overproduction of ROS, triggered by HBx, L-HDAg and S-HDAg, promote DNA damage and genomic instability. ROS also promote cancer cell survival, angiogenesis and invasiveness through upregulation of the NF-ĸB pathway, which is also stimulated by L-HDAg in a TNFα-dependent manner, and STAT-3 signaling, which is further activated by Th17 (via IL-22), pre-malignant cells and Kupffer cells (via IL-6). Image created with Biorender.com. Abbreviations: CDH, chronic delta hepatitis; CHB, chronic hepatitis B; DC, dendritic cells; DNMT-3b, DNA methyltransferase-3b; Elk-1, E26 transformation-specific like-1 protein; ER, endoplasmic reticulum; GSTP1, glutathione S-transferase P1; HBV, hepatitis B virus; HBx, X protein; HCC, hepatocellular carcinoma; HDV, hepatitis delta virus; HURP, hepatoma upregulated protein; IL-6, interleukin-6; IL-10, interleukin-10; IL-22, interleukin-22; JNK, c-Jun N-terminal kinase; L-HDAg, large hepatitis D antigen; MDSC, myeloid-derived suppressor cells; NK, natural killer cells; Nox-4, NADPH Oxidase 4; NF-ĸB, nuclear factor ĸB; ROS, reactive oxygen species; SATB1, special AT-rich sequence-binding protein-1; S-HDAg, small hepatitis D antigen; Smad, small mother against decapentaplegic proteins; SRE, serum response element; SRF, serum response factor; STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor-β; TGFβR, transforming growth factor-β receptor; Th1, T-helper 1 cells; Th17, T-helper 17 cells; TNFα, tumor necrosis factor α; TNFαR, tumor necrosis factor α receptor; Treg, regulatory T cells; Wnt, wingness-related integration site. |
Most often, acute HBV infection is asymptomatic or paucisymptomatic. Once chronic infection occurs, it typically progresses in an asymptomatic manner, so that the diagnosis is often established by chance during laboratory tests.15 The immune reaction to HBV foreign antigens, rather than the virus itself, promotes inflammation, necrosis and eventually fibrosis of liver parenchyma. However, the immunological activity against HBV is not continuous, so that this process proceeds intermittently; indeed, cirrhosis and HCC development result from repeated periods of immunological activity over several years. Hence, chronic HBV infection has been schematically divided into five, not necessarily sequential, phases, according to the presence of the following biomarkers: HBeAg, HBV-DNA levels, alanine aminotransferase value, and liver necroinflammation and fibrosis. The combination of these indicators reflects the degree of hepatic immunological activity, thus guiding the choice to begin treatment.16
Epidemiological and Microbiological Characteristics of HDV
HDV infection affects about 12 million people worldwide, with an estimated prevalence of 4.5% among HBsAg-positive individuals. Prevalence rates are higher in Africa and America (slightly under 6%), although the highest one is registered in Mongolia (36.9%). Also, HCV-positive or human immunodeficiency virus (HIV)-positive subjects, persons who inject drugs and haemodialysis recipients show higher prevalence rates compared to the general population. HDV genotype 1 prevails globally (89.9%), while other genotypes are more regionally specific, such as genotypes 2 and 4 in Asia, genotype 3 in Latin America, genotypes 5, 6 and 8 in Africa (Table 1).12
 |
Table 1 Estimated Prevalence of HDV Infection in HBsAg-Positive General Population and HDV Genotypes Distribution by Country |
HDV is made up of an inner nucleocapsid carrying a single-stranded circular RNA combined with roughly 200 molecules of hepatitis D antigen (HDAg), and an outer lipid envelope embedded with the HBsAg derived from HBV.13 In fact, HDV is considered a faulty virus, requiring the simultaneous presence of HBV to be a human pathogen; specifically, it needs HBsAg to assemble virions and penetrate into hepatocytes.14 After entering the cell via a HBsAg-mediated receptor binding, HDV-RNA is translocated into the nucleus and uses host RNA polymerase for replication. Following viral genome replication, synthesis of HDAg (the only protein encoded by HDV genome, in 2 isoforms: large-HDAg or L-HDAg and small-HDAg or S-HDAg) and post-translational HDAg modifications, HDAg must combine with HBsAg to generate an infectious virion that can exit the hepatocyte.15
Pathogenesis and Clinical Evolution of HBV-HDV Dual Infection
Unlike the other main hepatotropic viruses, the pathogenesis of HDV-related liver damage is still unclear. Analogous to HBV infection, HDV infection seems to cause liver damage predominantly via a host-mediated immune response, as suggested by both the presence of HDAg-specific CD4+ T-cells in peripheral blood,16 and the pronounced necroinflammatory activity at liver histology17,18 of affected patients. Furthermore, in subjects with HDV/HIV co-infection, the reduction in CD4+ T-lymphocyte counts due to HIV activity has been associated with higher HDV-RNA levels, indicating that adaptive immunity may play a relevant role in the containment of HDV replication.19 The existence of HDV cytopathic activity is still debated, as some studies support direct virus-mediated hepatocellular damage,20 while others do not.21
Because HDV is a defective virus that requires the concomitant presence of HBV to establish infection, which can occur at the same time as HBV infection (co-infection) or in the context of chronic HBV infection (superinfection). Compared with acute HBV monoinfection, HBV-HDV coinfection is associated with a higher mortality rate in the short term, as it can result in life-threatening acute or fulminant hepatitis with acute liver failure.22 However, 90–95% of HBV-HDV coinfections resolve spontaneously, as a consequence of HBV clearance.23 In contrast, HDV superinfection often results in exacerbation of hepatitis24 and evolves into chronic HDV infection in the vast majority of cases.23 Chronic delta hepatitis (CDH) is considered the most aggressive form of hepatitis, as it is associated with a more rapid progression to cirrhosis23,25 and a higher rate of liver decompensation leading to death23,26 than chronic HBV infection alone.
Since HBV infection is mandatory to allow HDV to initiate the infection and subsequently accomplish an oncogenic role, the role of HDV in promoting the development of HCC is still controversial, because it is difficult to specifically weigh the respective contributions of the two viruses. In fact, as of 1994, HDV is still included in International Agency for Research on Cancer (IARC) Group 3 (not sufficient evidence of carcinogenicity), while HBV and HCV are classified in Group 1 (high evidence of carcinogenicity).27 Although some studies have found no statistically significant difference in HCC incidence rates between patients with dual HBV-HDV infection and those mono-infected with HBV,28,29 many others have found a higher cancer risk in the former population than in the latter.26,30–32 Among HBV-HDV-infected individuals, the development of HCC has been associated with higher levels of HDV-RNA in peripheral blood,33 which could be a further indication of the oncogenic effect of HDV. In addition, two very recent meta-analyses comparing dual-infected and single-infected patients confirmed the oncogenic potential of HDV, pointing to a higher incidence rate of HCC in the former group.34,35 Furthermore, subgroup analyses performed by Chang et al34 found that the risk of HCC was higher in HBV-HDV dual infection, regardless of ethnic group or concomitant infection with HIV or HCV, with no statistically significant difference according to the severity of liver fibrosis.
Impact of HBV and HDV on Hepatocarcinogenesis
Liver Tumor Microenvironment in Chronic Hepatitis B
HBV conventionally does not exhibit cytopathic activity, and liver tissue damage results from the immune response against surface and core viral antigens. Over time, this leads to a state of chronic inflammation and promotes immune depression, which in turn promotes hepatocarcinogenesis. Specifically, immunological alterations in the tumor microenvironment involve dysregulation of specific key signaling pathways, cytokine production, and functional impairment of T cells and innate immune cells (Figure 2).
 |
Figure 2 Natural history of HBV and HBV/HDV infected patients. Chronic HBV infection occurs in 90% of infected children and up to 10% of infected adults, while HDV superinfection orcoinfection lead to chronic disease in 10% and 77% of exposed patients, respectively. Compared with HBV mono-infection, HDV/HBV coinfection more frequently leads to liver cirrhosis (OR 3.84), and in a shorter time. The risk of HCC development is increased in both HBV mono-infection and HBV/HDV coinfection (OR 1.66). Image created with Biorender.com. Abbreviations: HBV, hepatitis B virus; HDV, hepatitis delta virus; OR, odds ratio. |
Inflammation promotes the activation and interaction between nuclear factor (NF)-ĸB and signal transducer and activator of transcription 3 (STAT3), which act preventing pre-malignant and tumor cells from apoptosis and promoting tumor immune escape, angiogenesis and invasiveness.36,37 STAT3 activation is mainly mediated by interleukin (IL)-6,38 which is initially secreted in an autocrine manner by pre-malignant cells.39 Once HCC is developed, Kupffer cells become critical in the tumor growth process, through IL-6 release.40 The STAT3 signaling cascade can also be triggered by IL-22 produced by T helper (Th) 17 cells,41 the role of which in HCC progression has recently been underlined.42 As for the NF-ĸB pathway, some evidence has revealed that it is also involved in anti-oncogenic processes;35 hence, a strategy to inhibit NF-ĸB signalling may be a double-edged sword.
Recent evidence has linked a higher concentration of tumor-associated macrophages in HCC microenvironment, with a poorer prognosis.43 Specifically, M2-phenotype tumor-associated macrophages (a pro-oncogenic subtype of macrophages) release transforming growth factor-β (TGF-β), which triggers CD8+ T-cells to expressmarkers of “exhaustion”,44 such as programmed cell death-1 (PD-1), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), lymphocyte-activation gene-3 (LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT).45 Exhausted CD8+ T-cells exhibit defective cytotoxic activity and impaired ability to release antiviral cytokines,46 thus promoting the development of HCC. TGF-β negatively affects the host immune response through several other mechanisms; in particular, it inhibits the anti-oncogenic M1-macrophages subtype, reduces the antitumor function of Th1 cells by inducing their differentiation to Th2 cells, and suppresses the cytotoxic activity of natural killer (NK) and dendritic cells.47,48 In addition to M2-macrophages, myeloid-derived suppressor cells and tumor cells also secrete TGF-β and IL-10; IL-10 exerts analogous inhibitory effects on CD8 T-cells and NK cells.38,49
Furthermore, TGF-β and IL-10 enable the activation of regulatory T cells (Treg), which represent a subpopulation of T cells (expressing CD4, CD25 and the forkhead box P3 transcriptional regulator [Foxp3]) with tolerogenic and immunosuppressive properties, mostly antagonizing the killing function of CD8+ T-cells. Their role in promoting immune escape in HBV-related HCC is well documented.50 Treg can be activated by TGF-β and IL-10 both directly (receptor–ligand interaction)38 and indirectly, as high levels of TGF- β (typically found in chronic hepatitis B) down-regulate the transcription of microRNA-34a, eliciting the synthesis of chemokine C-C Motif Chemokine Ligand 22 (CCL22) and the subsequent Treg recruitment.51
Oncogenic HBV Mechanisms
In addition to chronic liver inflammation, virus-related factors significantly contribute to tumorigenesis in chronic hepatitis B (CHB)-related HCC (Figure 2).
The integration of viral DNA is one of the most well-known oncogenic factors, since its integration in the host genome is thought to provoke deletions, translocations, chromosomal aberrations, fusion transcript synthesis and generalized genomic instability.52 Moreover, the presence of viral genetic material in the nucleus of hepatocytes elicits the activation of DNA repair mechanisms, increasing the DNA recombination rate.53 Accordingly, HBV-DNA incorporation occurs more commonly in cancer cells than in healthy hepatocytes.54 In addition, chronic inflammation leads to hepatocyte death and subsequent compensatory proliferation, which increases the viral DNA insertion rate, and exacerbates the potential direct oncogenic effects of HBV-DNA.55
HBx is another viral factor that strongly promotes HCC tumorigenesis, as suggested by its significantly higher expression in HBV-related HCC as compared to first stages of HBV infection.56 HBx acts by altering several pathways; for example, it prevents cancer cells from apoptosis by increasing the expression of the hepatoma upregulated protein (HURP) and special AT-rich binding -1 (SATB1) gene, resulting in the overproduction of the anti-apoptotic protein survivin.57 Moreover, HBx upregulates the Wingness-related integration site (Wnt)/β-catenin signaling pathway, favouring the adhesion of epithelial cells.58 HBx can also interfere with transcriptional regulation and DNA-binding of p53, which acts as regulator of apoptosis, cell cycle arrest and DNA repair.59 Also, it can hinder the activity of mitochondria by altering their membrane potential60 and increasing their calcium content,61 both resulting in the overproduction of reactive oxygen species (ROS) and oxidative stress, whose carcinogenic potential is well established.62 HBx promotes HCC metastatization too, by inducing epithelial-mesenchymal transition.63 These are only a small subset of the multitude of molecular mechanisms through which HBx can boost hepatocarcinogenesis, as it is also implicated in promoting genetic instability,64 preventing cellular senescence65 and inducing epigenetic modifications.66
Further potential viral factors implied in tumorigenesis are envelope proteins, such HBsAg and others encoded by specific mutant variants of preS/S genes. PreS mutants can lead to the overproduction of mutated envelope proteins, which accumulate into the endoplasmic reticulum (ER), resulting in ROS release with subsequent DNA damage and genomic instability. Furthermore, ER stress can trigger the activation of signaling pathways leading to hepatocyte proliferation, angiogenesis and eventually HCC development.67
HDV Infection Influences the Molecular Profile of HCC
Although HDV infection seems to increase the risk of HCC in individuals already infected by HBV, data concerning the oncogenic molecular mechanisms specifically related to HDV are limited and fragmentary (Figure 2).
A significant contribution comes from Choi et al,68 who described one of the potential oncogenic properties of L-HDAg. They demonstrated that L-HDAg upregulates TGF-β-mediated transcriptional activity of c-Jun, which modulates cell growth, proliferation and apoptosis.69 The same signaling cascade is activated by the interaction between HBx and Small mother against decapentaplegic (Smad) proteins, which was found to be enhanced by L-HDAg in a dose-dependent manner;68 this synergistic molecular mechanism could, at least partially, explain why HBV-HDV dual infection is associated with a higher HCC incidence rate, as compared with HBV mono-infection. The activation of TGF-β and c-Jun pathways is mainly related to the post-transcriptional isoprenylation of a cysteine residue at the C-terminal of L-HDAg;68 indeed, S-HDAg lacks both the isoprenylated cysteine and the TGF-β-stimulation activity. Goto et al70 provided another example of molecular synergy between HBV and HDV in promoting HCC onset, demonstrating that HBx can trigger the transcriptional ability of E26 transformation-specific like-1 (Elk-1) protein, and that L-HDAg can activate the transcriptional function of the serum response factor (SRF). Both Elk-1 and SRF can enhance the serum response element (SRE)-dependent pathway, which in turn mediates the c-fos proto-oncogene transcription. L-HDAg can also enhance the activation of NF-ĸB and STAT3 signaling cascades, which are already overstimulated in CHB. Indeed, L-HDAg can elicit the expression of Nox4 gene, leading to ROS overproduction, which is a trigger for the abovementioned pathways.71 NF-ĸB can also be upregulated by L-HDAg via the induction of TNFα; however, this mechanism is not related to the isoprenylated cysteine residue.72 STAT3 activation induced by HDV also accounts for the overexpression of DNA methyltransferase-3b, which is involved in potentially oncogenic methylation events occurring during DNA replication.73 HDAg-mediated histone acetylation is another epigenetic alteration possibly involved in tumorigenesis. L-HDAg and S-HDAg can induce histone H3 hyperacetylation within the clusterin promoter,74 resulting in increased expression of clusterin, a molecule involved in cell death control.75
A further oncogenic mechanism, promoted by S-HDAg, involves the glutathione S-transferase P1 (GSTP1) gene. S-HDAg can bind GSTP1 mRNA, thus decreasing the GSTP1 protein expression, leading to ROS accumulation and subsequent rise in cellular apoptosis and DNA damage in proliferative cells.76
More recently, Yu et al77 performed a sophisticated study based on microarray dataset analysis, comparing cancerous and para-cancerous specimens from individuals with CHB or CDH-related HCC. Seven genes closely involved in mitotic cell cycle and DNA replication (CDC6, CDC45, CDCA5, CDCA8, CENPH, MCM4, MCM7) were found to be differently expressed (mostly upregulated) only in the CDH-driven HCC subgroup; therefore, the alteration of pathways in which these genes are involved seems selectively mediated by HDV. Overall, these findings show that the molecular profile of CDH-related HCC is characterized by an overexpression of genes involved in cell cycle and DNA replication/repair, highlighting genomic instability as a key mechanism of hepatocarcinogenesis.
Epidemiology of HCC in CBH and CDH
In the past, chronic HDV infection was thought to be at minor HCC risk, due to its rapidly progressive liver disease pattern, while nowadays evidence shows that CDH leads to early cirrhosis and a higher risk of HCC development. Even if cirrhosis decompensation, rather than incidence of HCC, is the main clinical endpoint that occurs during HDV infection, some clinical studies suggest that HCC and cirrhosis could be secondary effects of the same necro-inflammatory process.78
However, evidence-based clinical data point to a significant increase in the global risk of HCC development in patients with chronic HDV infection compared to those with chronic HBV mono-infection. Among CDH patients, HCC cumulative incidence was, respectively, 2.3% at 1 year, 5.4% at 3 years, and 7.5% at 5-year in non-cirrhotic patients, rising to 5.4% at 1 year, 15.9% at 3 years and 23.1% at 5 years in cirrhotic patients.79 Major risk factors are liver cirrhosis (hazard ratio [HR] 9.98), higher HDV-RNA serum level (HR 5.73), age >50 years (HR 3.64), male gender (HR 2.69), higher body mass index (BMI) (HR 1.11), and lower platelet count.30,79,80
In a systematic review and meta-analysis from Alfaiate et al, the correlation between CDH and HCC was proven to be clinically relevant in cohort studies (pooled odds ratio [OR] 1.67; 95% confidence interval [CI] 1.28–2.1, p-value [p] <0.001), and even stronger in prospective studies (pooled OR 2.77; 95% CI 1.79–4.28, p < 0.001).35 According to a recent study, the risk of HCC is increased in patients with CDH when compared to those with CHB (standardized incidence ratio [SRI] = 137.17, 95% CI: 62.19 to 261.51) or with HBV mono-infected patients (SRI = 6.11, 95% CI: 2.77 to 11.65).78 Another study also found an increased HCC risk in acute (relative risk [RR] 6.1) or chronic (RR 3.9) HDV infection when compared toHBV mono-infection.81 Other studies identified a 3-fold increase in the risk of CDH-related HCC, and a 2-fold increase in overall mortality compared to HBV mono-infected patients.82 Moreover, a further increase in the risk of HCC development has been reported in human immunodeficiency virus (HIV)/HBV/HDV coinfection.83
Clinical Presentation of HCC in CBH and CDH
Clinical features of HBV-HDV-related HCC are summarized in Figure 3. Usually, CHB-related HCC frequently presents with large tumor size, vascular invasion, aggressive growth pattern and high AFP serum levels at the time of diagnosis when compared to other aetiologies of liver disease.81,84,85
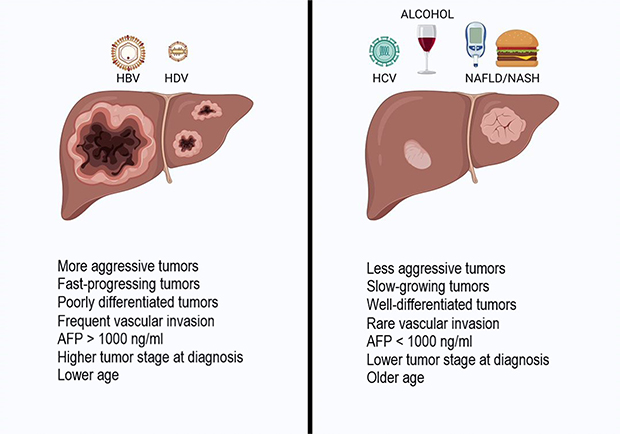 |
Figure 3 HCC clinical features at diagnosis in HBV/HDV patients and other aetiologies of liver disease. HBV/HDV-related HCC shows a more aggressive behavior compared with HCC related to other risk factors (such as alcohol, NAFLD, HCV, autoimmune diseases, biliary diseases), with a faster growth rate, larger nodule size, higher AFP serum levels, and a more advanced tumor stage. Image created with Biorender.com. Abbreviations: AFP, alpha-fetoprotein; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HDV, hepatitis delta virus. |
Concerning the clinical differences between HBV-related and HDV-related HCC, one study from Abbas et al showed a decreased liver size in patients affected by CDH compared to those affected by CHB (p < 0.05), associated with lower platelet count (p = 0.05) and larger varices (P < 0.05), underlining that HCC occurs in CDH with a higher burden of portal hypertension.84 Moreover, multifocal tumors (p < 0.05), a higher TNM stage (p < 0.05) and an alpha-fetoprotein level >1000 IU/mL (p = 0.06) were more common in the CHB group than in coinfected patients.84 These data are explained by a lead time bias, probably due to earlier diagnosis made while liver decompensation is investigated.78,84
Prevention of HCC in CBH and CDH
Worldwide hepatitis B vaccination and antiviral maternal prophylaxis in selected patients aim to achieve the goal of primary prevention of HBV infection and HBV-related HCC.86 Some studies confirmed the lower infection and HCC development rate in populations undergoing vaccination and maternal prophylaxis programs.87,88
Secondary prevention of HBV-related HCC can be achieved by treating CHB patients, monitoring for drug resistance, and establishing tailored surveillance programs (based on age, familial history of HCC, HBV viral load, and PAGE B score).86,89 Some consistent evidence has shown that the risk of HCC occurrence can be reduced, even if not eliminated, by current antiviral treatment in CHB patients. However, HCC still occurs in a percentage of patients obtaining a long-term virological suppression, and, the presence of persistent intrahepatic cccDNA and a high viral relapse rate after discontinuation of nucleoside analogues (NAs) represent still a challenge for these patients.86 The use of new drugs such as bulevirtide, alone or in combination with pegylated interferon alpha, has been shown to reduce or eliminate HDV viremia in addition to normalising transaminases.90 Although little is known about the long-term effects, especially on complications such as HCC, it can be expected that bulevirtide will lead to a reduction in the incidence of HCC, as already occurred with the use of antivirals in patients with HBV mono-infection. Schedule and duration of treatment are currently under investigation, and especially in the setting of HBV/HDV infection a “learning- by-doing” approach is currently adopted.
Moreover, tertiary prevention can lower HBV-related HCC recurrence after curative treatment.91 The protective effect of NAs in reducing HCC recurrence in CHB patients was also confirmed by a meta-analysis, showing a better recurrence free survival (HR: 0.66; P < 0.0001).92
Conclusion
In conclusion, the most recent data suggest that HDV infection is a major risk factor for HCC occurrence worldwide, in overt contradiction with the old hypothesis proposing that the rapid progression towards decompensation, leading to early liver transplantation or death, minimizes the risk of HCC development. This increment seems to be more clinically significant in high-quality studies with a robust design, in particular prospective studies adjusted for confounders with well-established inclusion criteria.
While HBV molecular pathways are well-known, the comprehension of HDV molecular pathways and its effect on cell microenvironment and epigenetic modifications have strengthened the evidence of its oncogenic role in acutely and chronically infected patients only in the recent years. Indeed, the increasing evidence on the oncogenic role of chronic HDV infection should be a reason to implement screening policies worldwide. In reason of the elevated biological risk of HCC occurrence in CDH patients, these patients should be considered as a very high-risk subgroup, suggesting that a personalized screening schedules should be adopted to detect HCC at its earlier stages.
HBV infection and HBV-related HCC primary prevention through vaccination programs and maternal prophylaxis, as well as CHB treatment before or after the first HCC occurrence (secondary and tertiary HCC prevention) have shown an incredible efficacy in lowering HCC occurrence rates. Concerning HDV, bulevirtide effect on HCC development is still under investigation. We therefore look forward to seeing the forthcoming data on how bulevirtide may change the natural history of HCC associated with CDH-related HCC.
Acknowledgments
We thank Fondazione Roma for the continuous support to our research.
Funding
No specific funding was identified for the study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Petrick JL, Florio AA, Znaor A, et al. International trends in hepatocellular carcinoma incidence, 1978–2012. Int J Cancer. 2020;147(2):317–330. PMID: 31597196 PMCID: PMC7470451. doi:10.1002/ijc.32723
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. PMID: 33479224. doi:10.1038/s41572-020-00240-3
3. Anthony PP. Hepatocellular carcinoma: an overview. Histopathology. 2001;39(2):109–118. PMID: 11493326. doi:10.1046/j.1365-2559.2001.01188.x
4. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–232. PMID: 29379119. doi:10.1038/s41590-018-0044-z
5. Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, Ioannou GN. Trends in burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US veterans, 2001–2013. Gastroenterology. 2015;149(6):1471–1482.e5; quiz e17–e18. doi:10.1053/j.gastro.2015.07.056
6. Romeo R, Petruzziello A, Pecheur EI, et al. Hepatitis delta virus and hepatocellular carcinoma: an update. Epidemiol Infect. 2018;146(13):1612–1618. PMID: 29991359 PMCID: PMC9507952. doi:10.1017/S0950268818001942
7. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl1):4–13. PMID: 32319693 PMCID: PMC7577946. doi:10.1002/hep.31288
8. Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75(6):1476–1484. PMID: 34453963. doi:10.1016/j.jhep.2021.08.012
9. Akinyemiju T, Abera S, Ahmed M, et al.; Global Burden of Disease Liver Cancer Collaboration. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the Global, Regional, and National Level: results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3(12):1683–1691. PMID: 28983565 PMCID: PMC5824275. doi:10.1001/jamaoncol.2017.3055
10. Liang TJ. Hepatitis B: the virus and disease. Hepatology. 2009;49(5 Suppl):S13–S21. PMID: 19399811 PMCID: PMC2809016. doi:10.1002/hep.22881
11. European Association for the Study of the Liver. Electronic address: [email protected]; European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370–398. PMID: 28427875. doi:10.1016/j.jhep.2017.03.021
12. Stockdale AJ, Kreuels B, Henrion MYR, et al. The global prevalence of hepatitis D virus infection: systematic review and meta-analysis. J Hepatol. 2020;73(3):523–532. PMID: 32335166 PMCID: PMC7438974. doi:10.1016/j.jhep.2020.04.008
13. Bonino F, Heermann KH, Rizzetto M, Gerlich WH. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J Virol. 1986;58(3):945–950. PMID: 3701932 PMCID: PMC253003. doi:10.1128/JVI.58.3.945-950.1986
14. Taylor JM. Hepatitis delta virus. Virology. 2006;344(1):71–76. PMID: 16364738. doi:10.1016/j.virol.2005.09.033
15. Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378(9785):73–85. PMID: 21511329. doi:10.1016/S0140-6736(10)61931-9
16. Nisini R, Paroli M, Accapezzato D, et al. Human CD4+ T-cell response to hepatitis delta virus: identification of multiple epitopes and characterization of T-helper cytokine profiles. J Virol. 1997;71(3):2241–2251. PMID: 9032359 PMCID: PMC191332. doi:10.1128/JVI.71.3.2241-2251.1997
17. Liao B, Zhang F, Lin S, et al. Epidemiological, clinical and histological characteristics of HBV/HDV co-infection: a retrospective cross-sectional study in Guangdong, China. PLoS One. 2014;9(12):e115888. PMID: 25532128 PMCID: PMC4274124. doi:10.1371/journal.pone.0115888
18. Govindarajan S, De Cock KM, Redeker AG. Natural course of delta superinfection in chronic hepatitis B virus-infected patients: histopathologic study with multiple liver biopsies. Hepatology. 1986;6(4):640–644. PMID: 3525368. doi:10.1002/hep.1840060415
19. Roingeard P, Dubois F, Marcellin P, et al. Persistent delta antigenaemia in chronic delta hepatitis and its relation with human immunodeficiency virus infection. J Med Virol. 1992;38(3):191–194. PMID: 1287132. doi:10.1002/jmv.1890380307
20. Bichko V, Netter HJ, Wu TT, Taylor J. Pathogenesis associated with replication of hepatitis delta virus. Infect Agents Dis. 1994;3(2–3):94–97. PMID: 7812659.
21. Guilhot S, Huang SN, Xia YP, La Monica N, Lai MM, Chisari FV. Expression of the hepatitis delta virus large and small antigens in transgenic mice. J Virol. 1994;68(2):1052–1058. PMID: 8289334 PMCID: PMC236543. doi:10.1128/JVI.68.2.1052-1058.1994
22. Rizzetto M. Hepatitis D: thirty years after. J Hepatol. 2009;50(5):1043–1050. PMID: 19285743. doi:10.1016/j.jhep.2009.01.004
23. Buti M, Homs M, Rodriguez-Frias F, et al. Clinical outcome of acute and chronic hepatitis delta over time: a long-term follow-up study. J Viral Hepat. 2011;18(6):434–442. PMID: 20546496. doi:10.1111/j.1365-2893.2010.01324.x
24. Grabowski J, Wedemeyer H. Hepatitis delta: immunopathogenesis and clinical challenges. Dig Dis. 2010;28(1):133–138. PMID: 20460901. doi:10.1159/000282076
25. Saracco G, Rosina F, Brunetto MR, et al. Rapidly progressive HBsAg-positive hepatitis in Italy. The role of hepatitis delta virus infection. J Hepatol. 1987;(3):274–281. PMID: 3429834. doi:10.1016/s0168-8278(87)80032-6
26. Romeo R, Del Ninno E, Rumi M, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136(5):1629–1638. PMID: 19208358. doi:10.1053/j.gastro.2009.01.052
27. Humans, I.W.G.o.t.E.o.C.R.t. Hepatitis Viruses. Vol. 59. World Health Organization; 1994.
28. Cross TJ, Rizzi P, Horner M, et al. The increasing prevalence of hepatitis delta virus (HDV) infection in South London. J Med Virol. 2008;80(2):277–282. PMID: 18098143. doi:10.1002/jmv.21078
29. Manesis EK, Vourli G, Dalekos G, et al. Prevalence and clinical course of hepatitis delta infection in Greece: a 13-year prospective study. J Hepatol. 2013;59(5):949–956. PMID: 23850875. doi:10.1016/j.jhep.2013.07.005
30. Brancaccio G, Fasano M, Grossi A, Santantonio TA, Gaeta GB. Clinical outcomes in patients with hepatitis D, cirrhosis and persistent hepatitis B virus replication, and receiving long-term tenofovir or entecavir. Aliment Pharmacol Ther. 2019;49(8):1071–1076. PMID: 30793345. doi:10.1111/apt.15188
31. Fattovich G, Giustina G, Christensen E, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut. 2000;46(3):420–426. PMID: 10673308 PMCID: PMC1727859. doi:10.1136/gut.46.3.420
32. Amougou MA, Noah DN, Moundipa PF, Pineau P, Njouom R. A prominent role of hepatitis D virus in liver cancers documented in Central Africa. BMC Infect Dis. 2016;16(1):647. PMID: 27821080 PMCID: PMC5100184. doi:10.1186/s12879-016-1992-2
33. Romeo R, Foglieni B, Casazza G, Spreafico M, Colombo M, Prati D. High serum levels of HDV RNA are predictors of cirrhosis and liver cancer in patients with chronic hepatitis delta. PLoS One. 2014;9(3):e92062. PMID: 24658127 PMCID: PMC3962389. doi:10.1371/journal.pone.0092062
34. Chang TE, Su CW, Huang YS, Huang YH, Hou MC, Wu JC. Hepatitis D virus dual infection increased the risk of hepatocellular carcinoma compared with hepatitis B virus mono infection: a meta-analysis. J Chin Med Assoc. 2022;85(1):30–41. PMID: 35006125. doi:10.1097/JCMA.0000000000000606
35. Alfaiate D, Clément S, Gomes D, Goossens N, Negro F. Chronic hepatitis D and hepatocellular carcinoma: a systematic review and meta-analysis of observational studies. J Hepatol. 2020;73(3):533–539. PMID: 32151618. doi:10.1016/j.jhep.2020.02.030
36. Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. 2014;20(33):11630–11640. PMID: 25206269 PMCID: PMC4155355. doi:10.3748/wjg.v20.i33.11630
37. He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21(1):159–168. PMID: 21187858 PMCID: PMC3193410. doi:10.1038/cr.2010.183
38. Lawal G, Xiao Y, Rahnemai-Azar AA, et al. The immunology of hepatocellular carcinoma. Vaccines. 2021;9(10):1184. PMID: 34696292 PMCID: PMC8538643. doi:10.3390/vaccines9101184
39. He G, Dhar D, Nakagawa H, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155(2):384–396. PMID: 24120137 PMCID: PMC4015514. doi:10.1016/j.cell.2013.09.031
40. Yang YM, Kim SY, Seki E. Inflammation and liver cancer: molecular mechanisms and therapeutic targets. Semin Liver Dis. 2019;39(1):26–42. PMID: 30809789 PMCID: PMC6616367. doi:10.1055/s-0038-1676806
41. Jiang R, Tan Z, Deng L, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54(3):900–909. PMID: 21674558. doi:10.1002/hep.24486
42. Lan YT, Fan XP, Fan YC, Zhao J, Wang K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine. 2017;96(32):e7704. PMID: 28796055 PMCID: PMC5556221. doi:10.1097/MD.0000000000007704
43. Yeung OW, Lo CM, Ling CC, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62(3):607–616. PMID: 25450711. doi:10.1016/j.jhep.2014.10.029
44. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med. 2019;25(11):1010–1023. PMID: 31353124. doi:10.1016/j.molmed.2019.06.007
45. Blank CU, Haining WN, Held W, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19(11):665–674. PMID: 31570879 PMCID: PMC7286441. doi:10.1038/s41577-019-0221-9
46. Fisicaro P, Barili V, Rossi M, et al. Pathogenetic mechanisms of T cell dysfunction in chronic HBV infection and related therapeutic approaches. Front Immunol. 2020;11:849. PMID: 32477347 PMCID: PMC7235343. doi:10.3389/fimmu.2020.00849
47. An Y, Gao S, Zhao WC, et al. Transforming growth factor-β and peripheral regulatory cells are negatively correlated with the overall survival of hepatocellular carcinoma. World J Gastroenterol. 2018;24(25):2733–2740. PMID: 29991878 PMCID: PMC6034152. doi:10.3748/wjg.v24.i25.2733
48. Achyut BR, Yang L. Transforming growth factor-β in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology. 2011;141(4):1167–1178. PMID: 21839702 PMCID: PMC6644047. doi:10.1053/j.gastro.2011.07.048
49. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–692. PMID: 25501578 PMCID: PMC4334715. doi:10.1158/1078-0432.CCR-14-1860
50. Zhang HH, Mei MH, Fei R, et al. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J Viral Hepat. 2010;17(Suppl 1):34–43. PMID: 20586932. doi:10.1111/j.1365-2893.2010.01269.x
51. Yang P, Li QJ, Feng Y, et al. TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell. 2012;22(3):291–303. PMID: 22975373 PMCID: PMC3443566. doi:10.1016/j.ccr.2012.07.023
52. Ringelhan M, O’Connor T, Protzer U, Heikenwalder M. The direct and indirect roles of HBV in liver cancer: prospective markers for HCC screening and potential therapeutic targets. J Pathol. 2015;235(2):355–367. PMID: 25196558. doi:10.1002/path.4434
53. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64(12):1972–1984. PMID: 26048673. doi:10.1136/gutjnl-2015-309809
54. Sung WK, Zheng H, Li S, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44(7):765–769. PMID: 22634754. doi:10.1038/ng.2295
55. Chemin I, Zoulim F. Hepatitis B virus induced hepatocellular carcinoma. Cancer Lett. 2009;286(1):52–59. PMID: 19147276. doi:10.1016/j.canlet.2008.12.003
56. Hwang GY, Lin CY, Huang LM, et al. Detection of the hepatitis B virus X protein (HBx) antigen and anti-HBx antibodies in cases of human hepatocellular carcinoma. J Clin Microbiol. 2003;41(12):5598–5660. PMID: 14662947 PMCID: PMC309044. doi:10.1128/JCM.41.12.5598-5603.2003
57. Kuo TC, Chao CC. Hepatitis B virus X protein prevents apoptosis of hepatocellular carcinoma cells by upregulating SATB1 and HURP expression. Biochem Pharmacol. 2010;80(7):1093–1102. PMID: 20541537. doi:10.1016/j.bcp.2010.06.003
58. Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 2011;300(2):162–172. PMID: 20971552. doi:10.1016/j.canlet.2010.09.018
59. Chan C, Wang Y, Chow PK, Chung AY, Ooi LL, Lee CG. Altered binding site selection of p53 transcription cassettes by hepatitis B virus X protein. Mol Cell Biol. 2013;33(3):485–497. PMID: 23149944 PMCID: PMC3554210. doi:10.1128/MCB.01189-12
60. Clippinger AJ, Bouchard MJ. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J Virol. 2008;82(14):6798–6811. PMID: 18448529 PMCID: PMC2446973. doi:10.1128/JVI.00154-08
61. Yang B, Bouchard MJ. The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J Virol. 2012;86(1):313–327. PMID: 22031934 PMCID: PMC3255901. doi:10.1128/JVI.06442-11
62. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Oxidative stress and cancer: an overview. Ageing Res Rev. 2013;12(1):376–390. PMID: 23123177. doi:10.1016/j.arr.2012.10.004
63. Jin Y, Wu D, Yang W, et al. Hepatitis B virus x protein induces epithelial-mesenchymal transition of hepatocellular carcinoma cells by regulating long non-coding RNA. Virol J. 2017;14(1):238. PMID: 29258558 PMCID: PMC5735895. doi:10.1186/s12985-017-0903-5
64. Wen Y, Golubkov VS, Strongin AY, Jiang W, Reed JC. Interaction of hepatitis B viral oncoprotein with cellular target HBXIP dysregulates centrosome dynamics and mitotic spindle formation. J Biol Chem. 2008;283(5):2793–2803. PMID: 18032378. doi:10.1074/jbc.M708419200
65. Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle. 2005;4(12):1722–1724. PMID: 16294043. doi:10.4161/cc.4.12.2260
66. Sartorius K, An P, Winkler C, et al. The epigenetic modulation of cancer and immune pathways in hepatitis B virus-associated hepatocellular carcinoma: the influence of HBx and miRNA dysregulation. Front Immunol. 2021;12:661204. PMID: 33995383 PMCID: PMC8117219. doi:10.3389/fimmu.2021.661204
67. Pollicino T, Cacciola I, Saffioti F, Raimondo G. Hepatitis B virus PreS/S gene variants: pathobiology and clinical implications. J Hepatol. 2014;61(2):408–417. PMID: 24801416. doi:10.1016/j.jhep.2014.04.041
68. Choi SH, Jeong SH, Hwang SB. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology. 2007;132(1):343–357. PMID: 17241884. doi:10.1053/j.gastro.2006.10.038
69. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(Pt25):5965–5973. PMID: 15564374. doi:10.1242/jcs.01589
70. Goto T, Kato N, Yoshida H, et al. Synergistic activation of the serum response element-dependent pathway by hepatitis B virus x protein and large-isoform hepatitis delta antigen. J Infect Dis. 2003;187(5):820–828. PMID: 12599056. doi:10.1086/368389
71. Williams V, Brichler S, Khan E, et al. Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress. J Viral Hepat. 2012;19(10):744–753. PMID: 22967106. doi:10.1111/j.1365-2893.2012.01597.x
72. Park CY, Oh SH, Kang SM, Lim YS, Hwang SB. Hepatitis delta virus large antigen sensitizes to TNF-alpha-induced NF-kappaB signaling. Mol Cells. 2009;28(1):49–55. PMID: 19711042. doi:10.1007/s10059-009-0100-5
73. Benegiamo G, Vinciguerra M, Guarnieri V, Niro GA, Andriulli A, Pazienza V. Hepatitis delta virus induces specific DNA methylation processes in Huh-7 liver cancer cells. FEBS Lett. 2013;587(9):1424–1428. PMID: 23523924. doi:10.1016/j.febslet.2013.03.021
74. Liao FT, Lee YJ, Ko JL, Tsai CC, Tseng CJ, Sheu GT. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J Gen Virol. 2009;90(Pt 5):1124–1134. PMID: 19264665. doi:10.1099/vir.0.007211-0
75. Trougakos IP, Gonos ES. Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic Res. 2006;40(12):1324–1334. PMID: 17090421. doi:10.1080/10715760600902310
76. Chen M, Du D, Zheng W, et al. Small hepatitis delta antigen selectively binds to target mRNA in hepatic cells: a potential mechanism by which hepatitis D virus downregulates glutathione S-transferase P1 and induces liver injury and hepatocarcinogenesis. Biochem Cell Biol. 2019;97(2):130–139. PMID: 30153423. doi:10.1139/bcb-2017-0321
77. Yu Z, Ma X, Zhang W, et al. Microarray data mining and preliminary bioinformatics analysis of hepatitis D virus-associated hepatocellular carcinoma. Biomed Res Int. 2021;2021:1093702. PMID: 33564675 PMCID: PMC7867452. doi:10.1155/2021/1093702
78. Abbas Z, Abbas M, Abbas S, Shazi L. Hepatitis D and hepatocellular carcinoma. World J Hepatol. 2015;7(5):777–786. PMID: 25914778 PMCID: PMC4404383. doi:10.4254/wjh.v7.i5.777
79. Jang TY, Wei YJ, Liu TW, et al. Role of hepatitis D virus infection in development of hepatocellular carcinoma among chronic hepatitis B patients treated with nucleotide/nucleoside analogues. Sci Rep. 2021;11(1):8184. PMID: 33854160 PMCID: PMC8047028. doi:10.1038/s41598-021-87679-w
80. Russo FP, Zanetto A, Pinto E, et al. Hepatocellular carcinoma in chronic viral hepatitis: where do we stand? Int J Mol Sci. 2022;23(1):500. PMID: 35008926 PMCID: PMC8745141. doi:10.3390/ijms23010500
81. Philips CA, Rajesh S, Nair DC, Ahamed R, Abduljaleel JK, Augustine P. Hepatocellular carcinoma in 2021: an exhaustive update. Cureus. 2021;13(11):e19274. PMID: 34754704 PMCID: PMC8569837. doi:10.7759/cureus.19274
82. Botelho-Souza LF, Vasconcelos MPA, Dos Santos AO, Salcedo JMV, Vieira DS. Hepatitis delta: virological and clinical aspects. Virol J. 2017;14(1):177. PMID: 28903779 PMCID: PMC5597996. doi:10.1186/s12985-017-0845-y
83. Papatheodoridis A, Papatheodoridis G. Hepatocellular carcinoma: the virus or the liver? Liver Int. 2022. PMID: 35319167. doi:10.1111/liv.15253
84. Abbas Z, Qureshi M, Hamid S, Jafri W. Hepatocellular carcinoma in hepatitis D: does it differ from hepatitis B monoinfection? Saudi J Gastroenterol. 2012;18(1):18–22. PMID: 22249087 PMCID: PMC3271688. doi:10.4103/1319-3767.91731
85. Stella L, Santopaolo F, Gasbarrini A, Pompili M, Ponziani FR. Viral hepatitis and hepatocellular carcinoma: from molecular pathways to the role of clinical surveillance and antiviral treatment. World J Gastroenterol. 2022;28(21):2251–2281. PMID: 35800182 PMCID: PMC9185215. doi:10.3748/wjg.v28.i21.2251
86. Lin C-L, Kao J-H. Review article: the prevention of hepatitis B-related hepatocellular carcinoma. Aliment Pharmacol Ther. 2018;1:1–10. doi:10.1111/apt.14683
87. Chen DS, Hsu NHM, Sung JL, et al. A mass vaccination program in Taiwan against hepatitis B virus infection in infants of hepatitis B surface antigen-carrier mothers. JAMA. 1987;257:2597–2603. doi:10.1001/jama.1987.03390190075023
88. Stevens CE, Toy P, Kamili S, et al. Eradicating hepatitis B virus: the critical role of preventing perinatal transmission. Biologicals. 2017;50:3–19. doi:10.1016/j.biologicals.2017.08.008
89. Wu CY, Lin JT, Ho HJ, et al. Association of nucleos(t)ide analogue therapy with reduced risk of hepatocellular carcinoma in patients with chronic hepatitis B: a nationwide cohort study. Gastroenterology. 2014;147:143–151. doi:10.1053/j.gastro.2014.03.048
90. Ferenci P, Reiberger T, Jachs M. Treatment of chronic hepatitis D with bulevirtide-A fight against two foes-an update. Cells. 2022;11(22):3531. PMID: 36428959 PMCID: PMC9688382. doi:10.3390/cells11223531
91. Wu JC, Huang YH, Chau GY, et al. Risk factors for early and late recurrence in hepatitis B-related hepatocellular carcinoma. J Hepatol. 2009;51:890–897. doi:10.1016/j.jhep.2009.07.009
92. Sasaki K, Shindoh J, Nishioka Y, et al. Impact of viral etiology on postoperative de novo recurrence after hepatectomy for hepatocellular carcinoma in cirrhotic patients. J Gastrointest Surg. 2017;21:487–495. doi:10.1007/s11605-016-3344-3
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.