Back to Journals » Journal of Inflammation Research » Volume 16
Mitophagy, Inflammasomes and Their Interaction in Kidney Diseases: A Comprehensive Review of Experimental Studies
Authors Wang Y ![]() , Song D
, Song D ![]() , Tang L
, Tang L
Received 21 December 2022
Accepted for publication 17 March 2023
Published 5 April 2023 Volume 2023:16 Pages 1457—1469
DOI https://doi.org/10.2147/JIR.S402290
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yulin Wang, Dongxu Song, Lin Tang
Department of Nephrology, Zhengzhou University First Affiliated Hospital, Zhengzhou, Henan, 450052, People’s Republic of China
Correspondence: Lin Tang, Department of Nephrology, Zhengzhou University First Affiliated Hospital, 1 Jianshe Road, Zhengzhou, Henan, 450052, People’s Republic of China, Email [email protected]
Abstract: Mitophagy is an important mechanism for mitochondrial quality control by regulating autophagosome-specific phagocytosis, degradation and clearance of damaged mitochondria, and involved in cell damage and diseases. Inflammasomes are important inflammation-related factors newly discovered in recent years, which are involved in cell innate immunity and inflammatory response, and play an important role in kidney diseases. Based on the current studies, we reviewed the progress of mitophagy, inflammasomes and their interaction in kidney diseases.
Keywords: mitophagy, inflammasomes, kidney diseases, acute kidney injury, chronic kidney disease, diabetic kidney disease
Introduction
Mitochondria are the energy production center of cells and are involved in several biological processes, including steroid synthesis, lipid metabolism, calcium signal transduction, and apoptosis.1 Mitochondria are a vital organelle in mammalian cells. The kidney is an organ with a high energy metabolic demand, and its mitochondrial abundance is second only to the heart.2 Several studies show that mitochondrial dysfunction is one of the key factors in the pathogenesis and progression of different kidney diseases.2–5 Therefore, mitochondria quality control is necessary to maintain the kidney’s normal function.6,7 Mitochondrial quality control includes the regulation of mitochondrial proteases and molecular chaperones, mitochondrial dynamic homeostasis (fusion and fission), mitochondrial biogenesis, and mitophagy.8 Mitophagy is an important mechanism for mitochondrial quality control by regulating autophagosome-specific phagocytosis, degradation and clearance of damaged mitochondria. In recent years, mitophagy has been widely studied in tumors,9 neurological diseases,10 and heart diseases.11 Currently, mitophagy is also associated with both acute and chronic kidney diseases, and studies on the signaling pathways involved in the phenomenon are increasing. It has been found that inflammasomes can mediate mitophagy to promote the progression of various kidney diseases, which have been studied in acute kidney injury (AKI), chronic kidney disease (CKD), and diabetic kidney disease (DKD). This review aims to summarize the progress of the interaction between mitophagy and inflammasomes in kidney diseases.
Mitophagy
Overview of Mitophagy
Mitochondria are the main energy production site of eukaryotic cells and originated from α-Actinobacillus, which were engulfed by eukaryotes one billion years ago. Mitochondria are a double-layer membrane-wrapped organelle, having various functions such as producing adenosine triphosphate, regulating calcium signal and membrane potential, and participating in cell death and signal transduction.12 Mitochondria can only function normally by maintaining a steady state, that is, the quality control of mitochondria.
Mitophagy was first discovered by Lemasters in 2005.13 Mitophagy removes dysfunctional or redundant mitochondria in a cellular process that fine-tunes mitochondrial numbers and preserves energy metabolism.14 It belongs to selective autophagy and plays an important role in maintaining mitochondrial function and cell survival. Mitophagy is a complex process, including the initiation of mitophagy and mitochondria for autophagy mechanism recognition, phagocytosis of labeled mitochondria through the formation of autophagosomes, and the formation and hydrolytic degradation of lysosomes.
Mitophagy in mammals can be roughly divided into two categories: 1) ubiquitination dependent mitophagy. It uses P62, a neighbor of BRCA1 gene 1 (NBR1) and optineurin (OPTN) as autophagy receptors, and is mediated by phosphatase and tensin homolog (PTEN) induced putative kinase 1 (PINK1)/Parkinson protein 2 (parkin).15 PINK1 is a serine/threonine kinase and its level is usually very low. However, when mitochondria are damaged (mitochondrial DNA, mtDNA mutations), mitochondrial reactive oxygen species (mtROS) are increased, or misfolded proteins are accumulated, PINK1 will be stabilized and accumulated in the outer mitochondrial membrane (OMM).16 The accumulated PINK1 is automatically phosphorylated and activated, followed by phosphorylation of ubiquitin on serine 65, thereby recruiting parkin from the cytoplasm to the mitochondrial membrane. Parkin could drive the ubiquitination of mitochondrial proteins upon recruitment and activation, and then leads to autophagy.17,18 In mammals, PINK1/parkin-mediated mitophagy is dominant.19 2) Receptor-dependent mitophagy. At present, these autophagy receptors such as Bcl2/adenovirus E1B 19 ku interacting protein 3 (BNIP3)/BNIP3L (Bnip3-like, Nix),20 FUN14 domain-containing 1 (FUNDC1), FK506-binding protein 8 (FKBP8), Prohibitin 2 (PHB2), and cardiolipin (CL) have been found.21 BNIP3/BNIP3L can mediate the translocation of mitochondrial fission protein dynamin-related protein 1 (DRP 1) to mitochondria, split mitochondria into small fragments, then activate the PINK1/parkin pathway and initiate mitophagy.22,23 FUNDC1 is associated with hypoxia-induced mitophagy as a conserved mitophagy receptor. Its microtubule associated protein 3 (LC3)-binding activity is increased by phosphorylation and dephosphorylation, thereby enhancing its LC3-interacting region (LIR) to initiate mitophagy.24,25 The LC3 family plays an important role in the elongation and maturation of mitophagosomes.26 FKBP8 (also described as FKBP38) is a novel OMM mitophagy receptor that interacts with LC3 to initiate mitophagy without parkin attendance.27 Nonetheless, how it is activated is currently under investigation. PHB2, an inner mitochondrial membrane (IMM) scaffolding protein, has been suggested as a possible mitophagy receptor under stress conditions. PHB2 mediates the stabilization of PINK1 on the OMM following mitochondrial membrane depolarization or aggregation of misfolded proteins. Then PHB2 promotes the recruitment of mitochondrial parkin and leads to proteasome-dependent mitochondrial OMM rupture. PHB2 then externalizes to the OMM and interacts with LC3, leading to the formation of autophagosomes and clearance of dysfunctional mitochondria.21,28 CL is a phospholipid that supports the mitochondrial cristae and is distributed along healthy mitochondrial IMM.29 Upon mitochondrial damage, CL can be externalized to OMM and directly bind to LC3 to induce mitophagy29 (Figure 1).
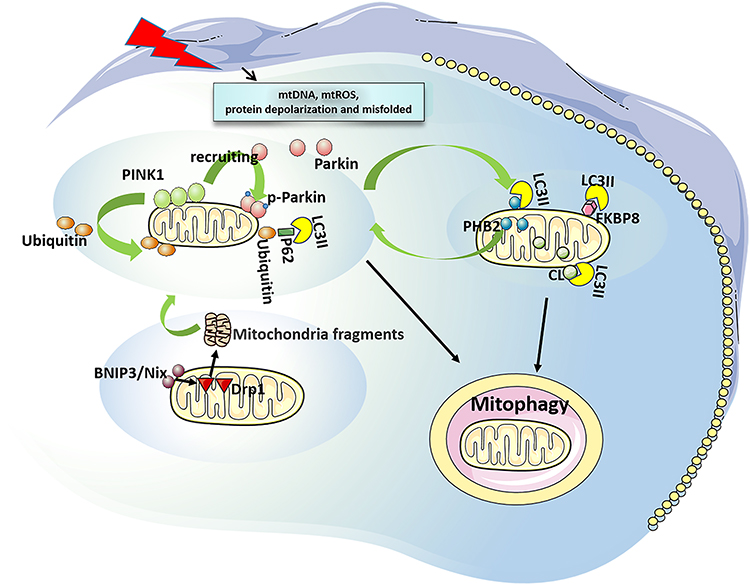 |
Figure 1 Overview of mitophagy. Ubiquitination dependent mitophagy: It is mediated by PINK1/ parkin. When mitochondrial damage, PINK1 stabilizes and accumulates at the OMM to be phosphorylated and activated, thus recruiting parkin from the cytoplasm to the mitochondrial membrane, which driving the ubiquitination of mitochondrial proteins, leading to autophagy. Receptor-dependent mitophagy: BNIP3/Nix mediates the translocation of DRP1 to mitochondria, split mitochondria into small fragments, then activate PINK1/Parkin pathway and initiate mitophagy. FUNDC1 and FKBP8 can interact with LC3 to initiate mitophagy. PHB2 mediates stabilization of PINK1 on the OMM, and recruits Parkin. Then, PHB2 is externalized to the OMM and combines to LC3, leading to mitophagy. CL can be externalized to the OMM and directly bind to LC3 to induce mitophagy. Abbreviations: PINK1, PTEN induced putative kinase 1; OMM, outer mitochondrial membrane; BNIP3, Bcl2/adenovirus E1B 19 ku interacting protein 3; Nix, Bnip3-like; FUNDC1, FUN14 domain-containing 1; FKBP8, FK506-binding protein 8; PHB2, Prohibitin 2; CL, cardiolipin; DRP1, dynamin-related protein 1; IMM, inner mitochondrial membrane. |
Mitophagy and Kidney Diseases
The role of mitophagy in daily life activities has received increasing emphasis as research progresses. Consistent with the abundance of the distribution of mitochondria, the nervous system, kidney, skeletal muscles, heart, and liver have a higher level of basal mitophagy than other tissues, for instance, the spleen and thymus.30 Therefore, prior studies of molecular and biochemical pathways involving mitophagy have primarily focused on models of aging,31 neuropsychiatric diseases,32 malignant tumors,33 and heart diseases.34 Since the kidney also has a high level of mitophagy, the action of mitophagy in kidney diseases has received frequent attention in recent years.20,21
Zhao et al35 observed increased expression of mitophagy-related proteins (PINK1, parkin, LC3) and enhanced immunofluorescence colocalization signal of mitochondrial and lysosomal proteins in cisplatin-treated human proximal tubular epithelial cells line (HK2). Knockdown of PINK1 and parkin could inhibit mitophagy and aggravated cisplatin-induced HK2 damage. Thus, it suggested that mitophagy is important in damaging and repairing renal tubular epithelial cells (RTECs) during AKI. Tang et al36 found that both PINK1 and parkin were up-regulated in RTECs of ischemic AKI, and lack of PINK1 and/or parkin led to mitochondrial damage, increased ROS production, and inflammation. Mitophagy mediated by PINK1/Parkin/OPTN pathway is also activated in RTECS of septic AKI, playing a role in protecting RTECs.37 Knock-down of PINK1 or parkin inhibited the accumulation of OPTN and led to mitophagy disorder.37 Fu et al38 showed that in ischemia/reperfusion (I/R)-induced AKI BNIP3 overexpression significantly reversed renal I/R injury (IRI), decreased mitophagy in renal tubular hypoxia-inducible factor-1α (HIF-1α) knockout mice, and prevented the aggravation of the renal injury. The aforementioned studies suggested that PINK1/parkin and BNIP3-mediated mitophagy have an important protective role in mitochondrial quality control and cell damage in the cell survival and function of AKI tubular.
CKD remains a major public health problem with poor outcomes.39 Eleven percent of individuals with stage 3 CKD will eventually progress to end-stage renal disease (ESRD), requiring dialysis or kidney transplantation.39 Mitophagy levels were increased in AKI, whereas they were lowered in CKD. In the 5/6 nephrectomy model, mitochondrial volume was increased, fission level was reduced due to hyperperfusion of the kidney, and mitophagy was reduced.40 A recent study found that PINK1/parkin-mediated mitophagy was attenuated in a cisplatin-induced CKD mouse model, and nuclear factor erythroid 2-related factor 2 (Nrf2) inducers could improve PINK1/parkin-mediated mitophagy and effectively inhibit cisplatin-induced inflammation and renal fibrosis, providing a potential therapeutic target for CKD.41 In obesity-related CKD, chicoric acid could activate Nrf2 pathway, increase the expressions of PINK and parkin, and then improve mitophagy to alleviate mitochondrial damage in RTECs.42 Another study explored the effect of the recently discovered mitophagy-inducing chemical UMI-77 on mitophagy activation in a mouse model of CKD with unilateral ureteral obstruction (UUO).43 In UUO mice, mitochondrial damage, ROS production, activation of the transforming growth factor (TGF)-β1/Smad pathway, and phenotypic transition of epithelial–mesenchymal cells and renal fibrosis were observed. These changes were ameliorated after using UMI-77 by enhancing mitophagy.43 The PI3K/AKT/mTOR pathway might also play an important role in the inflammatory response caused by mitophagy disorder in CKD.44,45
The studies on DKD also found that mitophagy disorder is the key pathological mechanism of the occurrence and development of DKD.46 In a streptozotocin (STZ)-induced diabetic rat model, the expression of PINK1 in the renal cortex of early diabetic rats was increased.47 As DKD progresses, mitophagy becomes overwhelmed, resulting in fragmented mitochondria accumulation and cell death.48 The in vitro study by Xiao et al found that PINK1 and parkin were significantly reduced in the tubules of diabetic mice and HK2 under high glucose conditions, resulting in mitophagy impairment of RTECs.49 Our preliminary study also confirmed these results.50 PINK1/Parkin-mediated mitophagy was also involved in mesangial cells apoptosis and mitigated by erythropoietin (EPO).51 Other studies found that disrupting PHB2 mediated mitophagy could aggravate RTECs injury52 and suppression of FUNDC1-mediated mitophagy-induced podocytes injury53 in DKD. Improving mitophagy by metformin or antioxidants may prevent DKD kidney damage.49,54,55
Recent studies have found that mitophagy is also involved in developing lupus nephritis (LN). In a model of LN podocytes in vitro, it was found that the expressions of PINK1 and LC3 were up-regulated, while the expression of p62 was down-regulated. PINK1-specific siRNA could reduce the expression of PINK1 and inhibit mitophagy.56 However, there are still few related studies. The kidney is particularly sensitive to different mutations in mitochondrial DNA.57 Mitochondrial dysfunction also plays an important role in inherited renal tubulointerstitial diseases.57,58
With the in-depth study of mitophagy, it has recently been found that it is also associated with the inflammatory response triggered by inflammasome activation.59 Damaged mitochondria release mtDNA, mtROS, CL, and other triggers under disease conditions, thereby activating the inflammasomes,59,60 which are involved in disease initiation and progression.
Inflammasomes
Overview of inflammasomes
The inflammasomes are multi-protein complexes composed primarily of receptor proteins, adaptor proteins, and downstream caspases that regulate cytokine maturation, inflammation, and cell death.61
According to the activation of cysteinyl aspartate-specific protease (caspase) during the formation of inflammasomes, they can be divided into two categories: “canonical” and “non-canonical”62 The “canonical” inflammasome mainly activates caspase-1, whereas the “non-canonical” inflammasome activates caspases other than caspase-1.62 The “canonical” inflammasome generally includes the NOD receptor (NLRs) family, AIM2, and the pyrin inflammasome.63 The NLRs family shares common structural features, including a carboxy-terminal leucine-rich repeat (LRR) domain, a central nucleotide-binding oligomerization domain (NOD), and a variable amino-terminal domain.64,65 NLRs can be divided into five subtypes and 22 members according to the different amino acid terminal domains.64–66 (1) NLRA (also known as MHC class II transactivator, CIITA), containing an acidic transactivation domain; (2) NLRB (also known as neuronal apoptosis inhibitory protein, NAIP), containing baculovirus inhibitor repeats (BIR); (3) NLRC, including NOD1, NOD2 and NLRC3-5, containing Caspase Activation and Recruitment Domain (CARD); (4) NLRP1-14, contains pyrin domain (PYD); (5) NLRX1, the domain is unknown. NOD1, NOD2, NLRP10, NLRC5, CIITA and NLRX1 do not form inflammasomes to activate caspase-1, but activate canonical signaling pathways, such as nuclear factor kappa-B (NF-κB), mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), p38 and extracellular regulated protein kinases (ERK), then promote tumor necrosis factor (TNF-α), interleukin (IL)-1β, IL-6, IL-18, IL-12, and other inflammatory factors to initiate immune responses.67 Activation of the ‘canonical’ inflammasome depends on pattern recognition receptors (PRRs), which play a pathophysiological role only upon detection of microbial pathogens or damage-associated molecular patterns. The identified PRR families are the followings: AIM2-like receptors (ALRs), NLRs, RIG-I-like receptors (RLRs), toll-like receptors (TLRs) and C-type lectin receptors (CLRs). In the structure of the inflammasome, PRRs are present in the cytoplasm and do not have a transmembrane domain. The inflammasome and its homologous proteins are used to hydrolyze related proteins through PYD and/or CARD, and finally activate caspase-1, thereby enabling the activation of caspase-1. Inflammatory factors, like IL-1β and IL-18, are matured and secreted extracellularly, and gasdermin D (GSDMD) is cleaved, eventually leading to inflammation and cell death (Figure 2). GSDMD is a key protein involved in pyroptosis and a substrate protein of caspase.
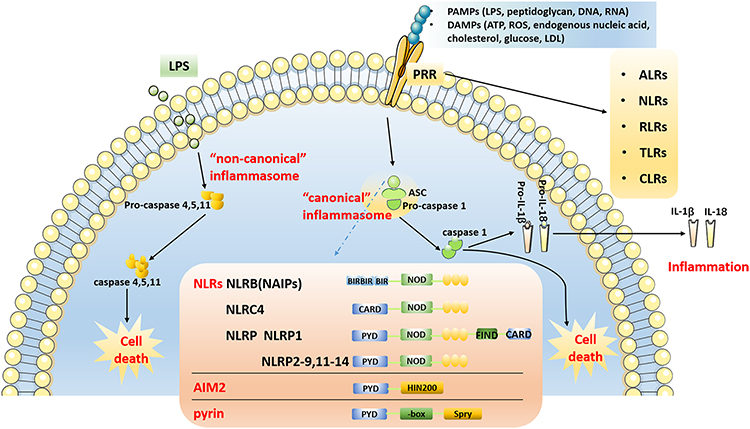 |
Figure 2 Overview of inflammasome pathway. The “canonical” inflammasome pathway: After PRRs recognize PAMPs and DAMPs, NLRs, AIM2 and pyrin form inflammasome complexes with ASC, pro-caspase 1 through PYD and/or CARD, and then activate caspase-1. Inflammatory factor such as IL-1β and IL-18 are matured and secreted, and eventually results in inflammation and cell death. The “non-canonical” inflammasome pathway: Inflammasomes directly bind to LPS to activate caspase-11, caspase-4 and caspase-5 through the CARD, resulting in cell death. Abbreviations: PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; LPS, lipopolysaccharide; LDL, low-density lipoprotein; PRRs, pattern recognition receptors; ALRs, AIM2-like receptors; NLRs, NOD-like receptors; RLRs, RIG-I-like receptors; TLRs, toll-like receptors; CLRs, C-type lectin receptors; ASC, apoptosis-associated speck-like protein CARD; CARD, caspase activation and recruitment domain; PYD, pyrin domain. |
The “non-canonical” inflammasomes primarily activate caspase-466 and caspase-5 in humans,68 and caspase-11 in mice.69 The activation of caspase-8 mediated by inflammation has also been identified as a “non-canonical” inflammasome pathway that may result in the maturation of IL-1β.70 Inflammasomes can activate caspase-11, caspase-4 and caspase-5 by interacting with lipopolysaccharide (LPS) and its component lipid A through the CARD.68,69 Subsequently, GSDMD is cleaved, resulting in cell death (Figure 2). Nevertheless, activation of these “non-canonical” inflammasomes may also induce activation of the canonical inflammasome, such as the NLRP3 inflammasome, leading to the “canonical” inflammasome pathway.63
Inflammasomes and Kidney Diseases
Inflammasomes attend in the pathogenesis and development of many kinds of diseases in humans, including metabolic diseases,71 cancer,72 lung injury,73 cardiovascular diseases (CVD),74 neurodegeneration75 and so on. Recent studies also showed that inflammasomes formation and activation occur not only in immune cells of the kidney, for instance, activated macrophages or dendritic cells, but also in renal tissue resident cells, such as RTECs and podocytes,63 resulting in histopathological damage. This showed the significance of inflammasomes in the pathogenesis and progression of kidney diseases.63
The NLRP3 inflammasome was discovered in 2002. The NLRP3 inflammasome was initially thought to be a high molecular weight protein oligomeric complex consisting of NLRP3, the CARD-containing apoptosis-associated speck protein (ASC), and caspase-1 that regulates IL-1β and IL-18 maturation. NLRP3 is the most widely studied inflammatory gene in kidney disease by far.61 Numerous studies had investigated the role of the NLRP3 inflammasome in various models of kidney diseases,61 including obstructive nephropathy,76 tubulointerstitial nephritis,77 metabolic syndrome-related kidney disease,78 hyperhomocysteinemia-induced glomerulonephritis,79 albumin overload-induced kidney damage,80 hypertension-related renal damage,81 DKD,82 AKI,83 immune complex-related glomerulonephritis,84 LN,85 5/6 nephrectomy,86 human immunodeficiency virus (HIV) infection-related nephropathy,87 and HIV infection-related nephropathy et al.88,89 Taken together, these studies reveal distinct roles for the NLRP3 inflammasome in kidney diseases.
AIM2 acts as a cytoplasmic innate immune receptor by recognizing double-stranded DNA (dsDNA) released during the damage of cells and pathogenesis.90,91 Sensing dsDNA in the cytoplasm by AIM2 is critical for mediating protection from invading pathogens including bacteria, viruses, fungi, and parasites.90 The dsDNA released from damaged host cells can also activate AIM2, leading to the secretion of inflammatory cytokines that promote the progression of different diseases, such as skin diseases, neurological diseases, CKD, CVD, and diabetes.90 AIM2 inflammasome is also involved in kidney disease. In rhabdomyolysis-induced acute kidney injury, dsDNA-induced AIM2 macrophages pyroptosis may play an important role in promoting the recovery of renal injury.92 Zhen et al found that AIM2 is related to hepatitis B virus (HBV)-related glomerulonephritis, and the expressions of AIM2 and inflammatory factors caspase-1, IL-1β and IL-18 were increased in the tissues and cell lines of patients with HBV-related glomerulonephritis.93 The expression of caspase-1, IL-1β and IL-18 were reduced in HBV-infected and uninfected human mesangial cells following AIM2 knockout.93 In 2018, Komada et al reported the role of AIM2 on kidney injury in CKD human and UUO mouse models.94 Compared with normal kidney tissue, the expression of AIM2 in RTECs and infiltrating leukocytes was increased in renal biopsies from patients with DKD or hypertensive nephrosclerosis.94 Immunofluorescence detection of renal tissue from patients with LN showed that the expression of AIM2 was significantly increased.95 These results suggested that AIM2 inflammasome activation may lead to renal injury. However, a recent study found that AIM2 expression was increased in parietal epithelial cells within the crescent in patients with anti-glomerular basement membrane (anti-GBM) disease.96 In the nephrotoxic serum (NTS)-induced glomerulonephritis model, glomerular crescent formation, tubular damage, inflammation and proteinuria were more severe in AIM2−/−(B6) mice.96 In addition, AIM2 may also be a therapeutic target for acute renal transplantation rejection.97 It was suggested that AIM2 has atypical effects of inhibiting inflammation and epithelial cell proliferation in the process of nephritis.96
NLRC4 can directly recruit and activate pro-caspase-1 in the absence of ASC. Current studies indicated that NLRC4 plays an important role in defense against enteric pathogens, while its role in non-enteric pathogen infection is still poorly understood. Yuan et al reported that the NLRC4 inflammasome is associated with human and mouse DKD. NLRC4-deficient diabetic mice had improved hyperglycemia and diabetic glomerulopathy than diabetic wild-type controls.98,99 Our study also demonstrated that the expression of NLRC4 was increased in the kidney tissue of DKD patients and HK2 cells under high glucose environment. The down-regulation of NLRC4 could alleviate the damage of HK2 cells under high glucose environment.100 However, further studies are needed to confirm the role of the NLRC4 inflammasome in kidney diseases.
NLRC5 is a transcriptional activator of histocompatibility complex (MHC) class I genes.101 In mice models of AKI induced by IRI or cisplatin, NLRC5 overexpression inhibited the expression of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) to active ERK1/2 and Akt, hence enhancing the renal damage.102 In animal models of AKI, the importance of NLRC5 was found by bone marrow chimera-related experiments.102 NLRC5 is also found to be expressed in the glomeruli of DKD patients. It could promote DKD by inducing inflammation and fibrosis in mesangial cells and macrophages of patients, partly by affecting NF-κB and TGF-β/Smad signaling pathways.103
NLRP1 plays a significant role in inflammation and autoimmunity, including vitiligo-related autoimmune disease, Addison disease, rheumatoid arthritis, and systemic lupus erythematosus (SLE).104 Studies have also found that NLRP1 and its gene polymorphisms are involved in the progression of DKD.104
NLRP6 is found to be a protective role in cisplatin-induced AKI.105 NLRP6 expression was downregulated in cisplatin-induced AKI, and NLRP6 deficiency increased the severity of AKI.105 Functional studies confirmed that, NLRP6 might regulate RTECs apoptosis by MAPK activation, inflammatory factor expression and cell death in AKI.105
NLRX1 (NOD-like receptor family member X1) is localized to mitochondria and has a pronounced anti-inflammatory effect.106,107 NLRX1 protected renal parenchymal cells in the renal IRI model by inhibiting oxidative phosphorylation and protecting the integrity of the cell membrane.108
In addition to the above inflammasomes, no other inflammasomes have been studied in renal disorders. Further research is needed to uncover the role of the inflammasomes in kidney diseases.
The Interaction Between Mitophagy and Inflammasomes May Be a New Target for the Treatment of Kidney Diseases
Despite accumulating evidence for the link between mitophagy and inflammasomes in human health and diseases, few studies have revealed how mitophagy and inflammasomes are reciprocally regulated.11 The balance of the inflammasome-mitochondrial pathway may help protect host immunity and maintain human health.11 However, dysregulation between inflammasomes and mitophagy may lead to pathological inflammatory responses. In this section, we mainly summarized the studies on the interaction between mitophagy and inflammasomes in different kidney diseases.
AKI
In the mice model of sepsis-related AKI, parkin knockout increased the expression of NLRP3, ASC, caspase-1, and IL-1β in renal tissue, suggesting that decreased mitophagy may activate the NLRP3 inflammasome.109 RTECs undergo mitophagy during both in vivo and in vitro contrast agent-induced AKI (CI-AKI).110 When PINK1 or parkin was silenced, contrast-induced mitophagy impairment, mitochondrial damage, and mtROS production increased, leading to the increase of NLRP3, cleaved caspase-1, and mature IL-1β in mice and in vitro RTECs.110 Thus, apoptosis of RTECs and renal injury were both increased.110 These results suggested that PINK1/parkin mediated mitophagy protects against AKI by inhibiting the activation of the NLRP3 inflammasome. Nonetheless, other studies indicated that the inflammasome seems to be sent back to regulate mitophagy. NLRP3 or caspase-1 knockout CI-AKI mice were found to up-regulate cellular responses to hypoxia, mitochondrial oxidation, and autophagy, indicating that inhibition of the NLRP3 inflammasome leads to up-regulation of hypoxia signaling pathways and mitophagy.111 In NLRP3 or caspase-1 knockout CI-AKI mice and iohexol-treated HK-2 cells, BNIP3 and LC3II were overexpressed, LC3II colocalized with BNIP3 and mitochondria, and mitochondria colocalized with lysosomes.111 These results indicated that inhibition of NLRP3 inflammasome activation up-regulated mitophagy and protected RTECs from iohexol-induced damage.111 In the cisplatin-induced AKI model, the expression of IL-1β, caspase-1, and NLRP3 was enhanced, and RTECs exhibited mitochondrial dysfunction.112 SS31 treatment could effectively inhibit mtROS, improve damage, and reduce the expression of NLRP3, IL-1β, and caspase-1.112 IL-22 can significantly attenuate the accumulation of ROS in acute pancreatitis (APAP) renal injury both in vitro and in vivo, improve mitochondrial dysfunction, downregulate APAP-induced NLRP3 inflammasome activation and release of mature IL-1β, and ultimately reduce APAP-induced RTECs death.113 Consequently, the mitophagy-NLRP3 inflammasome pathway may become a target for AKI therapy.
CKD
Proteinuria and hypertension are two important factors in the progression of CKD. In 2014, Zhuang et al of Nanjing University found that proteinuria may result in the activation of the NLRP3 inflammasome and the marked abnormalities of mitochondria, manifesting as functional and morphological impairments.80 This mitochondrial abnormality could be reversed by marked blockade of the NLRP3/caspase-1 signaling pathway.80 These studies suggested that the NLRP3 inflammasome/caspase-1/mitochondrial axis contributed to albumin-induced tubular injury. Another study found that albumin stimulation could increase the production of mtROS and the expression levels of NLRP3, IL-1β, and IL-18 in cultured HK2 cells, whereas the ROS scavenger N-acetyl-l-cysteine (NAC) inhibited the expression of NLRP3, IL-1β, and IL-18.114 In aldosterone-treated mice kidney tissue, mtROS production, mitochondrial dysfunction, and NLRP3 inflammasome activation were increased. In vivo, the application of mitochondria-targeted antioxidants could significantly improve renal function and mitochondrial dysfunction, reduce NLRP3 inflammasome activation, and alleviate renal injury.115 In hyperuricemia-induced CKD, PINK1/parkin-mediated mitophagy was impaired, thereby activating NLRP3, resulting in increased production of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α, leading to RTECs injury.116 Mitophagy was also activated in angiotensin II-treated RTECs; PHB2 knockdown decreased mitophagy levels and increased cell death, whereas PHB2 overexpression enhanced mitochondrial dysfunction and inhibited NLRP3 inflammasome activation.117 This suggested that PHB2-mediated mitophagy via the inflammasome may be a future therapeutic target for CKD.117 Florfenidone, a new pyridone, reduced renal fibrosis by inhibiting the overproduction of mtROS and activation of the NLRP3 inflammasome in activated peritoneal-derived macrophages and RTECs.118 Compound K significantly reduced mitochondrial damage, mtROS generation, and mtDNA release in LPS-activated macrophages, inhibited NLRP3 inflammasome activation, and alleviated renal interstitial tubule lesions.119 Based on the preceding, it can be observed that targeting mitophagy-mtROS-inflammasome may be an effective strategy to prevent CKD.
DKD
In kidney tissues of DKD patients and db/db mice, mtROS was overproduced and the expression of NLRP3/IL-1β was increased.120 Intraperitoneal injection of the mitoquinone mesylate (mitoQ, a kind of mitochondria-targeted antioxidant) reversed the increased expression of NLRP3/IL-1β in db/db mice.120 Thioredoxin-interacting protein (TXNIP) can bind to NLRP3 and trigger its activation after dissociating from thioredoxin (TRX) in response to different stimuli.121 MitoQ treatment of HK-2 cells under a high glucose environment could inhibit the dissociation of TRX and TXNIP, thereby blocking the interaction between TXNIP and NLRP3. Then, NLRP3 inflammasome activation and IL-1β maturation were inhibited.120 TXNIP siRNA pretreatment enhanced the effect of mitoQ, while monosodium urate (MSU, a kind of cellular inflammation inducer) and TRX siRNA pretreatment eradicated the effect of mitoQ.120 These results suggested that mtROS-TXNIP/NLRP3/IL-1β axis activation is associated with tubular oxidative damage in DKD. One study found that under high glucose conditions, overexpression of OPTN increased the co-staining of LC3II and OMM 20 translocase in mice RTECs cultured in vitro, while enhancing mitophagy, and significantly reducing the expression of NLRP3, the cleavage levels of caspase-1 and IL-1β, and the releases of IL-1β and IL-18.122 Furthermore, the high glucose (HG)-induced NLRP3 inflammasome activation could be recovered by mitochondrial division inhibitor 1(Mdivi-1) by blocking the overexpression of OPTN.122 This suggested that OPTN inhibited the activation of the NLRP3 inflammasome by promoting mitophagy and ameliorated RTECs injury in DKD. As a class B scavenger receptor, CD36, inhibited mitochondrial fatty acid oxidation (FAO) in RTECs of DKD, stimulated the production of mtROS, and then activated the NLRP3 inflammasome.123 Mitochondrial antioxidants could reduce the activation effect of CD36 overexpression on NLRP3. Inhibition of CD36 could increase intracellular adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK, a key molecule in the regulation of biological energy metabolism) activity and mitochondrial FAO level, while inhibiting the expression of NLRP3 and IL-1 mRNA, thereby decreasing tubulointerstitial inflammation and RTECs apoptosis in DKD.123 Thus, due to the abundance of mitochondria in renal tubular epithelial cells, the mitophagy-inflammasome axis plays a crucial role in the RTECs injury of DKD. Moreover, Ding et al found that the mitophagy-inflammasome axis may be involved in the mechanism of podocyte injury in DKD.124 Icariin could promote mitophagy in STZ-treated rats and HG-treated mice podocyte cell line (MPC-5) to inhibit NLRP3 inflammasome activation via the Nrf2 pathway.124 Several studies have shown that the role of the mitophagy-inflammasome axis in DKD requires further exploration.
The Potential Interaction Between Mitophagy and Inflammasomes in Kidney Diseases
Currently, the NLPR3 inflammasome is most studied in the kidney disease involved in mitophagy. Nevertheless, in other disease models, we found that the NLRC4, AIM2, and NLRX1 inflammasomes may be involved in mitophagy. Induction of myocardial infarction in a mouse model of type 2 diabetes (T2 DM) may result in impaired mitophagy and increased mtDNA release in the peri-infarct area of the left ventricle.125 This resulted in hyperactivation of AIM2 and NLRC4 inflammasomes and caspase-1 in cardiomyocytes and macrophages in the infarcted area, cardiomyocyte death, and increased IL-18 secretion.125 It is suggested that mitophagy disorder may contribute to T2DM-induced myocardial infarction through AIM2 and NLRC4 inflammasome. In an Alzheimer’s disease (AD) mouse model of, the mitophagy receptor OPTN was found to inhibit the activity of caspase-1 and IL-1β in microglia by inhibiting the expression of AIM2 and ASC.126 Then it blocked the action of Aβ oligomer (Aβo) to activate the AIM2 inflammasome, thereby reducing inflammation.126 NLRX1 is the only NLR family member with a mitochondrial targeting sequence, which contains an LIR and directly binds to LC3 via LIR.127 NLRX1 and its LIR sequence are required for the induction of mitophagy by Listeria.127 Both NLRX1 deficiency and the use of mitophagy inhibitors increased mtROS production, thereby inhibiting Listeria survival.127 Mechanistically, Listeria and the virulence factor listeriolysin O (LLO) induce oligomerization of NLRX1, which promotes the binding of its LIR sequence to LC3 and induces mitophagy.127 In a study of intestinal IR injury, it was found that the expression of NLRX1 was significantly down-regulated after intestinal IR injury.128 Intestinal IR injury and mitochondrial dysfunction were ameliorated in rats with overexpressing NLRX1.128 Functional studies showed that IR injury decreased the expression of NLRX1 and promoted the phosphorylation of FUNDC1.128 Immunoprecipitation studies showed that phosphorylated FUNDC1 could not combine to the mitophagy related signaling protein non-neuronal SNAP25-like protein homolog 1 and 2 (NIPSNAP 1 and 2) on the OMM of damaged mitochondria, thereby failing to initiate mitophagy, leading to accumulation of damaged mitochondria and apoptosis of epithelial cells.128 These results suggested that NLRX1 may be involved in cell damage by regulating mitophagy. The AIM2, NLRC4 and NLRX1 inflammasome in the above study are all involved in the pathogenesis of kidney diseases, so whether they interact with mitophagy through different signaling pathways to participate in the pathogenesis of kidney disease is worthy of further exploration.
Conclusion and Future Prospects
Mitophagy belongs to specific autophagy, which can selectively clean up damaged mitochondria, engulf them into autophagosomes, fuse with lysosomes, and then start to cleave, and finally maintain the stability of the intracellular environment. Mitophagy disorder is indispensable in cell damage and disease progression. Therefore, studies on mitophagy can not only provide more information on cellular regulation, but also further reveal the relationship between mitophagy and disease. In recent years, more and more attention has been paid to the study of inflammasomes in the progression of diseases. At present, the role of the interaction between mitophagy and inflammasomes activation in diseases has become a hot spot in the field of medical biology. There is increasing evidence that the interaction between mitophagy and inflammasomes is closely related to the pathogenesis of kidney disease. However, the current studies are mainly focused on mitophagy and NLRP3 inflammasome, while studies on the interaction between AIM2, NLRC4, NLRX1 inflammasome and mitophagy in kidney diseases is still less. We need to conduct more in-depth studies in order to discover more pathogenic mechanisms of kidney diseases and formulate more effective treatment strategies.
Acknowledgments
Thanks to the support of Translational Medicine Center, the First Affiliated Hospital of Zhengzhou University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work was supported by the National Natural Science Foundation of China (grant no. U1904134).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Yang J, Xiao L, Sun L. Research progress of mitochondria, endoplasmic reticulum and their tight junctions in the pathogenesis of diabetic nephropathy. Antioxidants. 2021;101(10):750–754.
2. Emma F, Montini G, Parikh SM, et al. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12(5):267–280.
3. Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29(5):1261–1273. doi:10.1016/j.celrep.2019.09.050
4. Granata S, Dalla GA, Tomei P, et al. Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab. 2015;12:49.
5. Parikh SM, Yang Y, He L, et al. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol. 2015;35(1):108–119. doi:10.1016/j.semnephrol.2015.01.011
6. Ahmad AA, Draves SO, Rosca M. Mitochondria in diabetic kidney disease. Cells. 2021;10(11):11. doi:10.3390/cells10112945
7. Dai W, Lu H, Chen Y, et al. The loss of mitochondrial quality control in diabetic kidney disease. Front Cell Dev Biol. 2021;9:706832. doi:10.3389/fcell.2021.706832
8. Tang C, Cai J, Yin X-M, et al. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17(5):299–318. doi:10.1038/s41581-020-00369-0
9. Panigrahi DP, Praharaj PP, Bhol CS, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020;66:45–58. doi:10.1016/j.semcancer.2019.07.015
10. Kesharwani R, Sarmah D, Kaur H, et al. Interplay between mitophagy and inflammasomes in neurological disorders. ACS Chem Neurosci. 2019;10(5):2195–2208. doi:10.1021/acschemneuro.9b00117
11. Yuk JM, Silwal P, Jo EK. Inflammasome and mitophagy connection in health and disease. Int J Mol Sci. 2020;21(13):4714. doi:10.3390/ijms21134714
12. Mcbride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16(14):R551–R560. doi:10.1016/j.cub.2006.06.054
13. Guan X, Qian Y, Shen Y, et al. Autophagy protects renal tubular cells against ischemia / reperfusion injury in a time-dependent manner. Cell Physiol Biochem. 2015;36(1):285–298. doi:10.1159/000374071
14. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. doi:10.1038/s41556-018-0176-2
15. Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73(4):775–795. doi:10.1007/s00018-015-2087-8
16. Doblado L, Lueck C, Rey C, et al. Mitophagy in human diseases. Int J Mol Sci. 2021;22(8):8. doi:10.3390/ijms22083903
17. Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–1737. doi:10.1093/hmg/ddr048
18. Matsuda N. Phospho-ubiquitin: upending the PINK-Parkin-ubiquitin cascade. J Biochem. 2016;159(4):379–385. doi:10.1093/jb/mvv125
19. Zachari M, Ktistakis NT. Mammalian mitophagosome formation: a focus on the early signals and steps. Front Cell Dev Biol. 2020;8:171. doi:10.3389/fcell.2020.00171
20. Bhatia D, Choi ME. The emerging role of mitophagy in kidney diseases. J Life Sci. 2019;1(3):13–22.
21. Zuo Z, Jing K, Wu H, et al. Mechanisms and functions of mitophagy and potential roles in renal disease. Front Physiol. 2020;11:935. doi:10.3389/fphys.2020.00935
22. Lin Q, Ni Z. 线粒体自噬在肾脏疾病中的研究进展 [Research advances in mitophagy in kidney disease]. Chin J Blood Purif. 2019;18(03):197–200. Chinese.
23. Choi GE, Lee HJ, Chae CW, et al. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat Commun. 2021;12(1):487. doi:10.1038/s41467-020-20679-y
24. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16(1):3–17. doi:10.1080/15548627.2019.1603547
25. Wu W, Tian W, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15(5):566–575. doi:10.1002/embr.201438501
26. Killackey SA, Philpott DJ, Girardin SE. Mitophagy pathways in health and disease. J Cell Biol. 2020;219(11). doi:10.1083/jcb.202004029
27. Bhujabal Z, Birgisdottir AB, Sjottem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–961. doi:10.15252/embr.201643147
28. Yan C, Gong L, Chen L, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2020;16(3):419–434. doi:10.1080/15548627.2019.1628520
29. Kagan VE, Jiang J, Huang Z, et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016;23(7):1140–1151. doi:10.1038/cdd.2015.160
30. Sun N, Yun J, Liu J, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–696. doi:10.1016/j.molcel.2015.10.009
31. Bakula D, Scheibye-Knudsen M. MitophAging: mitophagy in aging and disease. Front Cell Dev Biol. 2020;8:239. doi:10.3389/fcell.2020.00239
32. Zhang Y, Zhang M, Zhu W, et al. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biol. 2020;28:101365. doi:10.1016/j.redox.2019.101365
33. Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017;36(10):1315–1327. doi:10.1038/onc.2016.302
34. Zhou H, He L, Xu G, et al. Mitophagy in cardiovascular disease. Clin Chim Acta. 2020;507:210–218. doi:10.1016/j.cca.2020.04.033
35. Zhao C, Chen Z, Xu X, et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp Cell Res. 2017;350(2):390–397. doi:10.1016/j.yexcr.2016.12.015
36. Tang C, Han H, Yan M, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14(5):880–897. doi:10.1080/15548627.2017.1405880
37. Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. doi:10.1016/j.redox.2020.101767
38. Fu ZJ, Wang ZY, Xu L, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020;36:101671. doi:10.1016/j.redox.2020.101671
39. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309–326. doi:10.1146/annurev-physiol-022516-034227
40. Aparicio-Trejo OE, Tapia E, Molina-Jijon E, et al. Curcumin prevents mitochondrial dynamics disturbances in early 5/6 nephrectomy: relation to oxidative stress and mitochondrial bioenergetics. Biofactors. 2017;43(2):293–310. doi:10.1002/biof.1338
41. Ma N, Wei Z, Hu J, et al. Farrerol ameliorated cisplatin-induced chronic kidney disease through mitophagy induction via Nrf2/PINK1 pathway. Front Pharmacol. 2021;12:768700. doi:10.3389/fphar.2021.768700
42. Ding XQ, Jian TY, Gai YN, et al. Chicoric acid attenuated renal tubular injury in HFD-induced chronic kidney disease mice through the promotion of mitophagy via the Nrf2/PINK/Parkin pathway. J Agric Food Chem. 2022;70(9):2923–2935. doi:10.1021/acs.jafc.1c07795
43. Jin L, Yu B, Liu G, et al. Mitophagy induced by UMI-77 preserves mitochondrial fitness in renal tubular epithelial cells and alleviates renal fibrosis. FASEB J. 2022;36(6):e22342. doi:10.1096/fj.202200199RR
44. Liu B, Cao Y, Wang D, et al. Zhen-Wu-Tang induced mitophagy to protect mitochondrial function in chronic glomerulonephritis via PI3K/AKT/mTOR and AMPK pathways. Front Pharmacol. 2021;12:777670. doi:10.3389/fphar.2021.777670
45. Wu X, Li J, Wang S, et al. 2-undecanone protects against fine particle-induced kidney inflammation via inducing mitophagy. J Agric Food Chem. 2021;69(17):5206–5215. doi:10.1021/acs.jafc.1c01305
46. Yi X, Guo T, Zhang M, et al. 线粒体自噬紊乱在糖尿病肾病发病机制中的作用研究进展 [Research progress in role of mitophagy dysfunction in pathogenesis of diabetic nephropathy]. Chin J Pharmacol Toxicol. 2019;33(11):994–999. Chinese.
47. Smith MA, Covington MD, Schnellmann RG. Loss of calpain 10 causes mitochondrial dysfunction during chronic hyperglycemia. Arch Biochem Biophys. 2012;523(2):161–168. doi:10.1016/j.abb.2012.04.020
48. Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. 2014;171(8):1917–1942. doi:10.1111/bph.12503
49. Xiao L, Xu X, Zhang F, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi:10.1016/j.redox.2016.12.022
50. Yu L, Wang Y, Guo YH, et al. HIF-1alpha alleviates high-glucose-induced renal tubular cell injury by promoting Parkin/PINK1-mediated mitophagy. Front Med. 2021;8:803874. doi:10.3389/fmed.2021.803874
51. Yi X, Yan W, Guo T, et al. Erythropoietin mitigates diabetic nephropathy by restoring PINK1/Parkin-mediated mitophagy. Front Pharmacol. 2022;13:883057. doi:10.3389/fphar.2022.883057
52. Liu L, Bai F, Song H, et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol. 2022;50:102260. doi:10.1016/j.redox.2022.102260
53. Zheng T, Wang HY, Chen Y, et al. Src activation aggravates podocyte injury in diabetic nephropathy via suppression of FUNDC1-mediated mitophagy. Front Pharmacol. 2022;13:897046. doi:10.3389/fphar.2022.897046
54. Song YM, Lee WK, Lee YH, et al. Metformin restores Parkin-mediated mitophagy, suppressed by cytosolic p53. Int J Mol Sci. 2016;17(1):122. doi:10.3390/ijms17010122
55. Sun J, Zhu H, Wang X, et al. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J Endocrinol. 2019. doi:10.1530/JOE-18-0578
56. Tian Y, Guo H, Miao X, et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. 2020;11(5):319. doi:10.1038/s41419-020-2547-4
57. Connor TM, Hoer S, Mallett A, et al. Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genet. 2017;13(3):e1006620. doi:10.1371/journal.pgen.1006620
58. Viering D, Schlingmann KP, Hureaux M, et al. Gitelman-like syndrome caused by pathogenic variants in mtDNA. J Am Soc Nephrol. 2022;33(2):305–325. doi:10.1681/ASN.2021050596
59. Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol. 2018;9:1283. doi:10.3389/fimmu.2018.01283
60. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21(3):193–201. doi:10.1016/j.molmed.2014.11.008
61. Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol. 2019;15(8):501–520. doi:10.1038/s41581-019-0158-z
62. Zhang WJ, Chen SJ, Zhou SC, et al. Inflammasomes and fibrosis. Front Immunol. 2021;12:643149. doi:10.3389/fimmu.2021.643149
63. Chi K, Geng X, Liu C, et al. Research progress on the role of inflammasomes in kidney disease. Mediators Inflamm. 2020;2020:8032797. doi:10.1155/2020/8032797
64. Wang X. NOD1,NOD2和NLRP3炎症小体与牙髓炎 [NOD1, NOD2 and NLRP3 inflammasome in pulpitis]. J Clin Pathol Res. 2017;37(04):849–854. Chinese.
65. Corridoni D, Arseneau KO, Cifone MG, et al. The dual role of nod-like receptors in mucosal innate immunity and chronic intestinal inflammation. Front Immunol. 2014;5:317. doi:10.3389/fimmu.2014.00317
66. Schmid-Burgk JL, Gaidt MM, Schmidt T, et al. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. 2015;45(10):2911–2917. doi:10.1002/eji.201545523
67. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16(1):35–50. doi:10.1038/nri.2015.8
68. Vigano E, Diamond CE, Spreafico R, et al. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. 2015;6(1):8761. doi:10.1038/ncomms9761
69. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
70. Tummers B, Mari L, Guy CS, et al. Caspase-8-dependent inflammatory responses are controlled by its adaptor, FADD, and necroptosis. Immunity. 2020;52(6):994–1006. doi:10.1016/j.immuni.2020.04.010
71. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516
72. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. 2018;15(12):721–737. doi:10.1038/s41575-018-0054-1
73. Pinkerton JW, Kim RY, Robertson A, et al. Inflammasomes in the lung. Mol Immunol. 2017;86:44–55. doi:10.1016/j.molimm.2017.01.014
74. van Hout G, Bosch L. The inflammasomes in cardiovascular disease. Exp Suppl. 2018;108:9–40. doi:10.1007/978-3-319-89390-7_2
75. Silvis M, Demkes EJ, Fiolet A, et al. Immunomodulation of the NLRP3 inflammasome in atherosclerosis, coronary artery disease, and acute myocardial infarction. J Cardiovasc Transl Res. 2021;14(1):23–34. doi:10.1007/s12265-020-10049-w
76. Pulskens WP, Butter LM, Teske GJ, et al. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One. 2014;9(1):e85775. doi:10.1371/journal.pone.0085775
77. Correa-Costa M, Braga TT, Semedo P, et al. Pivotal role of toll-like receptors 2 and 4, its adaptor molecule MyD88, and inflammasome complex in experimental tubule-interstitial nephritis. PLoS One. 2011;6(12):e29004. doi:10.1371/journal.pone.0029004
78. Bakker PJ, Butter LM, Kors L, et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014;85(5):1112–1122. doi:10.1038/ki.2013.503
79. Zhang C, Boini KM, Xia M, et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. 2012;60(1):154–162. doi:10.1161/HYPERTENSIONAHA.111.189688
80. Zhuang Y, Ding G, Zhao M, et al. NLRP3 inflammasome mediates albumin-induced renal tubular injury through impaired mitochondrial function. J Biol Chem. 2014;289(36):25101–25111. doi:10.1074/jbc.M114.578260
81. Wen Y, Liu Y, Tang T, et al. NLRP3 inflammasome activation is involved in Ang II-induced kidney damage via mitochondrial dysfunction. Oncotarget. 2016;7(34):54290–54302. doi:10.18632/oncotarget.11091
82. Shahzad K, Bock F, Dong W, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87(1):74–84. doi:10.1038/ki.2014.271
83. Huang G, Bao J, Shao X, et al. Inhibiting pannexin-1 alleviates sepsis-induced acute kidney injury via decreasing NLRP3 inflammasome activation and cell apoptosis. Life Sci. 2020;254:117791. doi:10.1016/j.lfs.2020.117791
84. Andersen K, Eltrich N, Lichtnekert J, et al. The NLRP3/ASC inflammasome promotes T-cell-dependent immune complex glomerulonephritis by canonical and noncanonical mechanisms. Kidney Int. 2014;86(5):965–978. doi:10.1038/ki.2014.161
85. Wu D, Ai L, Sun Y, et al. Role of NLRP3 inflammasome in lupus nephritis and therapeutic targeting by phytochemicals. Front Pharmacol. 2021;12:621300. doi:10.3389/fphar.2021.621300
86. Gong W, Mao S, Yu J, et al. NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am J Physiol Renal Physiol. 2016;310(10):F1081–F1088. doi:10.1152/ajprenal.00534.2015
87. Chivero ET, Guo ML, Periyasamy P, et al. HIV-1 tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci. 2017;37(13):3599–3609. doi:10.1523/JNEUROSCI.3045-16.2017
88. Mulay SR, Anders HJ. Crystal nephropathies: mechanisms of crystal-induced kidney injury. Nat Rev Nephrol. 2017;13(4):226–240. doi:10.1038/nrneph.2017.10
89. Prencipe G, Caiello I, Cherqui S, et al. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol. 2014;25(6):1163–1169. doi:10.1681/ASN.2013060653
90. Sharma BR, Karki R, Kanneganti TD. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur J Immunol. 2019;49(11):1998–2011. doi:10.1002/eji.201848070
91. Du L, Wang X, Chen S, et al. The AIM2 inflammasome: a novel biomarker and target in cardiovascular disease. Pharmacol Res. 2022;186:106533. doi:10.1016/j.phrs.2022.106533
92. Baatarjav C, Komada T, Karasawa T, et al. dsDNA-induced AIM2 pyroptosis halts aberrant inflammation during rhabdomyolysis-induced acute kidney injury. Cell Death Differ. 2022. doi:10.1038/s41418-022-01033-9
93. Zhen J, Zhang L, Pan J, et al. AIM2 mediates inflammation-associated renal damage in hepatitis B virus-associated glomerulonephritis by regulating caspase-1, IL-1beta, and IL-18. Mediators Inflamm. 2014;2014:190860. doi:10.1155/2014/190860
94. Komada T, Chung H, Lau A, et al. Macrophage uptake of necrotic cell DNA activates the AIM2 inflammasome to regulate a proinflammatory phenotype in CKD. J Am Soc Nephrol. 2018;29(4):1165–1181. doi:10.1681/ASN.2017080863
95. Majidpoor J, Khezri Z, Rostamzadeh P, et al. The expressions of NLRP1, NLRP3, and AIM2 inflammasome complexes in the contusive spinal cord injury rat model and their responses to hormonal therapy. Cell Tissue Res. 2020;381(3):397–410. doi:10.1007/s00441-020-03250-5
96. Chung H, Komada T, Lau A, et al. AIM2 suppresses inflammation and epithelial cell proliferation during glomerulonephritis. J Immunol. 2021;207(11):2799–2812. doi:10.4049/jimmunol.2100483
97. Franchon MTN, Ziroldo LJ, Duarte GL, et al. AIM2 as a putative target in acute kidney graft rejection. Front Immunol. 2022;13:839359. doi:10.3389/fimmu.2022.839359
98. Yuan F, Kolb R, Pandey G, Li W, Sun L, Liu F, Sutterwala F S, Liu Y, Zhang W. (2016). Involvement of the NLRC4-Inflammasome in Diabetic Nephropathy. PLoS One, 11(10), e0164135 10.1371/journal.pone.0164135
99. Li Y, Yu W, Xiong H and Yuan F. (2022). Circ_0000181 regulates miR-667-5p/NLRC4 axis to promote pyroptosis progression in diabetic nephropathy. Sci Rep, 12(1), 10.1038/s41598-022-15607-7
100. Wang Y, Gou R, Yu L, et al. Activation of the NLRC4 inflammasome in renal tubular epithelial cell injury in diabetic nephropathy. Exp Ther Med. 2021;22(2):814. doi:10.3892/etm.2021.10246
101. Meissner TB, Li A, Biswas A, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. 2010;107(31):13794–13799. doi:10.1073/pnas.1008684107
102. Li Q, Wang Z, Zhang Y, et al. NLRC5 deficiency protects against acute kidney injury in mice by mediating carcinoembryonic antigen-related cell adhesion molecule 1 signaling. Kidney Int. 2018;94(3):551–566. doi:10.1016/j.kint.2018.02.031
103. Luan P, Zhuang J, Zou J, et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. 2018;32(2):1070–1084. doi:10.1096/fj.201700511RR
104. Soares J, Fernandes FP, Patente TA, et al. Gain-of-function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin Immunol. 2018;187:46–49. doi:10.1016/j.clim.2017.10.003
105. Valino-Rivas L, Cuarental L, Nunez G, et al. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol Dial Transplant. 2020;35(4):587–598. doi:10.1093/ndt/gfz169
106. Song L, Gao C, Xue T, et al. Characterization and expression analysis of mitochondrial localization molecule: NOD-like receptor X1 (Nlrx1) in mucosal tissues of turbot (Scophthalmus maximus) following bacterial challenge. Dev Comp Immunol. 2021;116:103944. doi:10.1016/j.dci.2020.103944
107. Moore CB, Bergstralh DT, Duncan JA, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451(7178):573–577. doi:10.1038/nature06501
108. Stokman G, Kors L, Bakker PJ, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. 2017;214(8):2405–2420. doi:10.1084/jem.20161031
109. Gao Y, Dai X, Li Y, et al. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med. 2020;18(1):114. doi:10.1186/s12967-020-02283-2
110. Lin Q, Li S, Jiang N, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254. doi:10.1016/j.redox.2019.101254
111. Lin Q, Li S, Jiang N, et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2021;17(10):2975–2990. doi:10.1080/15548627.2020.1848971
112. Yang S-K, Han Y-C, He J-R, et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed Pharmacother. 2020;130:110521. doi:10.1016/j.biopha.2020.110521
113. Shen Y, Jin X, Chen W, et al. Interleukin-22 ameliorated Acetaminophen-induced kidney injury by inhibiting mitochondrial dysfunction and inflammatory responses. Appl Microbiol Biotechnol. 2020;104(13):5889–5898. doi:10.1007/s00253-020-10638-4
114. Liu D, Xu M, Ding LH, et al. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: a novel mechanism of albumin-induced tubulointerstitial inflammation. Int J Biochem Cell Biol. 2014;57:7–19. doi:10.1016/j.biocel.2014.09.018
115. Ding W, Liu T, Bi X, et al. Mitochondria-targeted antioxidant mito-tempo protects against aldosterone-induced renal injury in vivo. Cell Physiol Biochem. 2017;44(2):741–750. doi:10.1159/000485287
116. Zhang C, Song Y, Chen L, et al. Urolithin A attenuates hyperuricemic nephropathy in fructose-fed mice by impairing STING-NLRP3 axis-mediated inflammatory response via restoration of Parkin-dependent mitophagy. Front Pharmacol. 2022;13:907209. doi:10.3389/fphar.2022.907209
117. Xu Y, Wang J, Xu W, et al. Prohibitin 2-mediated mitophagy attenuates renal tubular epithelial cells injury by regulating mitochondrial dysfunction and NLRP3 inflammasome activation. Am J Physiol Renal Physiol. 2019;316(2):F396–F407. doi:10.1152/ajprenal.00420.2018
118. Liao X, Jiang Y, Dai Q, et al. Fluorofenidone attenuates renal fibrosis by inhibiting the mtROS-NLRP3 pathway in a murine model of folic acid nephropathy. Biochem Biophys Res Commun. 2021;534:694–701. doi:10.1016/j.bbrc.2020.11.017
119. Hsu WH, Hua KF, Tuan LH, et al. Compound K inhibits priming and mitochondria-associated activating signals of NLRP3 inflammasome in renal tubulointerstitial lesions. Nephrol Dial Transplant. 2020;35(1):74–85. doi:10.1093/ndt/gfz073
120. Han Y, Xu X, Tang C, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018;16:32–46. doi:10.1016/j.redox.2018.02.013
121. Lerner AG, Upton JP, Praveen PV, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250–264. doi:10.1016/j.cmet.2012.07.007
122. Chen K, Feng L, Hu W, et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 2019;33(3):4571–4585. doi:10.1096/fj.201801749RRR
123. Hou Y, Wang Q, Han B, et al. CD36 promotes NLRP3 inflammasome activation via the mtROS pathway in renal tubular epithelial cells of diabetic kidneys. Cell Death Dis. 2021;12(6):523. doi:10.1038/s41419-021-03813-6
124. Ding X, Zhao H, Qiao C. Icariin protects podocytes from NLRP3 activation by Sesn2-induced mitophagy through the Keap1-Nrf2/HO-1 axis in diabetic nephropathy. Phytomedicine. 2022;99:154005. doi:10.1016/j.phymed.2022.154005
125. Durga DT, Babu M, Makinen P, et al. Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am J Pathol. 2017;187(12):2659–2673. doi:10.1016/j.ajpath.2017.08.023
126. Cao LL, Guan PP, Zhang SQ, et al. Downregulating expression of OPTN elevates neuroinflammation via AIM2 inflammasome- and RIPK1-activating mechanisms in APP/PS1 transgenic mice. J Neuroinflammation. 2021;18(1):281. doi:10.1186/s12974-021-02327-4
127. Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446. doi:10.1038/s41590-019-0324-2
128. Li S, Zhou Y, Gu X, et al. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021;54(3):e12986. doi:10.1111/cpr.12986
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Disrupted Alpha-Ketoglutarate Homeostasis: Understanding Kidney Diseases from the View of Metabolism and Beyond
Guo L, Chen S, Ou L, Li S, Ye ZN, Liu HF
Diabetes, Metabolic Syndrome and Obesity 2022, 15:1961-1974
Published Date: 27 June 2022
Clinical Efficacy, Safety, Tolerability, and Real-World Data of Patiromer for the Treatment of Hyperkalemia
Colbert G, Sannapaneni S, Lerma EV
Drug, Healthcare and Patient Safety 2022, 14:87-96
Published Date: 14 July 2022
Mesenchymal Stem Cell-Derived Small Extracellular Vesicles: A Novel Approach for Kidney Disease Treatment
Lu Y, Wang L, Zhang M, Chen Z
International Journal of Nanomedicine 2022, 17:3603-3618
Published Date: 13 August 2022
Risk Factors for Left Ventricular Hypertrophy in Patients with Diabetic Kidney Disease: A Multi-Center Study
Wang X, Zhu D, Peng L, Gao Y, Li X
International Journal of General Medicine 2023, 16:1705-1712
Published Date: 8 May 2023
Impact of Acute Kidney Injury, Co-Existing with and without Chronic Kidney Disease on the Short‐Term Adverse Outcomes Following Atherosclerotic Cardiovascular Disease Events in Patients with Diabetes
Chou CL, Zheng CM, Chiu HW, Tsou LLC, Kao PF, Hsu YH, Lin CL, Sung LC
Journal of Multidisciplinary Healthcare 2025, 18:2019-2037
Published Date: 12 April 2025