Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Mitophagy Disequilibrium, a Prominent Pathological Mechanism in Metabolic Heart Diseases
Authors Li Y ![]() , Zheng N
, Zheng N ![]() , Ding X
, Ding X ![]()
Received 30 August 2021
Accepted for publication 9 November 2021
Published 25 November 2021 Volume 2021:14 Pages 4631—4640
DOI https://doi.org/10.2147/DMSO.S336882
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Yunhao Li,1,* Ningning Zheng,2,* Xudong Ding3
1The First Clinical College, China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Department of Pathophysiology, College of Basic Medical Science, China Medical University, Shenyang, Liaoning, People’s Republic of China; 3Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xudong Ding
Department of Anesthesiology, Shengjing Hospital of China Medical University, No. 36 Sanhao Street, Shenyang, 110004, People’s Republic of China
Tel +8618940257698
Email [email protected]
Abstract: With overall food intake among the general population as high as ever, metabolic syndrome (MetS) has become a global epidemic and is responsible for many serious life-threatening diseases, especially heart failure. In multiple metabolic disorders, maintaining a dynamic balance of mitochondrial number and function is necessary to prevent the overproduction of reactive oxygen species (ROS), which has been proved to be one of the important mechanisms of cardiomyocyte injury due to the mismatching of oxygen consumption and mitochondrial population and finally to heart failure. Mitophagy is a process that eliminates damaged or redundant mitochondria. It is mediated by a series of signaling molecules, including PINK, parkin, BINP3, FUNDC1, CTSD, Drp1, Rab9 and mTOR. Meanwhile, increasing evidence also showed that the interaction between ferroptosis and mitophagy interfered with mitochondrial homeostasis. This review will focus on these essential molecules and pathways of mitophagy and cell homeostasis affected by hypoxia and other stimuli in metabolic heart diseases.
Keywords: mitophagy, metabolic heart diseases, metabolic syndrome, PTEN induced putative kinase, PINK, parkin, Bcl-2/E1B19kDa-interacting protein, BNIP3, FUN14 domain-containing protein 1, FUNDC1, ferroptosis
Introduction
Metabolic syndrome (MetS) refers to a group of cardiovascular risk factors that include insulin resistance, obesity, dyslipidemia, increased glucose intolerance, and increased blood pressure.1 MetS, caused by high caloric intake and lack of exercise, has become a global epidemic.2 The combination of two or more of these disorders can dramatically increase the risk for potentially life-threatening diseases, especially cardiovascular disease. Disorders of heart glucose and lipid metabolism inevitably lead to abnormal energy production and oxygen usage. It has been demonstrated that cells face severe oxidative stress in metabolic heart diseases. Mitochondria are considered the power-house of the cell and are involved in essential cellular functions, including ATP production, regulation of apoptosis, and especially the production and elimination of ROS. Mitochondrial abnormalities become the central pathogenesis of heart failure.3 Disorders of mitochondrial electron transfer typically lead to the accumulation of ROS, impaired mitochondrial oxidative phosphorylation, and imbalances in mitochondrial biogenesis, which cause irreversible damage. Glucose and lipid metabolism can be improved in MetS by reducing oxidative damage and facilitating mitochondria-network-dependent activities.4 A dynamic balance of mitochondrial population and function is essential for heart cell survival.5 Mitophagy, a process for clearing damaged or redundant mitochondria, is a primary method of protection from anoxic cellular stimuli in MetS heart. Under hypoxic conditions, reducing the number of mitochondria can prevent the production of ROS caused by the mismatching of oxygen consumption and mitochondrial population. Mitophagy was confirmed to be inhibited with several forms, like phosphatidylinositol 3-kinases and mTOR, in the hearts of diabetic patients.6,7 Furthermore, metabolic-stress-mediated vascular endothelial injury can be prevented by promoting mitophagy in obese and diabetic mice.8 Selective mitochondrial degradation was previously thought to be a random event, though recent evidence suggests that mitophagy is a targeted process.9
Mitophagy can be categorized into three forms, basal mitophagy, stress-induced mitophagy and programmed mitophagy, which are triggered by different conditions and manifested as dependence on different regulatory factors, like parkin or NIP3-like protein X (NIX).
The process of mitophagy relies on a series of steps: selection, tagging, phagosome formation, transportation, and lysosome attachment. Once targeted, vesicles are used to isolate and transport mitochondria. This process is mainly mediated by receptors, such as Bcl-2/E1B19kDa-interacting protein (BNIP3), NIX, FUN14 domain-containing protein 1 (FUNDC1), and microtubule-associated protein 1A/1B light chain 3 (LC3B; a binding protein that is located in the outer membrane of vesicle fragments), which are recruited to form phagosomes.10 Toshiro Saito el al11 carried researches in mice, which suggests that when suffering from myocardial ischemia, autophagy (mitophagy) is mediated by a protein consisting of unc-51 like kinase 1 (Ulk1), dynamin-1-like protein (Drp1) and some other receptors. Mitophagy process can be negatively regulated by mammalian target of rapamycin (mTOR) along with other key proteins, such as Beclin.
Here, we review current understandings and advances about these essential molecules and pathways of mitophagy in metabolic heart diseases (Figure 1).
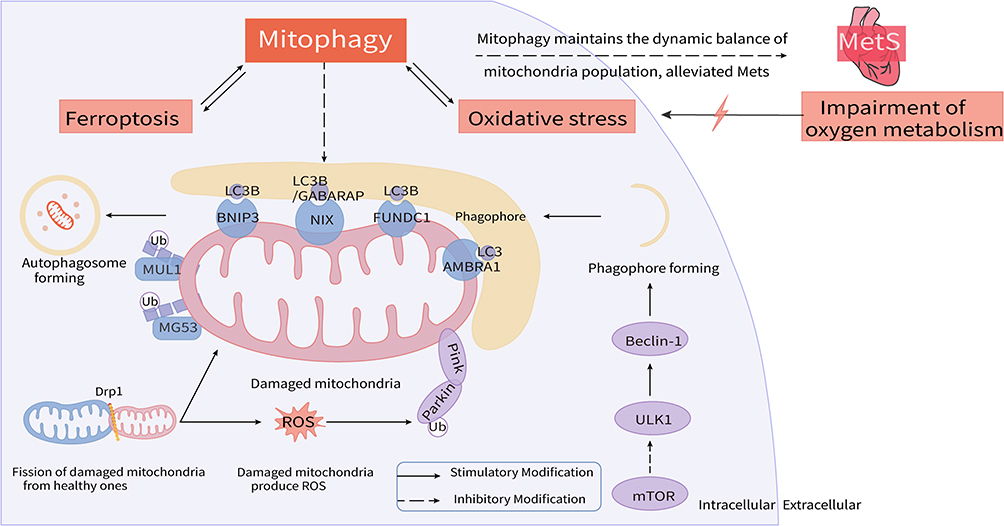 |
Figure 1 Mitophagy disequilibrium acts as a prominent pathological mechanism in metabolic heart diseases. Hypoxia and subsequent metabolic disorders contribute to oxidative stress injury and abnormal mitophagy in heart. Drp1 induces the fission of damaged mitochondria (in pink) from healthy mitochondria (in blue); Parkin and PINK response to the stimuli of ROS produced by damaged mitochondria and induce mitophagy; Receptors locating at the OMM, like BNIP3, NIX, FUNDC1 and AMBRA1, bind with LC3 proteins to mediate autophagosome formation; Ubiquitin ligases of MUL1 and MG53 recruit more phagophore to assist in mitophagy implement; mTOR plays a negative effect to regulate phagophore formation. In addition, ferroptosis has recently been found to be involved in and interact with mitophagy. Abnormalities of above molecules and pathways of mitophagy become critical nodes of heart dysfunction occurrence and development. Restoring mitochondrial homeostasis by modulating these targets may be an important strategy worthy of in-depth study and then accelerate the development of novel therapies for mitochondria-related metabolic heart diseases. |
PINK1 and Parkin
PTEN induced putative kinase 1(PINK1) is a Ser/Thr kinase encoded by PINK1 gene and consists of 581 amino acids. Parkin is a cytoplasmic protein with 465 amino acid residues. Under normal conditions, PINK1 is transported to the mitochondrial matrix and degraded. However, when mitochondria are damaged or redundant, damaged proteins located at the outer membrane of mitochondria are tagged by PINK1 or parkin—or various ubiquitin ligases such as mitochondrial ubiquitin ligase activator of NF-kB1 (MUL1), which is triggered by metabolic stress—thus promoting the degradation of damaged mitochondria by binding with lysosomes.12,13 PINK1 is thought to protect myocardial cells from stress-induced mitochondrial dysfunction. The binding of parkin and depolarized mitochondria caused by activated PINK1 induces mitophagy.14,15 It has been demonstrated in pancreatic beta-cells from patients with diabetes that PINK1/Parkin-mediated mitophagy has a protective role.16 The PINK1 pathway also plays an important role in MetS, where mitophagy is induced in response to environmental changes. Wu et al8 previously demonstrated that PINK1 and parkin pathways are activated by metabolic stress, protecting mitochondrial integrality in obese and diabetic mice. The absence of parkin partially inhibits mitochondrial autophagy, and increases lipid accumulation and exacerbates diastolic dysfunction.17 Inhibition of Sirt3-Foxo3A-Parkin signal-mediated down-regulation of mitochondrial autophagy may play an important role in the occurrence and development of diabetic cardiomyopathy in STZ (streptozocin)-induced diabetes mellitus accompanying cardiac dysfunction mouse model.18 Although the parkin/PINK1 axis is the main regulator of mitophagy, the process can also take place independent of parkin, suggesting that parkin is not unique.19 However, it is likely that PINK1 and parkin may have value in the treatment of metabolic heart failure, a hypothesis that requires further investigation. Contrary to the current opinion about the central role of parkin in the ubiquitination of mitochondrial proteins, in the Drp1KO heart, mitochondrial ubiquitination is independent of parkin, and the absence of Drp1 and parkin aggravates heart defects.20 Several ubiquitin ligases have been demonstrated as key players in the targeting process for mitophagy. These ubiquitin ligases include MUL1 and Mitsugumin 53 (MG53). MUL1 is a resident ubiquitin E3 ligase that is inserted into the mitochondrial outer membrane, which can stabilize Drp1.21,22 Several studies demonstrated that MUL1 not only plays a role as a ubiquitin ligase but also as a mitophagy receptor by binding with E3 ligases or E2 box enzymes.23,24 As a ubiquitin ligase, the complex MUL1-ubiquitin-conjugating enzyme E2 E3 (Ube2E3) associates with gamma aminobutyric acid receptor-associated protein (GABARAP, a protein of LC3 family) in HeLa cells. As a mitophagy receptor,25 it has the ability to recruit the mitophagy machinery directly into mature neurons.26,27 MG53, primarily expressed in striated muscle, is another ubiquitin ligase related to pathogenesis of MetS. Studies regarding the role of MG53 in MetS yielded conflicting results. Several studies have shown that overexpression of MG53 triggers muscle insulin resistance and MetS.28–30 Cardiac-specific transgenic expression of MG53 induces severe diabetic cardiomyopathy in mice, accompanied by insulin resistance, increased lipid accumulation, myocardial hypertrophy, fibrosis, and cardiac dysfunction,31 whereas others claimed that it has therapeutic applications, such as protecting injured mitochondria and inducing muscle regeneration.32,33 Mitophagy ubiquitin ligases appear to have complex functions, and a single ligase may not modulate every form of mitophagy, but the process of mitophagy and the formation of phagosomes are likely mediated and regulated by networks of these proteins.
BNIP3 and NIX/BNIP3L
BNIP3 is fundamental to the removal of mitochondria during the maturation of erythrocytes34,35 and may play a role in tumor suppression and heart disease.36,37 BNIP3L and NIX, two types of apoptotic proteins, function by binding to LC3B (microtubule-associated protein 1A/1B light chain 3B)38–40 or to GABARAP.41,42 Both BNIP3 and BNIP3L/NIX interact with LC3 and p62 during hypoxic stress,39–41 forming the LC3-p62 complex, which is partially involved in the process of mitophagy. Like many Bcl-2 family proteins, BNIP3 is also involved with mitochondrial outer membrane permeability.43 Upregulation of BNIP3 leads to a series of reactions, like decreased mitochondrial potential and increased ROS species.44 Moreover, BNIP3 can not only induce cell death but also aid in cell survival. Although few studies have been conducted on the clinical role of BNIP in heart disorder, it represents a potential target for therapeutic intervention requiring further investigation.
FUNDC1
FUNDC1 is another mitophagy receptor that has been intensely studied and is associated with LC3B. It can be positively or negatively modulated by phosphorylation, similar to BNIP3 and NIX/BNIP3L.19 In its basal state, FUNDC1 can be inhibited by phosphorylation, while during severe hypoxia, it can mediate mitophagy by binding with LC3B.45–48 FUNDC1 deficiency often leads to mitochondrial quality control disorders and mitochondrial redox imbalance,45 which, can lead to cell death in myocardial infarction.49 Moreover, FUNDC1 deficiency can also regulate ferroptosis leading to cardiac remodeling and dysfunction.50 Thus, maintaining the function of FUNDC1 plays an essential role in the treatment of metabolic heart diseases.51 Tong et al illustrated that mitophagy serves an essential role in maintaining cardiac function and that preserving or activating mitophagy can protect against HFD-induced diabetic cardiomyopathy.17 A similar method was used by Wu et al52 and Ren et al53 in a high fat diet-induced FUNDC1-deficient mouse model and FUNDC1−/− mice to demonstrate that FUNDC1 deficiency can promote obesity, insulin resistance, MetS, myocardial remodeling, and even cell death due to mitochondrial quality control dysregulation, suggesting a vital role for FUNDC1 in cardiac function in obese patients with MetS. However, consistent with previous findings, loss of FUNDC1 merely impaired hypoxia-induced mitophagy but had no influence on autophagy as a whole.54 These findings show that FUNDC1-mediated mitophagy is a key regulator of mitochondrial function, especially in response to hypoxia-mediated mitochondrial damage, which often occurs in cardiac metabolic disorders.
CTSD
Cathepsin D (CTSD), a major lysosomal protease that induces autophagosome removal and increases autophagic flux, is involved in heart failure. It has been shown that CTSD can prevent myocardial remodeling, which frequently occurs in patients with hypertension, a type of long-term MetS.55–59 Wu et al60 demonstrated in CTSD+/+ but not CTSD± mice that decreased autophagy can lead to restrictive cardiomyopathy, and protracted MetS can also cause heart failure.60,61 Overall, studies have shown that up-regulation of CTSD in the myocardium is an adaptive adjustment that protects the heart, at least in part by promoting mitophagy against cardiac remodeling and heart failure. Although CTSD has a clear role in several heart diseases associated with chronic MetS, a direct association with MetS has not yet been shown. Because of its function during autophagy,62–68 alterations in CTSD expression in the myocardium may potentially be used as an indicator of autophagic activity.
Drp1
Overexpression of Drp1 leads to increased mitochondrial number.69 Wada et al70 showed that Drp1 plays an important role during mitophagy in MetS, particularly in patients with diabetes. Smirnova et al71 showed that Drp1 is essential for mitochondrial fission using cells transiently transfected with mutant Drp1, which leads to an increase in the number of mitochondria. Thus, restricting Drp1 can prevent apoptosis and protect myocardial mitochondria from damage.72,73 While in contrast, studies from Xue et al74 argued that reduced oxidative damage and increased ATP generation may be triggered by decreasing cyclin-dependent protein kinase (CDK)-p-Drp1 protein levels in MetS mouse model.
The contradictory phenomenon above may be attributed to the phosphorylation of two different sites in Drp1, and the modification of one of them leads to both activation and inhibition of mitophagy. Failure to remove or antagonize Drp1 affects mitophagy, leading to abnormal ATP production, and may ultimately cause aseptic myocarditis, heart failure, and other disorders.75
mTOR
mTOR is a central modulator that promotes cellular growth and the synthesis of protein as well as regulating mitophagy. It responds to signals from growth factors, nutrients, energy levels, and stress. Experimental data from Zhang et al76 in mice showed that mTOR may interrupt autophagic flux. It regulates autophagy, especially inhibiting autophagy vesicle formation by Beclin.77 mTOR signaling also cross-talks with pathways modulated by branched-chain amino acids, insulin, oxidative stress, and the endoplasmic reticulum.78,79 mTORC1 is essential for adaptive cardiac hypertrophy in response to mechanical overload. And mTORC2 is required to maintain normal cardiac physiology and works for the survival of cardiomyocytes after pressure is overloaded.80 The progress of myocardial hypertrophy is often accompanied by excessive oxidative stress and chronic activation of mTOR. Empagliflozin application significantly alleviated myocardial hypertrophy and some other heart disorders in diet-induced obesity mice by activating AMPK and inhibiting Akt and mTOR.81 Studies have shown that vitamin D deficiency is closely related to metabolic cardiovascular diseases, and the protective effect of 1,25-(OH)2-D3, the active form of vitamin D, against cardiac hypertrophy and fibrosis in diabetic cardiomyopathy is also exerted by regulating the PARP1/SIRT1/mTOR signaling pathway.82 The mechanism of high protein diet-induced atherosclerosis and cardiovascular risk in mouse model was partially constituted by mTORC1-dependent inhibition of mitophagy, accumulation of dysfunctional mitochondria, and mitochondrial apoptosis.83 Okuno et al84 used mouse liver tumors with metabolic syndrome characteristics, found that mTOR activation suppressed mitophagy and apoptosis, which may cause tumorigenesis. The clinical trials carried out by De Matteis et al85 in 70 hepatocellular carcinoma (HCC) patients with MetS illustrated a close relationship between high levels of mTOR and poor prognosis, and listed mTOR as a potential target for the treatment of advanced-stage HCC. Khamzina et al86 demonstrated a relationship between mTOR activity and obesity-linked insulin resistance. Hyperglycemia inhibits the formation of cardiac autophagosomes and autolysosomes by regulating the mTOR-ULK1 signal. Antagonists of mTOR were previously used to ameliorate obesity in DahlS. Z-Leprfa /Leprfa (DS/obese) rats, a model of MetS.87 Alterations of autophagy activity also affect mitochondrial biosynthesis in diabetic heart. Maintaining mitophagy by inhibiting mTOR has become one of the most important strategies for the therapy of metabolic cardiac dysfunctions. What deserves our attention is that the role of mTOR in myocardial ischemia/reperfusion (MI/R) injury under diabetes has dual, cardiotoxic and cardioprotective effects. mTORC1 can be activated and increase the susceptibility to MI/R injury in obese and diabetic animals. While the cardioprotective effects mediated by mTOR are partly dependent on the activation of mTORC2, they are conducive to the survival of cardiomyocytes against MI/R injury and chronic ischemic remodeling.80
Ferroptosis
Myocardial cell death is the decompensated manifestation and the terminal stage of cardiac dysfunction. Loss of cardiomyocytes can be induced by ferroptosis. The study by Chen et al88 confirmed that overly activated autophagy and ferroptosis promote heart failure by inducing the loss of cardiomyocytes. Regulating ferroptosis is an important guarantee for maintaining the normal function of mitophagy receptors, such as FUNDC1. Pei’s study just indicates that FUNDC1 deficiency can activate cardiac remodeling and dysfunction through the regulation of ferroptosis.50 Mitophagy and ferroptosis can impact each other through lipid peroxidation and the parkin-dependent pathway.89,90 Some studies have also indicated ferroptosis as an autophagic cell death process, especially in metabolic heart diseases.91,92 Lee et al93 found that ferroptosis can be triggered by obesity or increased caloric intake, and inhibited by energy stress. Furthermore, ferroptosis is prominent in obese patients as well as those with insulin resistance and other diseases like rhabdomyolysis and radiation syndrome. Also, some heart disorders in diabetes mellitus have close relationships with ferroptosis and mitophagy. The study of Li et al94 showed that ferroptosis aggravates diabetes myocardial ischemia/reperfusion (I/R) injuries and was more serious in rat diabetes mellitus+ I/R or cell high-glucose+ hypoxia reoxygenation models. And the study carried out by Wang et al95 in diabetes mice further proved that diabetes aggravates myocardial I/R injuries through an AMPK-dependent pathway, which leads to ferroptosis. Ferroptosis is a key factor leading to I/R injury, and its chelator protects the heart against ex vivo I/R injuries, which was proved in mice.96,97 Obesity and type 2 diabetes mellitus-related cardiomyopathy can be contributed by palmitic acid (PA)-induced myocardial injury, and an underlying mechanism is that PA induces ferroptosis by decreasing the levels of ferroptosis inhibitive proteins, which was proved in a recent study by Wang et al.98 Ferroptosis can not only cause cell death but iron can trigger a variety of protective cellular cascades. Iron around the infarct zone is an important factor for reversing left ventricle remodeling, which is important for preventing heart failure after myocardial infarction.96 The management of iron in mitochondria may contribute to an effective treatment of myocardial I/R injury.99 Excessive production of ROS in diabetes mellitus enhances ferroptosis100 and mitophagy, the cell-protective progress against ROS. In return, mitophagy inhibits ROS production and thus inhibits ferroptosis, playing a protective role. While to some extent, mitophagy is triggered by energy stress,93 a withholder to ferroptosis can alleviate energy stress itself. The different functions of mitophagy under different conditions need to be further explored. Research on cardiomyocyte survival regulated by mitophagy and ferroptosis may be a potential tendency in the foreseeing future.
Other Regulators
Damaged mitochondria are transported to and fused with phagosomes and lysosomes, a process that is mediated by Beclin-1 and other molecules. Beclin-1 represents a “protein platform”, providing a framework to regulate the increase of mitophagy flux after myocardial fibrillation and is indispensable during autophagy.101,102 Liang et al103 found that, in yeast, Beclin-1 may facilitate mitochondrial fusion through binding with mammalian HOPS and UVRAG.104 Additionally, the LC3B-I/LC3B-II ratio and the total levels of LC3B-II can be used as markers of enhanced mitophagy in the myocardium after fibrillation.105 LC3 drives autophagosome vesicles into phagocytose fragments of mitochondria and subsequently fuses with lysosomes through LC3-I and LC3-II.106–108 Based on a short half-life, LC3B-II can be lysed and converted back to its original isoform, LC3-I.104 Recent studies in ob/ob mice found that an autophagy enhancer can promote the conversion from LC3-I to LC3-II without inhibiting mTOR, which suggests that autophagy enhancement may represent a therapeutic strategy to treat MetS.109 Autophagy and Beclin-1 regulator 1 (AMBRA1) is another receptor associated with Bcl2 that can induce mitophagy. AMBRA1 participates in the formation of Beclin-class-III-PI3K, a complex that protects against ROS dopaminergic cell death, as shown in SH-SY5Y cells.110 Unfortunately, although molecules such as Beclin, AMBRA1, and LC3B play key roles in mitophagy, there are few studies that have elucidated their roles in MetS.
Conclusion
The remodeling of cellular energy metabolism is a prominent manifestation of heart failure in MetS and finding the central regulatory points in such a complex unbalanced state is key to exploring its pathogenesis. Mitochondria are the central hub of the cellular metabolic network. The final stages of all carbohydrate, lipid, and protein metabolism are oxidized in the mitochondria through the tricarboxylic acid cycle. In addition to energy metabolism, mitochondria are also important organelles for regulating oxidative stress, autophagy, and apoptosis. Thus, mitochondria have become a reliable target to explore the central pathogenesis of multiple metabolic abnormalities, especially heart failure. Mitophagy is responsible for maintaining a healthy population of mitochondria and preventing the mismatching of oxygen and mitochondria number, which may lead to hypoxia and trigger the occurrence and development of heart disease. Mitochondrial quality control largely depends on the stability of mitophagy. Sufficient research evidence, as shown above from in vivo, in vitro, animals and clinical, indicates that mitophagy disequilibrium interferes with basic heart metabolic pattern. Regulators like parkin, BINP3 and FUNDC1 were shown to play remarkable roles in the regulation of mitophagy. Restoring mitochondrial homeostasis by modulating these key factors becomes an important strategy worthy of in-depth study. Drug development by targeting these candidates may accelerate to prevent and treat the mitochondria-related heart diseases.
Data Sharing Statement
All authors confirmed that all data and materials as well as software application support their published claims and comply with field standards.
Ethics Approval
The manuscript does not contain clinical studies or patient data.
Acknowledgments
We would like to thank Congcong Ma, Department of Pathophysiology, College of Basic Medical Science, China Medical University, who provided the revised version of Figure 1 (which is Yunhao Li’s original) to help us illustrate our abstract more clearly. Yunhao Li and Ningning Zheng are co-first authors of this study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Funding
The work was supported by the National Natural Science Foundation of China (FUND#81800763), the Natural Science Foundation of Liaoning Province of China (FUND#20180551076) and the Natural Science Foundation of Liaoning Province of China (FUND#2013021046).
Disclosure
All authors declare no competing interests.
References
1. Li A, Zheng N, Ding X. Mitochondrial abnormalities: a hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress. Heart Fail Rev. 2021. doi:10.1007/s10741-021-10109-6
2. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. doi:10.1007/s11906-018-0812-z
3. Morciano G, Vitto VAM, Bouhamida E, Giorgi C, Pinton P. Mitochondrial bioenergetics and dynamism in the failing heart. Life (Basel). 2021;11(5):436.
4. Cao K, Xu J, Zou X, et al. Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free Radic Biol Med. 2014;67:396–407. doi:10.1016/j.freeradbiomed.2013.11.029
5. Morciano G, Patergnani S, Pedriali G, et al. Impairment of mitophagy and autophagy accompanies calcific aortic valve stenosis favoring cell death and the severity of disease. Cardiovasc Res. 2021. doi:10.1093/cvr/cvab267
6. Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–1541. doi:10.1038/sj.cdd.4401728
7. Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med. 2006;27(5–6):411–425. doi:10.1016/j.mam.2006.08.002
8. Wu W, Xu H, Wang Z, et al. PINK1-parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One. 2015;10(7):e0132499. doi:10.1371/journal.pone.0132499
9. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462(2):245–253. doi:10.1016/j.abb.2007.03.034
10. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–795. doi:10.1038/cr.2014.75
11. Saito T, Nah J, Oka SI, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129(2):802–819. doi:10.1172/JCI122035
12. Moskal N, Riccio V, Bashkurov M, et al. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat Commun. 2020;11(1):88. doi:10.1038/s41467-019-13781-3
13. Roperto S, De Falco F, Perillo A, Catoi C, Roperto F. Mitophagy mediated by BNIP3 and BNIP3L/NIX in urothelial cells of the urinary bladder of cattle harbouring bovine papillomavirus infection. Vet Microbiol. 2019;236:108396. doi:10.1016/j.vetmic.2019.108396
14. Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi:10.1371/journal.pbio.1000298
15. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013;200(2):163–172. doi:10.1083/jcb.201210111
16. Hoshino A, Ariyoshi M, Okawa Y, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc Natl Acad Sci U S A. 2014;111(8):3116–3121. doi:10.1073/pnas.1318951111
17. Tong M, Saito T, Zhai P, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124(9):1360–1371. doi:10.1161/CIRCRESAHA.118.314607
18. Yu W, Gao B, Li N, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):1973–1983. doi:10.1016/j.bbadis.2016.10.021
19. Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without Parkin. Trends Cell Biol. 2018;28(11):882–895. doi:10.1016/j.tcb.2018.07.004
20. Kageyama Y, Hoshijima M, Seo K, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33(23):2798–2813. doi:10.15252/embj.201488658
21. Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10(7):748–754. doi:10.1038/embor.2009.86
22. Yun J, Puri R, Yang H, et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3:e01958. doi:10.7554/eLife.01958
23. Bae S, Kim SY, Jung JH, et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res. 2012;22(5):873–885. doi:10.1038/cr.2012.38
24. Li W, Bengtson MH, Ulbrich A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008;3(1):e1487. doi:10.1371/journal.pone.0001487
25. Ambivero CT, Cilenti L, Main S, Zervos AS. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal. 2014;26(12):2921–2929. doi:10.1016/j.cellsig.2014.09.004
26. Puri R, Cheng XT, Lin MY, Huang N, Sheng ZH. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. 2019;10(1):3645. doi:10.1038/s41467-019-11636-5
27. Puri R, Cheng XT, Lin MY, Huang N, Sheng ZH. Defending stressed mitochondria: uncovering the role of MUL1 in suppressing neuronal mitophagy. Autophagy. 2020;16(1):176–178. doi:10.1080/15548627.2019.1687216
28. Song R, Peng W, Zhang Y, et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature. 2013;494(7437):375–379. doi:10.1038/nature11834
29. Hu X, Xiao RP. MG53 and disordered metabolism in striated muscle. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5Pt B):1984–1990. doi:10.1016/j.bbadis.2017.10.013
30. Wu HK, Zhang Y, Cao CM, et al. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation. 2019;139(7):901–914. doi:10.1161/CIRCULATIONAHA.118.037216
31. Liu F, Song R, Feng Y, et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation. 2015;131(9):795–804. doi:10.1161/CIRCULATIONAHA.114.012285
32. Ma H, Liu J, Bian Z, et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS One. 2015;10(5):e0124128. doi:10.1371/journal.pone.0124128
33. Benissan-Messan DZ, Zhu H, Zhong W, Tan T, Ma J, Lee PHU. Multi-cellular functions of MG53 in muscle calcium signaling and regeneration. Front Physiol. 2020;11:583393. doi:10.3389/fphys.2020.583393
34. Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi:10.1038/nature07006
35. Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–19505. doi:10.1073/pnas.0708818104
36. Jung J, Zhang Y, Celiku O, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 2019;79(20):5218–5232. doi:10.1158/0008-5472.CAN-19-0198
37. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. doi:10.1038/cdd.2009.16
38. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287(23):19094–19104. doi:10.1074/jbc.M111.322933
39. Rogov VV, Suzuki H, Marinkovic M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7(1):1131. doi:10.1038/s41598-017-01258-6
40. Zhu Y, Massen S, Terenzio M, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288(2):1099–1113. doi:10.1074/jbc.M112.399345
41. Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi:10.1038/embor.2009.256
42. Kuhikar R, Khan N, Philip J, Melinkeri S, Kale V, Limaye L. Transforming growth factor beta1 accelerates and enhances in vitro red blood cell formation from hematopoietic stem cells by stimulating mitophagy. Stem Cell Res Ther. 2020;11(1):71. doi:10.1186/s13287-020-01603-z
43. Sassone J, Colciago C, Marchi P, et al. Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3). Cell Death Dis. 2010;1:e7. doi:10.1038/cddis.2009.6
44. Liu KE, Frazier WA, Zhang J. Phosphorylation of the BNIP3 C-terminus inhibits mitochondrial damage and cell death without blocking autophagy. PLoS One. 2015;10(6):e0129667. doi:10.1371/journal.pone.0129667
45. Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi:10.1038/ncb2422
46. Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–377. doi:10.1016/j.molcel.2014.02.034
47. Chen Z, Siraj S, Liu L, Chen Q. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy. 2017;13(7):1244–1245. doi:10.1080/15548627.2017.1310789
48. Chen Z, Liu L, Cheng Q, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18(3):495–509. doi:10.15252/embr.201643309
49. Lampert MA, Orogo AM, Najor RH, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15(7):1182–1198. doi:10.1080/15548627.2019.1580095
50. Pei Z, Liu Y, Liu S, et al. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism. 2021;122:154840. doi:10.1016/j.metabol.2021.154840
51. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415. doi:10.1016/j.redox.2019.101415
52. Wu H, Wang Y, Li W, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019;15(11):1882–1898. doi:10.1080/15548627.2019.1596482
53. Ren J, Sun M, Zhou H, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. 2020;6(38). doi:10.1126/sciadv.abc8561
54. Wu W, Lin C, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35(13):1368–1384. doi:10.15252/embj.201593102
55. Nadruz W. Myocardial remodeling in hypertension. J Hum Hypertens. 2015;29(1):1–6. doi:10.1038/jhh.2014.36
56. Poncelas M, Inserte J, Vilardosa U, et al. Obesity induced by high fat diet attenuates postinfarct myocardial remodeling and dysfunction in adult B6D2F1 mice. J Mol Cell Cardiol. 2015;84:154–161. doi:10.1016/j.yjmcc.2015.04.023
57. Saftig P, Hetman M, Schmahl W, et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14(15):3599–3608. doi:10.1002/j.1460-2075.1995.tb00029.x
58. Wu P, Yuan X, Li F, et al. Myocardial upregulation of cathepsin D by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ Heart Fail. 2017;10(7). doi:10.1161/CIRCHEARTFAILURE.117.004044
59. VanWagner LB, Wilcox JE, Colangelo LA, et al. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: a population-based study. Hepatology. 2015;62(3):773–783. doi:10.1002/hep.27869
60. McHugh K, DeVore AD, Wu J, et al. Heart failure with preserved ejection fraction and diabetes: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(5):602–611. doi:10.1016/j.jacc.2018.11.033
61. Bozkurt B, Aguilar D, Deswal A, et al.. Outcomes, contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation. 2016;134(23):e535–e578. doi:10.1161/CIR.0000000000000450
62. Conus S, Perozzo R, Reinheckel T, et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med. 2008;205(3):685–698. doi:10.1084/jem.20072152
63. Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU. Cathepsin D primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem. 2012;287(25):21142–21151. doi:10.1074/jbc.M111.306399
64. Laurent-Matha V, Huesgen PF, Masson O, et al. Proteolysis of cystatin C by cathepsin D in the breast cancer microenvironment. FASEB J. 2012;26(12):5172–5181. doi:10.1096/fj.12-205229
65. Vashishta A, Ohri SS, Vetvicka V. Pleiotropic effects of cathepsin D. Endocr Metab Immune Disord Drug Targets. 2009;9(4):385–391. doi:10.2174/187153009789839174
66. Di Domenico F, Tramutola A, Perluigi M. Cathepsin D as a therapeutic target in Alzheimer’s disease. Expert Opin Ther Targets. 2016;20(12):1393–1395. doi:10.1080/14728222.2016.1252334
67. Koch S, Scifo E, Rokka A, et al. Cathepsin D deficiency induces cytoskeletal changes and affects cell migration pathways in the brain. Neurobiol Dis. 2013;50:107–119. doi:10.1016/j.nbd.2012.10.004
68. Comitato A, Sanges D, Rossi A, Humphries MM, Marigo V. Activation of Bax in three models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55(6):3555–3562. doi:10.1167/iovs.14-13917
69. Zhao C, Chen Z, Qi J, et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget. 2017;8(13):20988–21000. doi:10.18632/oncotarget.15470
70. Wada J, Nakatsuka A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med Okayama. 2016;70(3):151–158. doi:10.18926/AMO/54413
71. Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143(2):351–358. doi:10.1083/jcb.143.2.351
72. Dominic EA, Ramezani A, Anker SD, Verma M, Mehta N, Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 2014;100(8):611–618. doi:10.1136/heartjnl-2013-304657
73. Karbowski M, Lee YJ, Gaume B, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159(6):931–938. doi:10.1083/jcb.200209124
74. Xue RQ, Yu XJ, Zhao M, et al. Pyridostigmine alleviates cardiac dysfunction via improving mitochondrial cristae shape in a mouse model of metabolic syndrome. Free Radic Biol Med. 2019;134:119–132. doi:10.1016/j.freeradbiomed.2019.01.011
75. Shirakabe A, Zhai P, Ikeda Y, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133(13):1249–1263. doi:10.1161/CIRCULATIONAHA.115.020502
76. Zhang Y, Xu X, Ren J. MTOR overactivation and interrupted autophagy flux in obese hearts: a dicey assembly? Autophagy. 2013;9(6):939–941. doi:10.4161/auto.24398
77. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi:10.1016/j.cell.2010.01.028
78. Bloomgarden Z. Diabetes and branched-chain amino acids: what is the link? J Diabetes. 2018;10(5):350–352. doi:10.1111/1753-0407.12645
79. Wang J, Yang X, Zhang J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic beta cells. Cell Signal. 2016;28(8):1099–1104. doi:10.1016/j.cellsig.2016.05.007
80. Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122(3):489–505. doi:10.1161/CIRCRESAHA.117.311147
81. Sun X, Han F, Lu Q, et al. Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes. 2020;69(6):1292–1305. doi:10.2337/db19-0991
82. Qu H, Lin K, Wang H, et al. 1,25(OH) 2D3 improves cardiac dysfunction, hypertrophy, and fibrosis through PARP1/SIRT1/mTOR-related mechanisms in type 1 diabetes. Mol Nutr Food Res. 2017;61(5):1600338. doi:10.1002/mnfr.201600338
83. Zhang X, Sergin I, Evans TD, et al. High-protein diets increase cardiovascular risk by activating macrophage mTOR to suppress mitophagy. Nat Metab. 2020;2(1):110–125. doi:10.1038/s42255-019-0162-4
84. Okuno T, Kakehashi A, Ishii N, Fujioka M, Gi M, Wanibuchi H. mTOR activation in liver tumors is associated with metabolic syndrome and non-alcoholic steatohepatitis in both mouse models and humans. Cancers (Basel). 2018;10(12):465. doi:10.3390/cancers10120465
85. De Matteis S, Scarpi E, Granato AM, et al. Role of SIRT-3, p-mTOR and HIF-1alpha in hepatocellular carcinoma patients affected by metabolic dysfunctions and in chronic treatment with metformin. Int J Mol Sci. 2019;20(6):1503. doi:10.3390/ijms20061503
86. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146(3):1473–1481. doi:10.1210/en.2004-0921
87. Uchinaka A, Yoneda M, Yamada Y, Murohara T, Nagata K. Effects of mTOR inhibition on cardiac and adipose tissue pathology and glucose metabolism in rats with metabolic syndrome. Pharmacol Res Perspect. 2017;5(4):e00331. doi:10.1002/prp2.331
88. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. 2019;516(1):37–43. doi:10.1016/j.bbrc.2019.06.015
89. Basit F, van Oppen LM, Schockel L, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3):e2716. doi:10.1038/cddis.2017.133
90. Liu M, Fan Y, Li D, et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 2021;15:2084–2105. doi:10.1002/1878-0261.12936
91. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
92. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
93. Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234. doi:10.1038/s41556-020-0461-8
94. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39(2):210–225. doi:10.1089/dna.2019.5097
95. Wang C, Zhu L, Yuan W, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. 2020;24(12):6670–6679. doi:10.1111/jcmm.15318
96. Baba Y, Higa JK, Shimada BK, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314(3):H659–H668. doi:10.1152/ajpheart.00452.2017
97. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
98. Wang N, Ma H, Li J, et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol. 2021;150:65–76. doi:10.1016/j.yjmcc.2020.10.010
99. Ravingerova T, Kindernay L, Bartekova M, et al. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int J Mol Sci. 2020;21(21):7889. doi:10.3390/ijms21217889
100. Ge ZD, Lian Q, Mao X, Xia Z. Current status and challenges of NRF2 as a potential therapeutic target for diabetic cardiomyopathy. Int Heart J. 2019;60(3):512–520. doi:10.1536/ihj.18-476
101. Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends Mol Med. 2009;15(3):129–138. doi:10.1016/j.molmed.2009.01.002
102. Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy. 2009;5(4):534–536. doi:10.4161/auto.5.4.8062
103. Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. doi:10.1038/ncb1740
104. Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16(7):984–990. doi:10.1038/cdd.2009.54
105. Meyer G, Czompa A, Reboul C, et al. The cellular autophagy markers Beclin-1 and LC3B-II are increased during reperfusion in fibrillated mouse hearts. Curr Pharm Des. 2013;19(39):6912–6918. doi:10.2174/138161281939131127122510
106. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi:10.1093/emboj/19.21.5720
107. Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279(46):47704–47710. doi:10.1074/jbc.M407016200
108. Wu J, Dang Y, Su W, et al. Molecular cloning and characterization of rat LC3A and LC3B–two novel markers of autophagosome. Biochem Biophys Res Commun. 2006;339(1):437–442. doi:10.1016/j.bbrc.2005.10.211
109. Lim H, Lim YM, Kim KH, et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat Commun. 2018;9(1):1438. doi:10.1038/s41467-018-03939-w
110. Di Rita A, D’Acunzo P, Simula L, Campello S, Strappazzon F, Cecconi F. AMBRA1-mediated mitophagy counteracts oxidative stress and apoptosis induced by neurotoxicity in human neuroblastoma SH-SY5Y cells. Front Cell Neurosci. 2018;12:92. doi:10.3389/fncel.2018.00092
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.