Back to Journals » OncoTargets and Therapy » Volume 16
MNK Proteins as Therapeutic Targets in Leukemia
Authors Mazewski C, Platanias LC
Received 13 January 2023
Accepted for publication 7 April 2023
Published 21 April 2023 Volume 2023:16 Pages 283—295
DOI https://doi.org/10.2147/OTT.S370874
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Arseniy Yuzhalin
Candice Mazewski,1,2 Leonidas C Platanias1– 3
1Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL, USA; 2Division of Hematology–Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA; 3Department of Medicine, Jesse Brown Veterans Affairs Medical Center, Chicago, IL, USA
Correspondence: Candice Mazewski; Leonidas C Platanias, Email [email protected]; [email protected]
Abstract: In leukemia, resistance to therapy is a major concern for survival. MAPK-interacting kinases (MNKs) have been identified as important activators of oncogenic-related signaling and may be mediators of resistance. Recent studies in leukemia models, especially acute myeloid leukemia (AML), have focused on targeting MNKs together with other inhibitors or treating chemotherapy-resistant cells with MNK inhibitors. The preclinical demonstrations of the efficacy of MNK inhibitors in these combination formats would suggest a promising potential for use in clinical trials. Optimizing MNK inhibitors and testing in leukemia models is actively being pursued and may have important implications for the future. These studies are furthering the understanding of the mechanisms of MNKs in cancer which could translate to clinical studies.
Keywords: MNK, resistance, MAPK, eIF4E, AML
Plain Language Summary
Identification and vetting of new targets in cancer medicine is essential for the development of treatments to improve survival of patients. MNKs are kinases involved in cancer promoting and cancer cell survival signaling. Preclinical evidence, especially using MNK inhibitors in combination with chemotherapy or other targeted therapy, shows promise for future clinical translational studies.
Introduction
With nearly three decades of research, the knowledge base on mitogen-activated protein kinase (MAPK) interacting kinases or MNKs and related translational efforts toward the development of MNK inhibitors has come a long way. Following the discovery of the eukaryotic translation initiation factor 4E (eIF4E) and its importance in protein translation in the 1980s, researchers identified its activation through the MAPK pathway, specifically by phosphorylation of serine 209 by MNKs.1–3 Subsequently in the early 2000s, a seminal study by Ueda et al demonstrated that both MNK1 and MNK2 were dispensable for normal cell growth, while this group and others later showed the significance of MNKs in tumorigenesis.4,5 These discoveries suggested that MNKs were potential viable targets in cancer therapy.
Realistically, a single-drug approach is not plausible for most cancer treatments and resistance is a common problem, so drug combinations are utilized. Combinations of mammalian target of rapamycin (mTOR) pathway inhibitors with MNK inhibitors have been previously identified as a potential therapeutic strategy in leukemia.6 The last few years of research surrounding MNK inhibitors had a plethora of studies on dual MNK inhibitors, such as MNK and fms-like tyrosine kinase 3 (FLT3) or MNK and proviral integration site for Moloney murine leukemia virus (PIM) kinase targeting.7,8 Alternatively, more specific MNK inhibitors, such as tomivosertib, are also being explored and have a potential place in combination treatments in efforts to overcome resistance.9 Many groups are still working to develop potent and selective MNK inhibitors with desirable pharmacokinetic properties, while others are focusing on understanding MNK mechanistic elements. There are only three clinical trials that have been initiated utilizing MNK inhibitors in hematological malignancies, but optimized inhibitors and preclinical evidence of combinatorial benefits of MNK inhibitors could promote further clinical research.10–12 In this review, we explore the latest updates in pharmacological MNK inhibitor research and how this may apply to clinical studies moving forward.
Acute and Chronic Leukemias
Leukemias are categorized as acute or chronic and based on the underlying white blood cell type, myeloid or lymphocytic. In general, acute leukemias have a worse prognosis and are more difficult to treat than chronic as evidenced by five-year survival rates for adults.13 The World Health Organization (WHO) 5th edition Haematolymphoid Tumours classification report was just released in 2022 with an aim to clarify myeloid neoplasm diagnosis parameters and categorization criteria to aid physicians in providing more appropriate treatments. Updates in the report for acute myeloid leukemia (AML) incorporate the inclusion of additional mutations, fusions, and rearrangements for defining genetic abnormalities, such as KMT2A, MECOM, and NUP98 rearrangements; additionally, the report explained an AML family restructure to include two categories of defining by genetic abnormalities and defining by differentiation, which allows for diagnosis with genetic abnormalities with less than 20% blasts.14
The standard of care differs amongst the leukemia subtypes. In general, the age and ability of the patient to tolerate intensive treatment stratify the approaches of therapy. Chronic lymphocytic leukemia (CLL), as the least aggressive, if not progressing, does not require treatment immediately and a watch-and-wait approach is used in most cases based on the 2018 guidelines.15,16 However, with a better understanding of the genomic landscape and the discovery of newer targeted therapies with fewer side effects, such as Bruton tyrosine kinase (BTK) inhibitors, there is an increasing number of clinical trials on early intervention strategies for CLL.17 For acute lymphocytic leukemia (ALL), typical treatment starts with chemotherapy regimens of vincristine, dexamethasone, and an anthracycline like doxorubicin, with the possible inclusion of cyclophosphamide and pegaspargase and alternating high-dose methotrexate.18 If it is a Philadelphia (Ph) chromosome positive case, a tyrosine kinase inhibitor may be included, and in T-ALL, nelarabine is sometimes added. In chronic myeloid leukemia (CML), a tyrosine kinase inhibitor such as imatinib or now, more commonly, the second-generation drugs dasatinib, nilotinib, or bosutinib, also BCR::ABL1 inhibitors, are used as first-line therapy in chronic phase CML since the Ph chromosome formation is a hallmark of CML.19 These tyrosine kinase inhibitors have generally been effective for treatment and help prevent progression to the blast phase, but resistance to these inhibitors is possible. For AML, which has the worst prognosis, the traditional treatment for favorable-risk patients is the 7+3 regimen, which consists of two chemotherapy drugs, cytarabine and an anthracycline (daunorubicin, idarubicin), with possible added gemtuzumab ozogamicin.20 However, since this is an intensive treatment regimen, elderly patients or patients with comorbidities may not be able to tolerate it and alternative regimens, such as a combination of a hypomethylating agent with venetoclax, a B-cell lymphoma 2 (BCL2) inhibitor, are often used. Especially with improved profiling techniques, molecular subtypes are even further stratifying treatments. With some of the latest drug developments, specific approvals are sought for certain mutations or patient conditions in AML. For example, olutasidenib, which targets mutant IDH1, showed clinical activity in a phase I/II trial with and without azacytidine and was FDA-approved in December 2022 for relapsed or refractory AML, specifically in patients with an IDH1 mutation.21,22 Similarly, midostaurin, a FLT3 inhibitor, was approved in 2017 for newly diagnosed AML patients with a FLT3 mutation.23 These targeted therapies can provide added benefits to patients that fit the specific criteria. Targeted therapy research, identifying meaningful targets and pursuing the translatability of inhibitors, is prevalent. FDA approvals for targeted drugs in the past five years have included, most recently, olutasidenib (2022), glasdegib targeting the Hedgehog pathway (2018), venetoclax (2018), ivosedinib targeting IDH1 (2018), and midostaurin (2017).24 However, as noted by Estey et al, more approvals are not always better, and the fitness of patients and trial randomization are amongst aspects that should be more rigorously factored in when determining approval and optimal treatment.24 Progress in understanding the genomic landscape, resistance tactics, and beneficial combinational targeting is helping push the field toward more effective leukemia treatments.
Chemistry of MAPK Interacting Kinases (MNKs)
MNK Pathway
MAPK pathways have been of significant interest in cancer research due to the diversity of functional responses mediated by the signaling of this multi-kinase cascade. Further downstream, MNKs are a particularly relevant target in malignancies, due to their vitality in cancer cell signaling but not in normal cell growth and development.4 MNKs are serine/threonine kinases.25 MNK1 and MNK2 both have α and β isoforms, resulting from alternative splicing where only the α isoforms have a MAPK binding domain and a nuclear export sequence.26 Generally, MNK2 has higher basal activity, while MNK1 is more inducible by MAPK signaling.27 Both can be phosphorylated by the MAPKs ERK and p38, but not JNK; MNK2 has preferential binding for ERK while MNK1 has comparable binding to ERK and p38.1 It should be noted that there are negative regulators of MNKs. It has been previously shown that the protein phosphatase 2A dephosphorylates and deactivates MNK1.28 Also, p21 activated kinase 2 (PAK2) phosphorylation was found to disturb the binding of MNK1 to eukaryotic initiation factor 4G (eIF4G) which is the scaffold protein of the eIF4F complex (eIF4G, eIF4E, eIF4A).29 Meanwhile, active MNKs were discovered to bind with mTORC1 and allow for the binding of TELO2 to the complex which instigates mTORC1 downstream substrate phosphorylation.30 To further add to the mTORC1 relation to MNKs, a recent study demonstrated phosphorylation of MNK2 at Ser74 by mTORC1.31 The group showed that this phosphorylation blocked MNK2 binding to eIF4G, thus inhibiting eIF4E phosphorylation by MNK2. This mTORC1-mediated Ser74 phosphorylation is a newly discovered mechanism linking the mTOR and MAPK pathways.
The mRNA 5’ cap-binding protein, eIF4E, the limiting factor of the eIF4F complex, is the most well-known target of MNKs and its phosphorylation at serine 209 is used as a readout for MNK activity. Higher phosphorylation of eIF4E correlates with worse prognosis in many cancers and appears to be involved in many oncogenic processes, thereby provoking research targeting the MNK/eIF4E axis.32 A recent study, specifically in AML, found higher nuclear p-eIF4E in primary patient samples was associated with higher tumor burden and worse clinical outcomes.33 Other downstream phosphorylation targets of both MNKs have been identified including Sprouty2, a negative feedback regulator of receptor tyrosine kinases that is phosphorylated at Ser 112 and 121; and the polypyrimidine tract-binding protein-associated splicing factor (PSF), a DNA/RNA binding protein involved in inflammatory cytokine signaling, that is phosphorylated at Ser 8 and 283.34–37 MNK1 specific substrates are heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), an AU-rich element binding protein involved in post-transcriptional regulation, phosphorylated at Ser 192 and Ser 310/311/312, and cytosolic phospholipase A2 (cPLA2), which regulates release of arachidonic acid from glycerophospholipids, phosphorylated at Ser 727 by MNK1.38,39 MNK2 has been shown to phosphorylate the cytoskeleton related protein plectin at Ser 4642.40 Additionally, a recent study demonstrated the translation function of MNK1 specifically in platelets and megakaryocytes where cPLA2 activity was regulated by MNK1, potentially via phosphorylation at serine 505.41 An overview of upstream effectors and downstream targets of MNKs is shown in Figure 1.
 |
Figure 1 Schematic of MNK signaling with the main upstream and downstream proteins and phosphorylation sites identified in the literature. Figure created with BioRender. Abbreviations: cPLA2, cytosolic phospholipase A2; eIF4E, eukaryotic translation initiation factor 4E; ERK, extracellular signal-regulated kinase; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; MNK, mitogen-activated protein kinase interacting protein kinase; mTORC1, mammalian target of rapamycin complex 1; PP2A, protein phosphatase 2A; PSF, polypyrimidine tract-binding protein-associated splicing factor; Spry2, sprouty2. |
MNK Structure and Unique Qualities
MNKs are part of the Ca2+/calmodulin-dependent kinase group based on structure elements despite not being regulated by either. MNKs have unique structural features that include three short alpha-helices in the catalytic domain, DFD (Asp-Phe-Asp) motifs in the activation loops instead of the usual DFG (Asp-Phe-Gly) motif for other kinases, and an inactive DFD-out conformation with Phe192 in the ATP-binding site which blocks ATP from the catalytic site.42 Asp228 of the DFD motif stabilizes the DFD-out conformation and is unique to MNKs.43 These structural features can be exploited in inhibitor design, especially in targeting the inactive MNK forms. MNK1 and MNK2 have many similarities; the catalytic domains share about 70% of the amino acid framework.44 Kinases most comparable in catalytic domain structure would be ribosomal s6 kinases (RSKs) which are also phosphorylated by ERK, but not p38.44 The unique qualities have led to some hurdles in drug design but that also benefit the development of more specific MNK inhibitors.
MNK Inhibitors
Commercially Available Synthetic Compounds
There are many MNK inhibitors, developed based on different design approaches, with a range of specificity that have been used in preclinical evaluations. Some MNK inhibitors are already commercially available for laboratory work and are highlighted in Table 1, most of which are type I inhibitors of MNKs. Tomivosertib (eFT508) from eFFECTOR therapeutics is perhaps the most commonly used in recent studies due to its high specificity for MNK1 and MNK2.45 The investigators optimized 4-aminopyrimidines leading to eFT508 which had inhibition of MNK1 and MNK2 and p-eIF4E reduction in the low nanomolar range, high selectivity with only DRAK1 and CLK4 in the off-target kinase profile, and efficacy at reducing tumors in three xenograft mouse models. Optimization of bicyclic fragments with an imidazopyridine derivative exhibiting the greatest MNK inhibition and best pharmacokinetic properties is how ETC-206, now AUM001, was synthesized.46 ETC-206 showed in vitro potency against CML lines and further reduced tumor size in combination with dasatinib in a model of blast crisis CML in mice. Another MNK inhibitor with low nanomolar range inhibition of both MNK1 and MNK2 is SEL201, developed by Selvita and reported in 2017.47 The group discovered this amino-1H-indazol-1,2-dihydropyridin-2-one derivative that they synthesized starting with 5-bromopyridin-2-ol to be an ATP-competitive MNK1/2 inhibitor. Bayer identified an amino-substituted imidazopyridazine by way of high-throughput screening and further optimized this base to design BAY 1142369 which has inhibition of MNK1 and MNK2, with greater activity against MNK1.48 PIM1 was amongst only four other kinases that BAY 1142369 had some inhibitory activity against in the selectivity screening. Another MNK inhibitor, CGP57380, is a pyrazolo-pyrimidine that has shown specificity for MNK1, but the IC50 of MNK1 and p-eIF4E reduction in cells is in the micromolar range.49 Earlier discovered MNK inhibitors were non-specific multi-kinase inhibitors, such as merestinib, which was designed as a type II MET inhibitor but had activity against eleven other kinases that included both MNKs.50 Merestinib mostly targets tyrosine kinases, with MNKs being the only serine/threonine kinases, which they proposed was because of their DFD-out conformation in the inactive form.
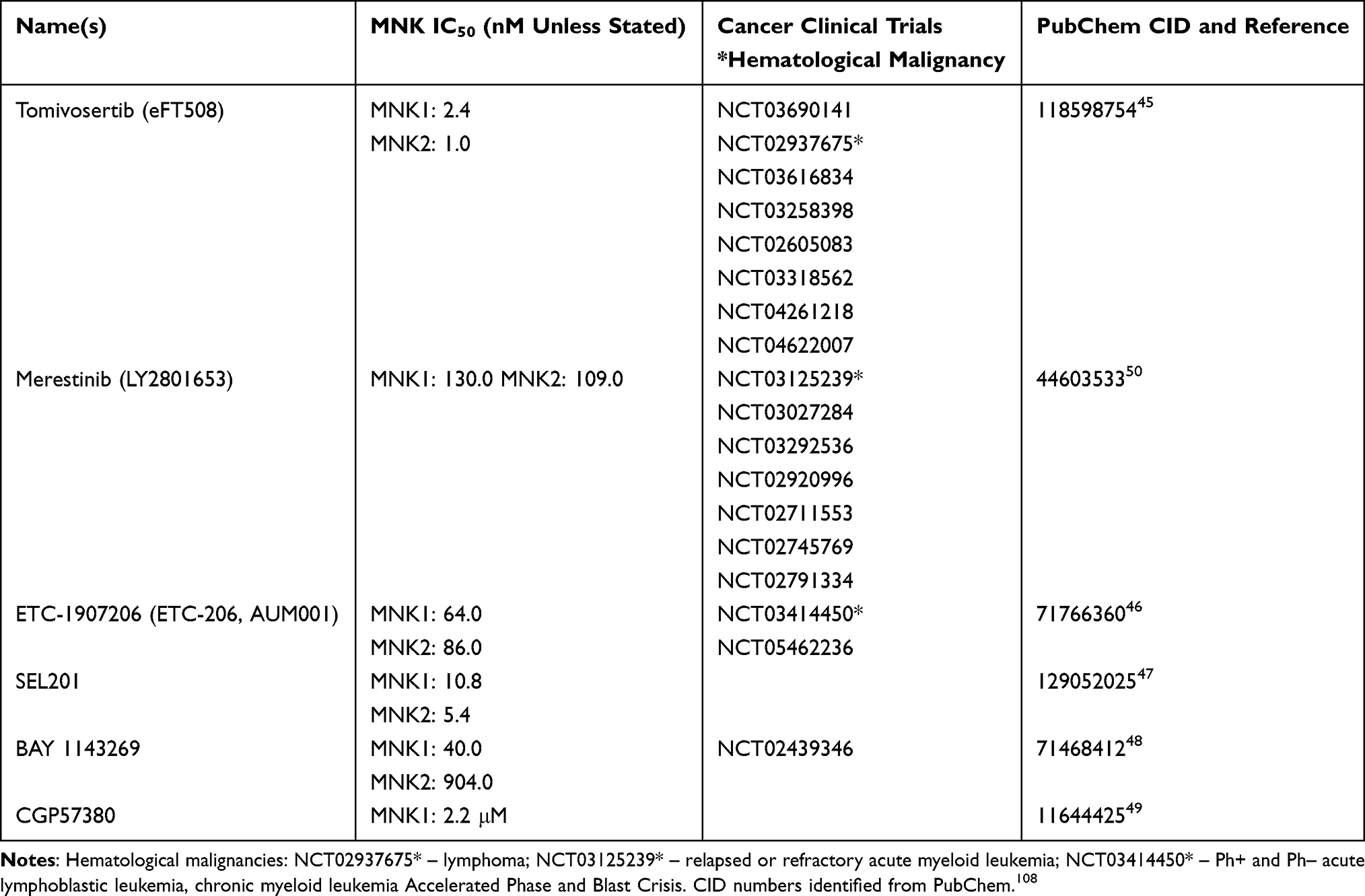 |
Table 1 Commercially Available Synthetically Derived MNK Inhibitors |
Experimental Design of Synthetic Compounds
Despite the existence of commercially available MNK inhibitors, many groups have still been on the hunt for more potent and selective inhibitors with the development of new inhibitors over the past five years. One study screened a fragment library and optimized compounds with a pyridine-(hetero)arylamide core using structure-guided expansion to create selective MNK1/2 inhibitors.51 In general, they found the compounds had greater activity against MNK2 than MNK1 with optimized compound 43 having IC50 values of 89 nM and 200 nM, respectively, and a favorable pharmacokinetic profile. Fragment based design was utilized by another group that designed MNK inhibitors by starting with a benzofuran scaffold and modified it to create 6-hydroxy-4-methoxy-3-methylbenzofuran-7-carboxamide derivatives with top compound 8k having a MNK2 IC50 of 0.27 μM and 41% inhibition of MNK1 at 1 μM.52 Our group at Northwestern utilized an in silico mutation-based induced-fit docking method and the MNK1 wild-type crystal structure to develop a series of MNK inhibitors that showed activity in the micromolar range against both MNK1 and MNK2 in vitro and reduction of AML cell progenitor growth.42 Another lab designed MNKi-8e, a 5-(2-(phenylamino)pyrimidin-4-yl)thiazole-2(3H)-one derivative, with potent MNK2 inhibition (IC50 = 0.37 μM) and antiapoptotic effects in AML cells.53 Jin et al developed 4-((4-fluoro-2-isopropoxyphenyl)amino)-5-methylthieno[2,3-d] pyrimidine derivatives that have MNK inhibitory activity with inhibition of p-eIF4E in cells at 0.1 μM and their compound MNK-7g having the most promise.54 These investigators had previously designed a MNK inhibitor, MNK-I1, and were using it as a comparison.
One group has gone through various stages of developing MNK inhibitors over the past ten years. They developed imidazopyridine and imidazopyrazine derivative type I inhibitors using molecular docking to show stable interactions with MNK1 and MNK2 and determined that the importance of MNK inhibitor efficacy was related to the hydrogen bond interaction of the inhibitors and the Lys and Ser residues in the MNK catalytic site.55 Subsequently, they identified type II inhibitors that showed promise through in silico docking simulations with MNK1 and MNK2 IC50 values below 100 nm and generally more potency towards MNK2.56 A majority of MNK inhibitors are type I inhibitors, meaning they are ATP-competitive with the active kinase conformation.57 A recent study took an alternative approach in designing a non-ATP-competitive MNK inhibitor. Bou-Petit et al designed EB1 which has a 4,6-diaryl-1H-pyrazolo[3,4-b]pyridin-3-amine core structure and binds to the inactive MNK1 with an IC50 of 0.69 μM (MNK2 IC50 = 9.4 μM) preventing transition to its active state and demonstrated reduction of phosphorylation of Ser209 on eIF4E in various cell lines including MV411 AML cells.58 Interestingly, the authors demonstrated that EB1 did not induce upstream activation and phosphorylation of MNK1 leading to enhanced binding to eIF4G as did the type I inhibitors tested in comparison. These results highlight the potential benefits of a type II inhibitor in that it would not lead to paradoxical target protein activation induced by some type I kinase inhibitors which occurs even with catalytic kinase activity inhibition.59 Another group designed non-ATP-competitive inhibitors that were 2′H-spiro[cyclohexane-1,3′-imidazo[1,5-a]pyridine]-1′,5′-dione derivatives, using tomivosertib as a comparison; their top prospects had nanomolar inhibition of MNK1 and MNK2 and showed antiproliferative activity in a variety of cancer cell lines.60 Abdelaziz et al also designed a MNK2 inhibitor, an N-phenyl-4-(1H-pyrrol-3-yl)pyrimidin-2-amine derivative, that showed antiproliferative and proapoptotic effects in AML cells.61 Another study designed a MNK1 inhibitor that stabilized the DFD-out conformation of the inactive protein, blocking its activation with a MNK1 IC50 of 21 nM. The reported crystal structure in the Protein Data Bank (5WVD) of the complex of the MNK1 kinase region and the inhibitor could be helpful for future MNK drug design.62 One group analyzed synthesized ferrocene-containing compounds from a known MNK inhibitor and while the added ferrocene made the compounds comparatively more potent at malignant cell inhibition, it negated their MNK1/2 blocking ability.63 Recent work by Halder and Cordeiro led to a multi-targeted in silico screening approach for MNK inhibitors.64 Exploratory model development such as this one could help in better identifying compounds with MNK inhibitory potential.
Natural MNK Inhibitors
Not all MNK inhibitors are synthetic in origin; some natural compounds have been discovered to have MNK targeting abilities and some have even been used in cancer clinical trials (Table 2). In general, it is not new to utilize natural compounds in cancer drug discovery, with previous reports indicating around 60% of new drugs having been of natural origin.65 A recent review highlighted over 20 different natural compounds, mostly in preclinical studies, with various antineoplastic effects in leukemia noting the need for therapeutic strategies with less side effects as reasoning for continued analysis of natural sources.66 Most abundantly are studies on various flavonoids which have high contents in fruits, vegetables, herbs, and other plant foods.67–69 One study analyzed flavones and flavonols to find kinase inhibitors with AML and ALL cell inhibitory activity and discovered an O-methylated flavonol, a precursor of fisetin, that inhibited MNK2 as well as FLT3, RSK, DYRK2, and JAK2.70 This compound had potent inhibitory effects on AML and ALL cell viability, and induced apoptosis and G0/G1 arrest of AML cells. Myricetin has been shown to inhibit CML cell viability.71 Additionally, a group found that myricetin targeted imatinib-resistant blast phase CML cells by inhibiting activation of eIF4E and demonstrated tumor reduction in a CML-resistant mouse model.72 Chen et al analyzed apigenin, hispidulin, and luteolin, also flavonoids, that all showed nanomolar level inhibition of MNK2, and luteolin also had MNK1 inhibition.73 Additionally, they showed luteolin and hispidulin reduced AML cell growth, increased markers of apoptosis (cleaved caspase 3 and PARP), and arrested cells in sub G1.
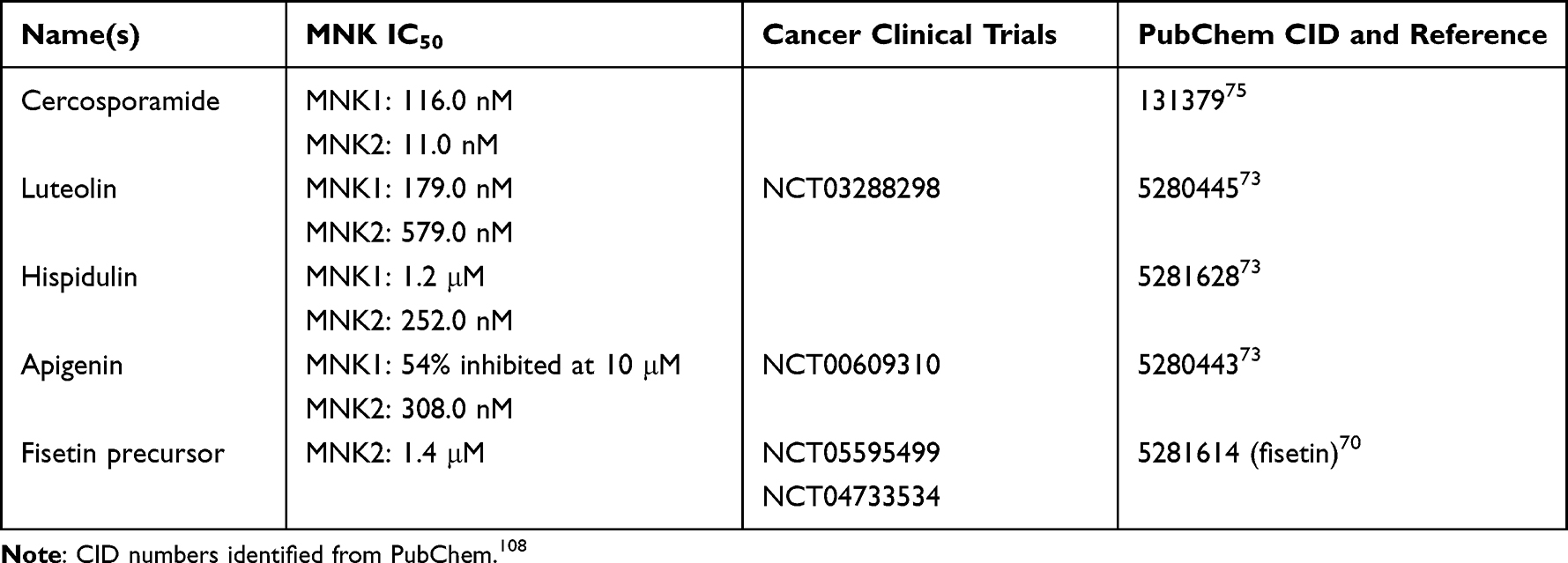 |
Table 2 Commercially Available MNK Inhibitors Found in Natural Sources |
Cercosporamide is an antifungal isolated from Cercosporidium henningsii of cassava plants.74 In 2011, it was discovered that it has potent MNK inhibitory activity and has been used in subsequent cancer studies.75 Usnic acid found in lichens has a similar structure to cercosporamide. A study analyzed usnic acid derivatives with a flavanone moiety in leukemia cells and found antiproliferative and antiapoptotic effects while seeing a reduction in phosphorylated eIF4E, but it appeared to be MNK pathway related, not MNK protein specific.76
MNK Inhibitors in Preclinical Leukemia Studies
There have been many studies on the activity of MNK inhibitors in AML models. Our group examined the antileukemic properties of four different commercially available MNK inhibitors, starting with cercosporamide in 2013 which was potent against AML cell lines (U937, MM6) and patient primitive leukemic progenitors while also reducing tumor growth in an MV411 xenograft mouse model.77 In subsequent studies, the lab investigated merestinib, demonstrating inhibition of AML cell progenitor colony formation and additionally saw efficacy in an MM6 mouse model.78 Later, SEL201 was studied, which exhibited antiproliferative and antiapoptotic effects in AML cells.79 Most recently, our group tested tomivosertib in the same established AML cell lines and also saw a reduction in cell viability and colony formation with the highest potency against MV411, MM6 cells, and KG-1 cells.9 Through these four studies, we have found that MNK inhibitors ranging from unselective multi-kinase to very MNK specific all have antineoplastic effects in AML models.
Although not as heavily studied as in AML, MNK inhibitors have been analyzed in CML models. ETC-206 has been identified as a potential MNK inhibitor for use in blast crisis CML with micromolar IC50 values against 25 hematological cell lines including five BCR::ABL1 expressing lines (K562, KCL-22, EM-2, BV-173, JURL-MK1) and enhancement of dasatinib tumor reduction in a blast crisis CML xenograft model.46 Another study analyzed blast crisis CML and leukemia stem cells which they identified to have self-renewal capabilities through β-catenin activation, and MNK inhibitors blocked that activation through eIF4E phosphorylation inhibition in vitro and in vivo.80 Resistance of blast crisis CML to tyrosine kinase inhibitors that are typically used for treatment is a major concern so demonstrating a way to target resistant populations is essential.
Preclinical Combination Studies Using MNK Inhibitors
Overcoming Therapeutic Resistance
Recent studies have shown that MNK targeting may be of importance in overcoming resistance in solid tumors both in targeted therapy related instances such as with anlotinib in lung cancer, as well as in chemotherapy related resistance such as with temozolomide in glioblastoma.81,82 Additionally, in gastric cancer, combining tomivosertib with 5-FU or paclitaxel showed benefits in vitro and in vivo, indicating a potential for this MNK inhibitor to sensitize gastric cancer cells to chemotherapy drugs.83 Similarly, in cervical cancer, combination of cercosporamide with chemotherapy drugs, doxorubicin and cisplatin, had increased efficacy in proliferation reduction and apoptosis induction; cercosporamide inhibited chemo-resistant cells and phosphorylation of eIF4E at serine 209 was shown to be induced with chemotherapy treatment.84 In AML, MNK inhibitors have similarly been shown to sensitize cells to the chemotherapy drug cytarabine. We previously reported the activation of the MNK pathway by cytarabine in AML cells and that the combination of the MNK inhibitor, CGP57380, or MNK knockdown with cytarabine enhances the inhibition of leukemic progenitor colony formation.85 Later studies also demonstrated that cytarabine treatment increases MNK activity in AML cells and that there are synergistic effects in AML cells with cytarabine and MNK inhibition (MNKI-8e) or MNK1 and MNK2 knockdown.86 Another group demonstrated that miR-134 was decreased in drug-resistant AML cells (K562/A02, HL-60/ADM) and that MNKs were a target of miR-134; when miR-134 was overexpressed, thereby blocking MNKs, these cells, as well as relapsed AML patient cells, were sensitized to cytarabine.87 Comparatively in CML studies, CML imatinib-resistant cells showed higher sensitivity to myricetin, a plant flavonoid, that reduced phosphorylation of eIF4E at serine 209.72 Similarly, a study showed synergy of CGP57380 with imatinib in inducing apoptosis in Ba/F3 and K562 CML cells.88 Dasatinib, another tyrosine kinase inhibitor used in CML, had synergistic antiproliferative effects in CML cells when combined with niclosamide, which targets the ERK/MNK/eIF4E axis, further demonstrating the importance of the MNK pathway in therapy resistance.89
Combinations with MNK Inhibitors
One of the most common approaches in combinations with MNK inhibitors has been co-targeting with mTOR inhibitors due to the reciprocity of these two pro-survival pathways.6 This has been shown to be a potentially promising approach for a variety of cancers, including in a recent study that demonstrated extended survival using rapamycin, an mTORC1 inhibitor, in combination with tomivosertib or MNK1/2 knockout in an APC KRAS mutated colorectal cancer model.90 In hematological malignancies, a group looking at hematopoietic stem cells found that mTOR deletion lead to increased proliferation and protein synthesis through adaptive signaling by way of MNKs, which may explain the resistance of leukemia cells to mTOR inhibitors and provides reasoning for combination with MNK inhibition; they found mTOR resistant leukemia cells were more sensitive to CGP57380.91 In another study, everolimus, a derivative of rapamycin and also an mTORC1 inhibitor, exhibited synergistic inhibitory effects with CGP57380 in T-ALL cell lines.92 Other work in myeloid leukemia cells showed that phosphorylation of the translational repressor 4EBP1 increases in MNK inhibitor treated cells and there is an enhancement of antiproliferative effects when the MNK1 and MNK2 inhibitors are combined with rapamycin.93 Additionally, a study from our lab showed that SEL201 enhanced the inhibitory effects of rapamycin on AML cells.79 Notably, in the same study it was demonstrated that SEL201 also promoted the effects of 5-azacytidine, a hypomethylating agent approved for use in AML. Cercosporamide was found to enhance rapamycin antileukemic effects in AML cells, while additionally showing synergistic effects with cytarabine both in vitro and in vivo.77 Tomivosertib showed synergistic effects in viability and colony formation reduction with venetoclax, a BCL2 inhibitor approved in CLL and AML in combination with hypomethylating agents or low-dose chemotherapy.9 Combination targets as well as dual MNK inhibitors are highlighted in Table 3.
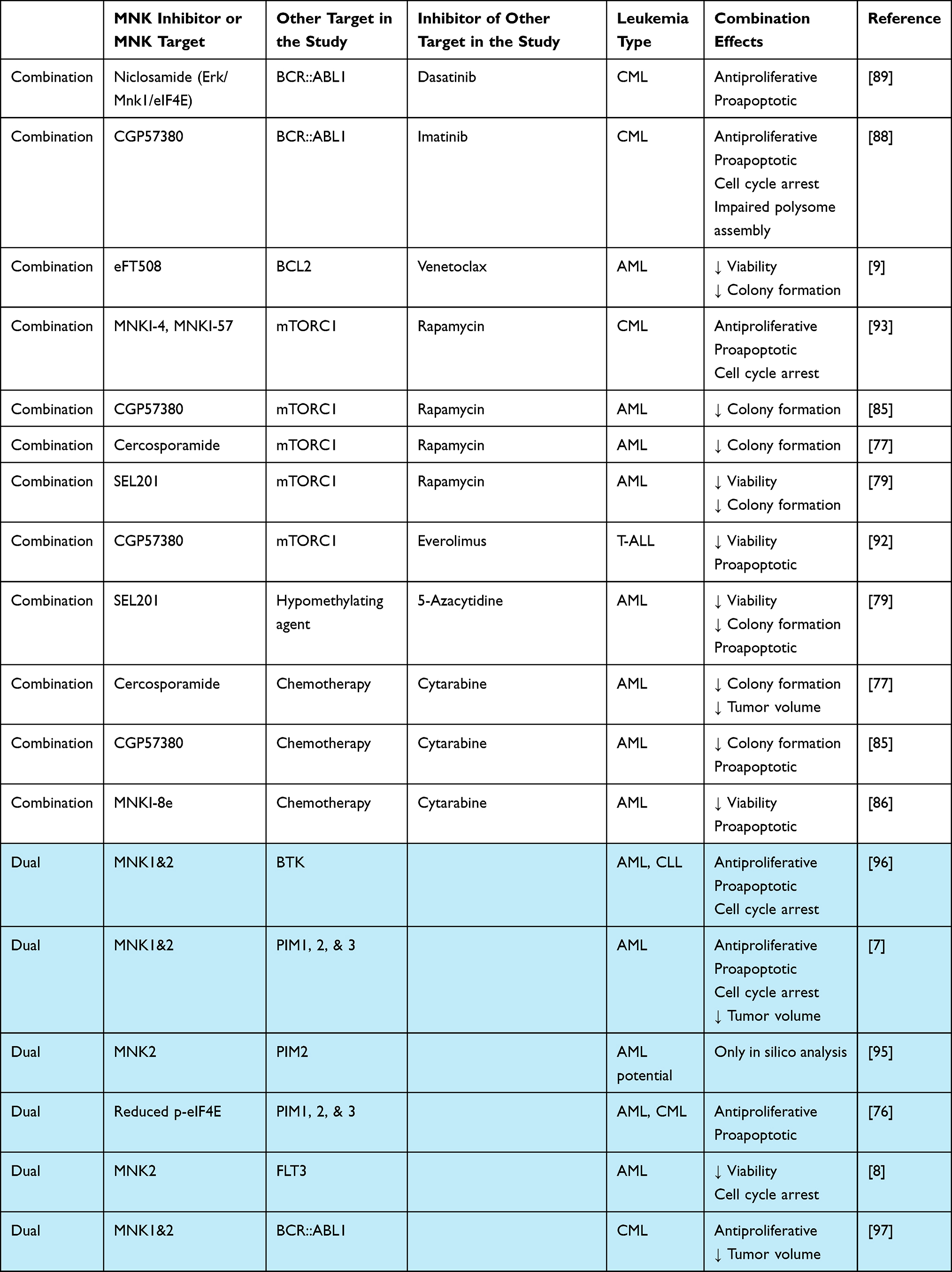 |
Table 3 Combination and Dual Targeting with MNK Inhibitors in Preclinical Studies |
Dual MNK and Secondary Kinase Inhibitors
Typically, in designing inhibitors, it is ideal to demonstrate specificity to one target as it shows minimization of potential off-target effects and mechanistically, higher confidence that an effect is due to the intended inhibited protein. However, in the recent literature surrounding MNKs, several studies are using purposefully designed dual inhibitors. The most common dual inhibition with MNK was with PIMs as the secondary kinase target. PIMs are pro-survival short-lived oncoproteins and are dependent on cap-dependent translation through the eIF4F complex.94 Han et al designed a 4,6-disubstituted pyrido[3,2-d]pyrimidine that had kinase inhibition in the nanomolar range for both MNKs and PIM1–3.7 One study used extensive in silico analysis to identify three natural compounds from the ZINC database that had high affinity and stable interaction to MNK2 and PIM2 which they suggested as potential therapeutics in AML.95 Usnic acid derivatives were demonstrated to reduce p-eIF4E as well as to target pan PIMs in K562 and HL-60 leukemia cells.76 Although it was not found that MNK was directly inhibited, the reduction to MNK/eIF4E signaling in combination with the PIM inhibition by these compounds further shows the efficacy in targeting these two axes. Virtual screening and docking analysis techniques identified a compound, K783-0308, with high potency and selectivity against MNK2 and FLT3 that was able to inhibit AML cell viability and promote cell cycle arrest.8 Additionally, a BTK/MNK inhibitor, QL-X-138, reduced proliferation in CLL and AML established lines and primary patient cells, noting however the need to improve the pharmacokinetics of the drug for in vivo studies.96 In a study using CML-derived cells, the investigators designed a MNK1/2 and BCR::ABL1 inhibitor with the idea that it would target leukemia stem cells with the MNK inhibition and promote cell death by blocking BCR::ABL1, and they demonstrated both in vitro viability and in vivo tumor reduction efficacy.97 Another group designed 4,6-disubstituted pyrido[3,2-d]pyrimidine derivatives that target both MNK and histone deacetylase (HDAC) which inhibited prostate cancer cell growth and could be tested in leukemia cells since HDAC inhibitors are also being explored individually in leukemia.98,99 The effectiveness of these dual inhibitors could be related to the links of MNKs to overcoming resistance as mentioned in the previous section.
Clinical Studies Using MNK Inhibitors
There are a few clinical trials for use of MNK inhibitors in hematological malignancies. Tomivosertib, ETC-1907206, and merestinib are the three MNK inhibitors that were in US clinical trials. Tomivosertib can be found in eight clinical trials with various cancers, with many being in combination studies in advanced cancer state or in combination with immunotherapy.100–105 A multicenter hematological malignancy (lymphoma) phase 1 and 2 trial was terminated (NCT02937675), but to the best of our knowledge, no further information has been published at this time.10 ETC-1907206 (ETC-206), now AUM001, was in a phase 1a/1b trial for Ph+ and Ph– ALL and Accelerated Phase and Blast Crisis CML in combination with chemotherapy drug dasatinib but was withdrawn (NCT03414450).11 For merestinib, the relapsed or refractory AML phase 1 clinical trial in combination with LY2874455, an FGFR inhibitor, was completed in 2020 with 16 patients (NCT03125239).12 They found merestinib was tolerable and one patient achieved complete remission on the merestinib monotherapy portion of the study.106 Merestinib is a multi-kinase inhibitor and confirmation of its biological activity was based on MET inhibition, so it is unclear as to specific effects of the MNK inhibition.
Conclusions
The current landscape of MNK inhibitor studies is mostly still in the preclinical stage. Only tomivosertib, eFT508, from eFFECTOR Therapeutics has an FDA orphan designation for diffuse large B-cell lymphoma.107 However, there is some promise for the potential future use of MNK inhibitors in combination studies with other agents. Many of the preclinical studies were done in AML models which alludes to a potential for AML clinical trials in the near future. The breadth of mechanistic studies implicates the MNK signaling as a targetable pathway due to its activation in resistant states. This raises the potential of unique clinical-translational approaches targeting MNKs to overcome resistance to chemotherapy and other antileukemia agents.
Acknowledgments
The research of Dr Platanias is supported by National Institutes of Health grants CA121192, CA77816, NS113425, NS113152 and by grant CX000916 from the Department of Veterans Affairs. Candice Mazewski was supported by NIH/NCI training grant T32 CA070085.
Disclosure
Dr Leonidas C Platanias reports issued patents 10,093,668 and 10,851,082.The authors report no conflicts of interest in this work.
References
1. Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16(8):1909–1920. doi:10.1093/emboj/16.8.1909
2. Bonneau AM, Sonenberg N. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J Biol Chem. 1987;262(23):11134–11139. doi:10.1016/S0021-9258(18)60935-4
3. Morley SJ, McKendrick L. Involvement of stress-activated protein kinase and p38/RK mitogen-activated protein kinase signaling pathways in the enhanced phosphorylation of initiation factor 4E in NIH 3T3 cells. J Biol Chem. 1997;272(28):17887–17893. doi:10.1074/jbc.272.28.17887
4. Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24(15):6539–6549. doi:10.1128/MCB.24.15.6539-6549.2004
5. Ueda T, Sasaki M, Elia AJ, et al. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A. 2010;107(32):13984–13990. doi:10.1073/pnas.1008136107
6. Kosciuczuk EM, Saleiro D, Platanias LC. Dual targeting of eIF4E by blocking MNK and mTOR pathways in leukemia. Cytokine. 2017;89:116–121. doi:10.1016/j.cyto.2016.01.024
7. Han Y, Zhang H, Wang S, et al. Optimization of 4,6-disubstituted pyrido[3,2-d]pyrimidines as dual MNK/PIM inhibitors to inhibit leukemia cell growth. J Med Chem. 2021;64(18):13719–13735. doi:10.1021/acs.jmedchem.1c01084
8. Yen SC, Chen LC, Huang HL, et al. Identification of a dual FLT3 and MNK2 inhibitor for acute myeloid leukemia treatment using a structure-based virtual screening approach. Bioorg Chem. 2022;121:105675. doi:10.1016/j.bioorg.2022.105675
9. Suarez M, Blyth GT, Mina AA, et al. Inhibitory effects of Tomivosertib in acute myeloid leukemia. Oncotarget. 2021;12(10):955–966. doi:10.18632/oncotarget.27952
10. Therapeutics E. A phase 1–2 dose-escalation and cohort-expansion study of oral tomivosertib (eFT-508) in subjects with hematological malignancies; 2017. Available from: https://ClinicalTrials.gov/show/NCT02937675.
11. EDDC ASRE, Inc. CI. Evaluation of ETC-1907206 with dasatinib in advanced haematologic malignancies; 2018. Available from: https://ClinicalTrials.gov/show/NCT03414450.
12. Jacqueline Garcia M, Lilly E; Company, Institute D-FC. Combination merestinib and LY2874455 for patients with relapsed or refractory acute myeloid leukemia; 2017. Available from: https://ClinicalTrials.gov/show/NCT03125239.
13. American Cancer Society. Cancer Facts & Figures. American Cancer Society; 2022.
14. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1
15. Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760. doi:10.1182/blood-2017-09-806398
16. Hampel PJ, Parikh SA. Chronic lymphocytic leukemia treatment algorithm 2022. Blood Cancer J. 2022;12(11):161. doi:10.1038/s41408-022-00756-9
17. Kay NE, Parikh SA, Parikh SA. Early intervention in asymptomatic chronic lymphocytic leukemia. Clin Adv Hematol Oncol. 2021;19(2):92–103.
18. American Cancer Society. Treating Acute Lymphocytic Leukemia (ALL). American Cancer Society; 2021.
19. Deininger MW, Shah NP, Altman JK, et al. Chronic myeloid leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2020;18(10):1385–1415. doi:10.6004/jnccn.2020.0047
20. Tallman MS, Wang ES, Altman JK, et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2019;17(6):721–749. doi:10.6004/jnccn.2019.0028
21. Watts JM, Baer MR, Yang J, et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: phase 1 results of a phase 1/2 trial. Lancet Haematol. 2022;10:e46–e58.
22. Administration FaD. FDA Approves Olutasidenib for Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation. Administration FaD; 2023:ed2022.
23. Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129(26):3403–3406. doi:10.1182/blood-2017-05-782292
24. Estey E, Karp JE, Emadi A, Othus M, Gale RP. Recent drug approvals for newly diagnosed acute myeloid leukemia: gifts or a Trojan horse? Leukemia. 2020;34(3):671–681. doi:10.1038/s41375-019-0704-5
25. Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16(8):1921–1933. doi:10.1093/emboj/16.8.1921
26. Buxade M, Parra-Palau JL, Proud CG. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front Biosci. 2008;13(14):5359–5374. doi:10.2741/3086
27. Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol. 2001;21(3):743–754. doi:10.1128/MCB.21.3.743-754.2001
28. Li Y, Yue P, Deng X, et al. Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eIF4F assembly through direct dephosphorylation of Mnk and eIF4E. Neoplasia. 2010;12(10):848–855. doi:10.1593/neo.10704
29. Orton KC, Ling J, Waskiewicz AJ, et al. Phosphorylation of Mnk1 by caspase-activated Pak2/gamma-PAK inhibits phosphorylation and interaction of eIF4G with Mnk. J Biol Chem. 2004;279(37):38649–38657. doi:10.1074/jbc.M407337200
30. Brown MC, Gromeier M. MNK controls mTORC1: substrate association through regulation of TELO2 binding with mTORC1. Cell Rep. 2017;18(6):1444–1457. doi:10.1016/j.celrep.2017.01.023
31. Xie J, Shen K, Jones AT, et al. Reciprocal signaling between mTORC1 and MNK2 controls cell growth and oncogenesis. Cell Mol Life Sci. 2021;78:249–270. doi:10.1007/s00018-020-03491-1
32. Yang X, Zhong W, Cao R. Phosphorylation of the mRNA cap-binding protein eIF4E and cancer. Cell Signal. 2020;73:109689. doi:10.1016/j.cellsig.2020.109689
33. Zhou H, Jia X, Yang FAN. Elevated nuclear phospho-eIF4E body levels are associated with tumor progression and poor prognosis for acute myeloid leukemia. Biocell. 2021;45(3):711–722. doi:10.32604/biocell.2021.014193
34. Joshi S, Platanias LC. Mnk kinase pathway: cellular functions and biological outcomes. World J Biol Chem. 2014;5(3):321–333. doi:10.4331/wjbc.v5.i3.321
35. DaSilva J, Xu L, Kim HJ, Miller WT, Bar-Sagi D. Regulation of sprouty stability by Mnk1-dependent phosphorylation. Mol Cell Biol. 2006;26(5):1898–1907. doi:10.1128/MCB.26.5.1898-1907.2006
36. Edwin F, Anderson K, Patel TB. HECT domain-containing E3 ubiquitin ligase Nedd4 interacts with and ubiquitinates Sprouty2. J Biol Chem. 2010;285(1):255–264. doi:10.1074/jbc.M109.030882
37. Buxade M, Morrice N, Krebs DL, Proud CG. The PSF.p54nrb complex is a novel Mnk substrate that binds the mRNA for tumor necrosis factor alpha. J Biol Chem. 2008;283(1):57–65. doi:10.1074/jbc.M705286200
38. Buxade M, Parra JL, Rousseau S, et al. The Mnks are novel components in the control of TNF alpha biosynthesis and phosphorylate and regulate hnRNP A1. Immunity. 2005;23(2):177–189. doi:10.1016/j.immuni.2005.06.009
39. Hefner Y, Borsch-Haubold AG, Murakami M, et al. Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J Biol Chem. 2000;275(48):37542–37551. doi:10.1074/jbc.M003395200
40. Bouameur JE, Schneider Y, Begre N, et al. Phosphorylation of serine 4642 in the C-terminus of plectin by MNK2 and PKA modulates its interaction with intermediate filaments. J Cell Sci. 2013;126(Pt 18):4195–4207. doi:10.1242/jcs.127779
41. Manne BK, Campbell RA, Bhatlekar S, et al. MAPK-interacting kinase 1 regulates platelet production, activation, and thrombosis. Blood. 2022;140(23):2477–2489. doi:10.1182/blood.2022015568
42. Mishra RK, Clutter MR, Blyth GT, et al. Discovery of novel Mnk inhibitors using mutation-based induced-fit virtual high-throughput screening. Chem Biol Drug Des. 2019;94(4):1813–1823. doi:10.1111/cbdd.13585
43. Jauch R, Jakel S, Netter C, et al. Crystal structures of the Mnk2 kinase domain reveal an inhibitory conformation and a zinc binding site. Structure. 2005;13(10):1559–1568. doi:10.1016/j.str.2005.07.013
44. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi:10.1128/MMBR.00031-10
45. Reich SH, Sprengeler PA, Chiang GG, et al. Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) Inhibition. J Med Chem. 2018;61(8):3516–3540. doi:10.1021/acs.jmedchem.7b01795
46. Yang H, Chennamaneni LR, Ho MWT, et al. Optimization of selective mitogen-activated protein kinase interacting kinases 1 and 2 inhibitors for the treatment of blast crisis leukemia. J Med Chem. 2018;61(10):4348–4369. doi:10.1021/acs.jmedchem.7b01714
47. Zhan Y, Guo J, Yang W, et al. MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J Clin Invest. 2017;127(11):4179–4192. doi:10.1172/JCI91258
48. Santag S, Siegel F, Wengner AM, et al. BAY 1143269, a novel MNK1 inhibitor, targets oncogenic protein expression and shows potent anti-tumor activity. Cancer Lett. 2017;390:21–29. doi:10.1016/j.canlet.2016.12.029
49. Knauf U, Tschopp C, Gram H. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol Cell Biol. 2001;21(16):5500–5511. doi:10.1128/MCB.21.16.5500-5511.2001
50. Yan SB, Peek VL, Ajamie R, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2013;31(4):833–844. doi:10.1007/s10637-012-9912-9
51. Kwiatkowski J, Liu B, Pang S, et al. Stepwise evolution of fragment hits against MAPK interacting kinases 1 and 2. J Med Chem. 2020;63(2):621–637. doi:10.1021/acs.jmedchem.9b01582
52. Wang S, Li B, Liu B, et al. Design and synthesis of novel 6-hydroxy-4-methoxy-3-methylbenzofuran-7-carboxamide derivatives as potent Mnks inhibitors by fragment-based drug design. Bioorg Med Chem. 2018;26(16):4602–4614. doi:10.1016/j.bmc.2018.05.004
53. Diab S, Teo T, Kumarasiri M, et al. Discovery of 5-(2-(Phenylamino)pyrimidin-4-yl)thiazol-2(3H)-one derivatives as potent Mnk2 inhibitors: synthesis, SAR analysis and biological evaluation. ChemMedChem. 2014;9(5):962–972. doi:10.1002/cmdc.201300552
54. Jin X, Merrett J, Tong S, et al. Design, synthesis and activity of Mnk1 and Mnk2 selective inhibitors containing thieno[2,3-d]pyrimidine scaffold. Eur J Med Chem. 2019;162:735–751. doi:10.1016/j.ejmech.2018.10.070
55. Kannan S, Poulsen A, Yang HY, et al. Probing the binding mechanism of Mnk inhibitors by docking and molecular dynamics simulations. Biochemistry. 2015;54(1):32–46. doi:10.1021/bi501261j
56. Kannan S, Pradhan MR, Cherian J, et al. Small molecules targeting the inactive form of the Mnk1/2 kinases. ACS Omega. 2017;2(11):7881–7891. doi:10.1021/acsomega.7b01403
57. Xu W, Kannan S, Verma CS, Nacro K. Update on the development of MNK inhibitors as therapeutic agents. J Med Chem. 2022;65(2):983–1007. doi:10.1021/acs.jmedchem.1c00368
58. Bou-Petit E, Hummer S, Alarcon H, et al. Overcoming paradoxical kinase priming by a novel MNK1 inhibitor. J Med Chem. 2022;65(8):6070–6087. doi:10.1021/acs.jmedchem.1c01941
59. Hantschel O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem Biol. 2015;10(1):234–245. doi:10.1021/cb500886n
60. Abdelaziz AM, Basnet SKC, Islam S, et al. Synthesis and evaluation of 2’H-spiro[cyclohexane-1,3’-imidazo[1,5-a]pyridine]-1’,5’-dione derivatives as Mnk inhibitors. Bioorg Med Chem Lett. 2019;29(18):2650–2654. doi:10.1016/j.bmcl.2019.07.043
61. Abdelaziz AM, Diab S, Islam S, et al. Discovery of N-phenyl-4-(1H-pyrrol-3-yl)pyrimidin-2-amine derivatives as potent Mnk2 inhibitors: design, synthesis, SAR analysis, and evaluation of in vitro anti-leukaemic activity. Med Chem. 2019;15(6):602–623. doi:10.2174/1573406415666181219111511
62. Matsui Y, Yasumatsu I, Yoshida KI, et al. A novel inhibitor stabilizes the inactive conformation of MAPK-interacting kinase 1. Acta Crystallogr Struct Biol Commun. 2018;74(Pt 3):156–160. doi:10.1107/S2053230X18002108
63. Sansook S, Lineham E, Hassell-Hart S, et al. Probing the anticancer action of novel ferrocene analogues of MNK inhibitors. Molecules. 2018;23(9):2126. doi:10.3390/molecules23092126
64. Halder AK, Cordeiro M. Multi-target in silico prediction of inhibitors for mitogen-activated protein kinase-interacting kinases. Biomolecules. 2021;11(11):1670. doi:10.3390/biom11111670
65. Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66:1022–1037. doi:10.1021/np030096l
66. Cotoraci C, Ciceu A, Sasu A, Miutescu E, Hermenean A. The anti-leukemic activity of natural compounds. Molecules. 2021;26(9):2709. doi:10.3390/molecules26092709
67. Kimira M, Arai Y, Shimoi K, Watanabe S. Japanese intake of flavonoids and isoflavonoids from foods. J Epidemiol. 1998;8(3):168–175. doi:10.2188/jea.8.168
68. Bower AM, Real Hernandez LM, Berhow MA, de Mejia EG. Bioactive compounds from culinary herbs inhibit a molecular target for type 2 diabetes management, dipeptidyl peptidase IV. J Agric Food Chem. 2014;62(26):6147–6158. doi:10.1021/jf500639f
69. Shankar E, Goel A, Gupta K, Gupta S. Plant flavone apigenin: an emerging anticancer agent. Curr Pharmacol Rep. 2017;3(6):423–446. doi:10.1007/s40495-017-0113-2
70. Yen SC, Wu YW, Huang CC, et al. O-methylated flavonol as a multi-kinase inhibitor of leukemogenic kinases exhibits a potential treatment for acute myeloid leukemia. Phytomedicine. 2022;100:154061. doi:10.1016/j.phymed.2022.154061
71. Pan H, Hu Q, Wang J, et al. Myricetin is a novel inhibitor of human inosine 5’-monophosphate dehydrogenase with anti-leukemia activity. Biochem Biophys Res Commun. 2016;477(4):915–922. doi:10.1016/j.bbrc.2016.06.158
72. Cai F, Li B, Li J, Ding Y, Xu D, Huang F. Myricetin is effective and selective in inhibiting imatinib-resistant chronic myeloid leukemia stem and differentiated cells through targeting eIF4E. Anticancer Drugs. 2022;34:620–626. doi:10.1097/CAD.0000000000001421
73. Chen LC, Huang HL, HuangFu WC, et al. Biological evaluation of selected flavonoids as inhibitors of MNKs targeting acute myeloid leukemia. J Nat Prod. 2020;83(10):2967–2975. doi:10.1021/acs.jnatprod.0c00516
74. Sugawara F, Strobel S, Strobel G. The structure and biological activity of cercosporamide from Cercosporidium henningsii. J Org Chem. 1991;56:909–910. doi:10.1021/jo00003a002
75. Konicek BW, Stephens JR, McNulty AM, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71(5):1849–1857. doi:10.1158/0008-5472.CAN-10-3298
76. Wang S, Zang J, Huang M, et al. Discovery of novel (+)-Usnic acid derivatives as potential anti-leukemia agents with pan-Pim kinases inhibitory activity. Bioorg Chem. 2019;89:102971. doi:10.1016/j.bioorg.2019.102971
77. Altman JK, Szilard A, Konicek BW, et al. Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood. 2013;121(18):3675–3681. doi:10.1182/blood-2013-01-477216
78. Kosciuczuk EM, Saleiro D, Kroczynska B, et al. Merestinib blocks Mnk kinase activity in acute myeloid leukemia progenitors and exhibits antileukemic effects in vitro and in vivo. Blood. 2016;128(3):410–414. doi:10.1182/blood-2016-02-698704
79. Kosciuczuk EM, Kar AK, Blyth GT, et al. Inhibitory effects of SEL201 in acute myeloid leukemia. Oncotarget. 2019;10(67):7112–7121. doi:10.18632/oncotarget.27388
80. Lim S, Saw TY, Zhang M, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A. 2013;110(25):E2298–2307. doi:10.1073/pnas.1301838110
81. Zhang Q, Li H, Li Q, Hu Q, Liu B. MNK/eIF4E inhibition overcomes anlotinib resistance in non-small cell lung cancer. In: Fundamental and Clinical Pharmacology. Wiley; 2022:1–8.
82. Grzmil M, Seebacher J, Hess D, et al. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell Signal. 2016;28(9):1412–1421. doi:10.1016/j.cellsig.2016.06.005
83. Yang X, Liu Z, Yin X, Zeng Y, Guo G. Inhibition MNK-eIF4E-beta-catenin preferentially sensitizes gastric cancer to chemotherapy. Fundam Clin Pharmacol. 2022;36(4):712–720. doi:10.1111/fcp.12759
84. Zhu Y, Wang C, Li M, Yang X. Targeting of MNK/eIF4E overcomes chemoresistance in cervical cancer. J Pharm Pharmacol. 2021;73(10):1418–1426. doi:10.1093/jpp/rgab094
85. Altman JK, Glaser H, Sassano A, et al. Negative regulatory effects of Mnk kinases in the generation of chemotherapy-induced antileukemic responses. Mol Pharmacol. 2010;78(4):778–784. doi:10.1124/mol.110.064642
86. Li PDS, Yu M, Adams J, et al. Inhibition of Mnk enhances apoptotic activity of cytarabine in acute myeloid leukemia cells. Oncotarget. 2016;7(35):56811–56825. doi:10.18632/oncotarget.10796
87. Chen K, Chen Y, Chen Z, et al. miR-134 increases the antitumor effects of cytarabine by targeting Mnks in acute myeloid leukemia cells. Onco Targets Ther. 2018;11:3141–3147. doi:10.2147/OTT.S143465
88. Zhang M, Fu W, Prabhu S, et al. Inhibition of polysome assembly enhances imatinib activity against chronic myelogenous leukemia and overcomes imatinib resistance. Mol Cell Biol. 2008;28(20):6496–6509. doi:10.1128/MCB.00477-08
89. Liu Z, Li Y, Lv C, Wang L, Song H. Anthelmintic drug niclosamide enhances the sensitivity of chronic myeloid leukemia cells to dasatinib through inhibiting Erk/Mnk1/eIF4E pathway. Biochem Biophys Res Commun. 2016;478(2):893–899. doi:10.1016/j.bbrc.2016.08.047
90. Knight JRP, Alexandrou C, Skalka GL, et al. MNK inhibition sensitizes KRAS-mutant colorectal cancer to mTORC1 inhibition by reducing eIF4E phosphorylation and c-MYC expression. Cancer Discov. 2021;11(5):1228–1247. doi:10.1158/2159-8290.CD-20-0652
91. Fan C, Zhao C, Zhang F, et al. Adaptive responses to mTOR gene targeting in hematopoietic stem cells reveal a proliferative mechanism evasive to mTOR inhibition. Proc Natl Acad Sci U S A. 2021;118(1). doi:10.1073/pnas.2020102118
92. Huang XB, Yang CM, Han QM, Ye XJ, Lei W, Qian WB. MNK1 inhibitor CGP57380 overcomes mTOR inhibitor-induced activation of eIF4E: the mechanism of synergic killing of human T-ALL cells. Acta Pharmacol Sin. 2018;39(12):1894–1901. doi:10.1038/s41401-018-0161-0
93. Teo T, Yu M, Yang Y, et al. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer Lett. 2015;357(2):612–623. doi:10.1016/j.canlet.2014.12.029
94. Schatz JH, Oricchio E, Wolfe AL, et al. Targeting cap-dependent translation blocks converging survival signals by AKT and PIM kinases in lymphoma. J Exp Med. 2011;208(9):1799–1807. doi:10.1084/jem.20110846
95. Mohamed LM, Eltigani MM, Abdallah MH, et al. Discovery of novel natural products as dual MNK/PIM inhibitors for acute myeloid leukemia treatment: pharmacophore modeling, molecular docking, and molecular dynamics studies. Front Chem. 2022;10:975191. doi:10.3389/fchem.2022.975191
96. Wu H, Hu C, Wang A, et al. Discovery of a BTK/MNK dual inhibitor for lymphoma and leukemia. Leukemia. 2016;30(1):173–181. doi:10.1038/leu.2015.180
97. Cherian J, Nacro K, Poh ZY, et al. Structure-activity relationship studies of mitogen activated protein kinase interacting kinase (MNK) 1 and 2 and BCR-ABL1 inhibitors targeting chronic myeloid leukemic cells. J Med Chem. 2016;59(7):3063–3078. doi:10.1021/acs.jmedchem.5b01712
98. Xing K, Zhang J, Han Y, Tong T, Liu D, Zhao L. Design, synthesis and bioactivity evaluation of 4,6-disubstituted pyrido[3,2-d]pyrimidine derivatives as Mnk and HDAC inhibitors. Molecules. 2020;25(18):4318. doi:10.3390/molecules25184318
99. San Jose-Eneriz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers. 2019;11(11):1794. doi:10.3390/cancers11111794
100. Oncology TRi, Therapeutics E, Cancer SUT. Safety, pharmacodynamics, pharmacokinetics, and efficacy of tomivosertib combined with paclitaxel in advanced breast cancer; 2020. Available from: https://ClinicalTrials.gov/show/NCT04261218.
101. Therapeutics E. An open-label study examining the effect of tomivosertib (eFT508) in patients with advanced castrate-resistant prostate cancer (CRPC); 2018. Available from: https://ClinicalTrials.gov/show/NCT03690141.
102. Therapeutics E. Tomivosertib (eFT-508) in combination with PD-1/PD-L1 inhibitor therapy; 2018. Available from: https://ClinicalTrials.gov/show/NCT03616834.
103. Therapeutics E, Medpace I. Tomivosertib combined with pembrolizumab in subjects with PD-L1 positive NSCLC (KICKSTART). Available from: https://ClinicalTrials.gov/show/NCT04622007;.
104. Therapeutics E, Merck KGaA D, Germany, Pfizer. A study to evaluate eFT508 alone and in combination with avelumab in subjects with MSS colorectal cancer; 2017. Available from: https://ClinicalTrials.gov/show/NCT03258398.
105. Therapeutics E. A PD study of oral eFT508 in subjects with advanced TNBC and HCC; 2017. Available from: https://ClinicalTrials.gov/show/NCT03318562.
106. Chen EC, Gandler H, Tosic I, et al. Targeting MET and FGFR in relapsed or refractory acute myeloid leukemia: preclinical and clinical findings, and signal transduction correlates. Clin Cancer Res. 2023;29(5):878–887. doi:10.1158/1078-0432.CCR-22-2540
107. eFFECTOR therapeutics’ lead product candidate, eFT508, receives orphan designation from FDA for treatment of diffuse large B-cell lymphoma [press release] eFFECTOR Therapeutics; 2017. Available from: https://www.prnewswire.com/news-releases/effector-therapeutics-lead-product-candidate-eft508-receives-orphan-designation-from-fda-for-treatment-of-diffuse-large-b-cell-lymphoma-300420884.html.
108. Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2019;49(D1):D1388–D1395. doi:10.1093/nar/gkaa971
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.