Back to Journals » ImmunoTargets and Therapy » Volume 14
Mitophagy: A Potential Therapeutic Target for Tuberculosis Immunotherapy
Authors Gao S ![]() , Yang Z, Yu J
, Yang Z, Yu J ![]() , Zhang F, Tang S
, Zhang F, Tang S ![]() , Pang Y
, Pang Y ![]()
Received 8 February 2025
Accepted for publication 11 July 2025
Published 22 July 2025 Volume 2025:14 Pages 773—786
DOI https://doi.org/10.2147/ITT.S518628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Siyu Gao,1,* Zeliang Yang,1,* Jiajia Yu,2 Fuzhen Zhang,1,3 Shenjie Tang,4 Yu Pang1
1Department of Bacteriology and Immunology, Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis & Thoracic Tumor Research Institute, Beijing, 101149, People’s Republic of China; 2Department of Infectious Diseases and Clinical Microbiology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, 100020, People’s Republic of China; 3Department of Epidemiology, School of Public Health, Cheeloo College of Medicine, Shandong University, Jinan, 250012, People’s Republic of China; 4Clinical Center on Tuberculosis, Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis & Thoracic Tumor Research Institute, Beijing, 101149, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yu Pang, Department of Bacteriology and Immunology, Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis & Thoracic Tumor Research Institute, No. 9 Beiguan Street, Tongzhou District, Beijing, 101149, People’s Republic of China, Tel +86 13810098209, Email [email protected] Shenjie Tang, Clinical Center on Tuberculosis, Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis & Thoracic Tumor Research Institute, No. 9 Beiguan Street, Tongzhou District, Beijing, 101149, People’s Republic of China, Tel +86 13621028338, Email [email protected]
Abstract: Mitophagy serves as a cytoprotective mechanism that is essential for eliminating dysfunctional or superfluous mitochondria, thereby fine-tuning mitochondrial quantity and maintaining cellular homeostasis. Recent studies underscore the critical role of mitophagy in determining the fate and function of host cells infected by Mycobacterium tuberculosis. The successful pathogen strategically integrates into the host’s mitochondrial network, manipulating processes such as apoptosis, metabolic reprogramming, mitochondrial fusion and fission, and reactive oxygen species production. Therefore, understanding those mechanisms is critical for the advancements of host-directed therapies against tuberculosis. This study offers a comprehensive overview of the interplay between Mycobacterium tuberculosis and mitophagy, emphasizing the associated signaling pathways and potential therapeutic targets involved in mitophagy in Mycobacterium tuberculosis infection. Activating mitophagy in infected host cells represents a promising avenue for improving therapeutic outcomes against tuberculosis. This review aims to summarize potential research direction for agents targeting induction of mitophagy. Notably, evidence suggests that BNIP3/NIX-mediated mitophagy may serve as a potential therapeutic target.
Keywords: mitophagy, Mycobacterium tuberculosis, tuberculosis, immunotherapy
Background
Tuberculosis (TB), a disease triggered by Mycobacterium tuberculosis (Mtb), mainly affects the lungs but can also invade other areas of the body. TB continues to be a significant global health concern and is the second leading cause of death due to a single infectious source, following COVID-19. According to the World Health Organization Global TB Report of 2023, approximately 10.6 million new TB cases were reported worldwide in 2022 (95% uncertainty interval [UI]: 9.9 million - 11.4 million), with about 1.3 million deaths attributed to TB globally (95% UI: 1.18 million - 1.43 million), exceeding prior years.1 The current TB treatment regimen heavily relies on combination chemotherapy with front-line drugs. However, there are many limitations, including prolonged treatment duration, high-dose medication requirements, complex adverse effects, and poor patient adherence. Additionally, the emergence of drug-resistant Mtb strains exacerbates the treatment burden. Despite the introduction of new antibiotics like bedaquiline, linezolid and pretomanid2–4 for drug-resistant TB, resistance has also emerged rapidly.5 Hence, the profound burden of TB and the rising incidence of drug resistance underscore the urgent need for alternative therapeutic strategies.
In this context, host-directed therapies (HDTs) that aim to reinforce the patient’s own immune defenses have gained considerable interest. Indeed, TB transcends the category of a mere infectious disease, with its progression intricately tied to the regulation by the immune system.6 The occurrence and progression of TB are significantly influenced by the host’s immune state. Upon Mtb infection, the immune system initiates a series of intricate defensive responses to get rid of various intruders. However, Mtb has possessed evasion strategies and manipulate the host’s immune response, which impedes effective pathogen clearance and even allows for reactivation after prolonged dormancy.7 During this process, immune system dysregulation may exacerbate disease progression. The innate immune assumes a dominant position in shielding the host during the initial phase of infection. Mtb is initially recognized by the pattern recognition receptors (PRRs) that belong to the innate immune system, which then initiate a cascade of cellular responses. These responses help the host initiate an effective defense against Mtb, including processes like autophagy and apoptosis.8
Autophagy is an evolutionarily conserved subcellular event with self-protection that satisfies the need of cellular and metabolic homeostasis by engulfment and degradation of dysfunctional organelles, proteins and lipids, as well as the recycling of their constituent.9 It targets endogenous and exogenous substrates, such as invading pathogens by delivering them to lysosomes. Mitochondrial autophagy (Mitophagy), a major type of selective autophagy, was initially presented by Lemasters in 2005.10 Although research on mitophagy is still in the early phases of discovery, mitochondria have been known to be important for regulating cellular energy balance and preventing cell death. They participate in signaling and coordinate the polarization and function of immune cells to enhance antimicrobial defense, as well as mediate appropriate inflammation.11,12
In recent years, mitophagy has become a key research focus in clinical disease pathogenesis.13 According to research, certain pathogens have the potential to initiate mitophagy, either through direct or indirect means, and modulate it through diverse mechanisms, thus impacting the immune response of the host and influencing pathogen clearance. For example, during Pseudomonas aeruginosa (P. aeruginosa) infection, the upregulation of microRNA-302/367 promotes a PHB2-dependent mitophagy pathway, facilitating the clearance of P. aeruginosa and reducing inflammation by downregulating NF-kB.14 Besides, the Edmonston B strain of the measles virus vaccine triggers mitophagy in infected lung cancer cells of the non-small cell type, leading to decreased cytochrome c (CYCS) release and consequently inhibition of the pro-apoptotic cascade. The suppression of apoptosis through mitophagy facilitates viral infection.15 Furthermore, transmissible gastroenteritis virus induces mitophagy and antioxidant responses in porcine intestinal epithelial cells, suppressing oxidative stress and potentially enhancing cell survival and viral replication.16
Like these pathogens, Mtb may also exploit mitophagy to regulate host immune responses, raising the following questions: How does Mtb infection affect the process of mitophagy? What role does mitophagy play in Mtb infection? Can mitophagy provide a new perspective for the treatment of tuberculosis? Given recent interest in mitophagy and HDTs in TB research, it is crucial to explore how Mtb engages mitophagy and whether targeting this process could offer an effective therapeutic strategy. Moreover, unlike broad HDT agents that risk systemic inflammation, mitophagy modulation selectively bolster host defense processes while limiting collateral damage. Here, we discuss the potential role and related signaling pathways involving mitophagy in the pathogenic stages of Mtb infection to date. We also provide an in-depth analysis of potential therapeutic targets in combating Mtb through mitophagy, which may aid in the development of new drugs targeting mitophagy.
Mechanism of Mitochondria and Mitophagy in Normal Cell Physiology
Mitochondria are multifunctional organelles that play a central role in cellular metabolism, acting as hubs for catabolic processes, regulating cell proliferation, and orchestrating apoptosis. Notably, they produce roughly 90% of cellular adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS).17 Beyond energy production, mitochondria also orchestrate signal transduction and cellular responses, thereby encouraging the immune cells’ activation and bolstering antimicrobial defenses. Mounting evidence demonstrates that mitochondria are involved in various processes closely associated with the immune system.12
As the first line of defense against external threats, the innate immune system uses pattern recognition receptors (PRRs) to identify damage-associated molecular patterns (DAMPs) from cellular injury and pathogen-associated molecular patterns (PAMPs) from invading pathogens.18 Mitochondria, believed to be descendants of ancient bacteria, contain their genetic material.19 Mitochondrial DNA (mtDNA) harbors unmethylated CpG motifs and formyl peptides, resembling bacterial features.20 Toll-like receptors 9 (TLR9) which is an intracellular or membrane-bound PRR can detect bacterial or viral DNA by binding to unmethylated CpG motifs, triggering a sequence of pro-inflammatory gene activation.21 Mitochondrial DAMPs, including mtDNA, mitochondrial ROS (mtROS), cardiolipin, CYCS, and mitochondrial Ca2+, are key initiators of innate immune responses, responding to stimuli such as infections, tissue injury, and genetic mutations.18,22
To maintain homeostasis, mitochondria undergo tightly regulated fission, fusion, and mitophagy, dynamically remodeling their morphology, abundance, and distribution to stabilize the intracellular environment and ensure cellular functionality. Mitochondrial fusion promotes energy synthesis during cellular proliferation through OXPHOS,23 whereas fission segregates damaged mitochondria for clearance.24 Together, these activities support mitochondrial quality and balance and immune functions.25,26 Mitophagy initiation is crucial for mitochondria quality control and holds promise as a therapeutic strategy to mitigate cell loss and tissue damage.
By selectively removing damaged mitochondria, mitophagy inhibits DAMPs release, which can trigger inflammatory responses27 and also regulates key physiological processes such as regulating intracellular pH, degrading abnormal proteins, facilitating energy metabolism, managing iron homeostasis, and promoting cell cycle control.8,24,26 For example, in the alveolar region of a murine pneumonia model caused by Staphylococcus aureus, mitophagy is extensively stimulated, enabling pneumocytes to clear and renew damaged mitochondria, and thus promote cell survival.28 The following sections will examine the molecular mechanisms involved in mitophagy and its role in Mtb infection.
Mechanism of Mitochondria and Mitophagy in Mtb Infection
There exists a threshold for the role of mitochondria in maintaining homeostasis, yet once their function becomes compromised, impaired mitophagy may lead to excessive activation of NLRP3 inflammasomes, thereby increasing mortality in septic animals.29 Mitochondrial function is significantly tied to their structural characteristics and distribution, both of which undergo changes following Mtb infection of macrophages.30,31 The virulence of Mtb strains further influences mitochondrial morphology,32 impacting cell survival and the host’s capacity to eradicate Mtb.30 Mtb disrupts mitochondrial function to its advantage by employing multiple virulence factors, many of which have mitochondrial targeting sequences that enable manipulation of the host cell’s mitochondrial machinery, ultimately impairing mitochondrial processes.30 So how does Mtb cause mitochondrial dysfunction? We highlight some interesting findings in this field, focusing on how Mtb infection of macrophages regulates host mitochondrial pathways such as apoptosis, ROS production, mitochondrial dynamics and metabolic reprogramming.
Apoptosis
Mtb infection may induce macrophage death by disrupting mitochondrial function. When the bacterial load in macrophages surpasses a certain threshold, two main types of cell death that are observed include necrosis, which is typified by cellular swelling and rupture, and apoptosis, which preserves cell membrane integrity. This process involves increased lysosomal permeability, leading to the release of hydrolytic enzymes that promote Bax/Bak-independent mitochondrial damage and necrosis, ultimately releasing CYCS and other pro-apoptotic factors.33,34
Avirulent Mtb species have consistently been observed to induce apoptosis more effectively than the pathogenic H37Rv strain in primary cultures, humans and murine cell lines, and bone marrow-derived macrophages. Thus, an interesting explanation is that virulent H37Rv strains are more likely to induce apoptosis, while virulent Mtb strains tend to activate necroptosis, a form of cell death that favors the pathogen and facilitates its spread.35 Furthermore, differentially expressed microRNAs (miRNAs) are present in infected macrophages, capable of specifically targeting the 3′-UTR of apoptotic genes, thereby modulating mitochondrial and death receptor pathways to regulate macrophage apoptosis.36 Moreover, Mtb-associated proteins such as PknG and PtpA may indirectly contribute to the initiation of apoptosis by inhibiting autophagosomes-lysosomes fusion and affecting mitochondrial function and stability.37,38
ROS
Mtb infection leads to the accumulation of mtROS. These ROS also infiltrate the phagosome from adjacent mitochondria, exposing the resident Mtb to oxidative stress.32 Excessive ROS that Mtb cannot effectively neutralize results in the activation of various immune responses, such as exogenous apoptosis and NLRP3 inflammasome activation, which in turn amplifies the local inflammatory response.39 However, other study concluded that NLRP3 activation may occur independently of mtROS.40 Notably, while mtROS by acting as a DAMP contributes to the pathogenesis of Mtb even though it possesses antibacterial properties. Host genetics and various signaling pathways also influence the regulation of mtROS production during host’s response to Mtb infections.41 During the initial phase of Mtb invasion, ROS deficiency results in heightened neutrophil inflammation and elevated levels of IL-1β.42 Additionally, mice deficient in NAD+-dependent protein deacetylase sirtuin 3 shows increased necrosis in bone marrow-derived macrophages (BMDMs) and elevated mtROS, correlating with increased bacterial load and macrophage death.43,44 The C-terminal region of the Mtb outer membrane channel protein CpnT, which encodes the tuberculosis necrotizing toxin (TNT), possesses NAD+ glycohydrolase activity. Even though certain ROS produced in the course of Mtb infection operates independently of TNT, removing the CpnT gene reduces ROS levels and prevents cell death upon infection.45 Some Mtb antigens including the 38 kDa antigen and ESAT-6 are implicated in ROS generation that induces macrophage apoptosis46,47 (Figure 1A). Increased ROS levels serve as an important antimicrobial effector, inhibiting Mtb replication and dissemination. However, Mtb can modulate host ROS production to evade immune responses.
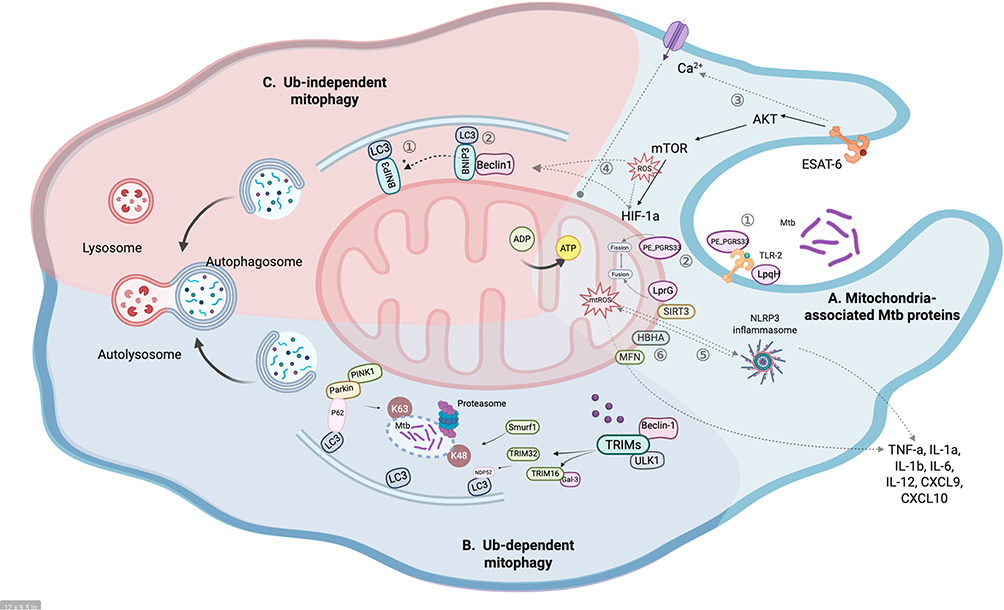 |
Figure 1 The molecular mechanism behind mitophagy in Mtb infection. (A) Mitochondria-associated Mtb proteins in an infected macrophage. Host macrophage is activated by sensing a wide range of bacterial proteins primarily through various receptors, such as TLR2. LpqH and PE_PGRS33 interact with TLR2 to enter cells (1). LprG induces mitochondrial fission. On the contrary, PE_PGRS33 promotes mitochondrial fusion (2). ESAT-6 could disrupt Ca2+ homeostasis that spirals into a disturbance in Ca2+ homeostasis of mitochondria with an increase in mitochondrial matrix Ca2+ leading to the loss of mitochondrial membrane potentials (3). ESAT-6, a virulence factor of M. tuberculosis, enhances ROS production via the AKT/mTOR signaling pathway. The increased ROS can directly stimulate mitophagy or indirectly induce HIF-1α expression, which in turn activates the HIF-1α/BNIP3 axis to drive mitophagy (4). The generation of mtROS activates NLRP3 inflammasome, which in turn can induce further mtROS generation, thereby triggering cytokines (5). Heparin-binding hemagglutinin (HBHA), a major secreted antigen during Mtb infection, localizes to mitochondria and is associated with apoptosis and autophagy (6). (B) Mitophagy pathway dependent on ubiquitin in Mtb infection. During Mtb infection of macrophages, Mtb-containing vacuoles are target by Parkin with K63-linked polyubiquitin, which is required for specific recruitment of the autophagy adaptors p62 to Mtb vacuoles. Concomitantly, Smurf1 targets Mtb vacuoles with K48-linked polyubiquitin which is required for specific recruitment of the proteasome to Mtb-containing vacuoles. TRIM family interact with key autophagy regulators ULK1 and Beclin1. TRIM32-triggered autophagy response to Mtb infection is associated with NDP52, and TRIM16 binds to galectin-3 and promotes Mtb ubiquitination and autophagosome engulfment. Adapter proteins are accumulated in the outer mitochondrial membrane after the ubiquitination, leading to ubiquitylated cargo recruited into autophagosomes by binding to LC3. The process of Smurf1-dependent proteasome and TRIM family recruitment to Mtb phagosomes in antimycobacterial autophagy is unknown. (C) Mitophagy pathway independent on ubiquitin in Mtb infection. BNIP3/NIX are multifunctional mitochondrial outer membrane proteins primarily involved in the ubiquitin-independent pathway of mitophagy. BNIP3/NIX sense distinct extra-/intracellular signaling to recruit autophagosomes by directly binding with LC3, via their LIR motif (1). Or it is possible that BNIP3/NIX may binding to Bcl-2 family proteins (including BCL2 and BCL-xL), which disrupts the BCL2-Beclin1 complex and releases Beclin-1, and also by repressing mTOR or regulating the production of ROS to activate mitophagy (2). The mature autophagosomes subsequently fuse with lysosomes to form the autophagolysosomes, where the mitochondria are degraded. Created in BioRender. Gao, (S) (2025) https://BioRender.com/p44hzi9. |
Metabolic Reprogramming
Metabolic reprogramming is the adaptive modifications in cellular energy production and utilization processes to optimize functionality in reaction to particular environmental signals or challenges.48 During Mtb infection, host cell metabolism is often altered, and this metabolic reprogramming may affect glycometabolism, lipid metabolism and OXPHOS, promoting rapid proliferation and effector functions by utilizing mitochondria to produce citrate succinate, fatty acid and ATP. These effects in neutrophils and macrophages are particularly pronounced.49 Previous studies have reported that Mtb infection triggers metabolic reprogramming in macrophages,48,50 which is considered a strategic host response. This response results in a marked up-regulation of glycolysis and down-regulation of OXPHOS, promoting macrophage differentiation into the M1 state. The primary source of energy for M1 macrophages is glycolysis, which produces energy quickly and gathers metabolites needed for synthesizing prostaglandins, nitric oxide (NO), ROS, and inflammatory mediators. This metabolic reprogramming during Mtb infection is thought to facilitate a protective immune response. Furthermore, glycometabolically enhanced M1 macrophages are believed to be better control of Mtb infections48,51 as they produce ATP and intermediates necessary for the synthesis of pro-inflammatory cytokines and antimicrobial peptides.52 Moreover, resistance mutations in Mtb can drive different metabolic reprogramming pathways, enhancing the ability to survive in host cells.53 For instance, a rifampicin-resistant Mtb strain with the H445Y mutation was found to modify host metabolic reprogramming and alter subsequent immune responses.54
Mitochondrial Dynamics
The remodeling of host cell metabolism can significantly impact mitochondrial function. Mtb infection may indirectly influence mitochondrial dynamics by altering the host cell’s metabolic state, which also encompasses responses, including changes in mitochondrial structure, morphology, and bioenergetics.8 Mitochondrial dynamics, involving fission and fusion, are mainly directed by mitofusins (MFNs) and dynamin-related protein 1 (DRP1).55 A study by Ning et al indicates that MFN1-drived mitochondrial fusion enhances ATP production and augments the bactericidal activity of macrophages against Mtb through enhanced autophagy.55 Lee et al revealed that MFN2 holds a key role in mitochondria disruption and inhibition of Mtb growth within macrophages.56 Mitochondrial dynamics are critical for regulating mitochondrial bioenergetics and other cellular processes including autophagy. In addition, Mtb virulence factors affect the metabolism of host cells by regulating mitochondrial kinetics. For example, LprG induces mitochondrial fission, which may support pathogen survival by reducing the host cell’s energy status. In contrast, PE_PGRS33, inducing mitochondrial fusion, may help maintain cellular metabolic activity57 (Figure 1A).
ROS overproduction, metabolic reprogramming, and imbalance of mitochondrial dynamics induced by Mtb infection do more than mark organelle dysfunction; they serve as the alarm signals of mitophagy. How, then, do these cues enlist the mitophagy pathways? In the next section, we explore the principal mitophagy pathways in Mtb infection.
Mitophagy in Mtb Infection and New Targets for Anti-Tuberculosis
The Ubiquitin-Dependent Mitophagy Pathways
PINK1, an enzyme with serine/threonine kinase activity, typically resides within the inner mitochondrial membrane. Under conditions of normal mitochondrial membrane potential, PINK1’s mitochondrial-targeting sequence (MTS) with positive charge facilitates its import into the mitochondrial matrix, where it undergoes stepwise cleavage. In impaired or depolarized mitochondria, PINK1 accumulated on the outer mitochondrial membrane (OMMs) and phosphorylates ubiquitin (Ub) molecules at S65.58,59
Upon Mtb infection of macrophages, the pathogen utilizes its primary virulence factor, the type VII secretion system ESX-1, to promote phagosome penetration.60 ESX-1-mediated permeabilization of the phagosome permits entry of the ubiquitination machinery, enabling ubiquitination of Mtb-containing phagosomes, as observed in infections involving both Salmonella61,62 and Mtb.63 Additionally, it is possible that segments of the Mtb surface can also undergo ubiquitination.63,64
Subsequently, PINK1, as a detector for mitochondrial damage, phosphorylates OMM protein, recruiting additional Parkin into the mitochondria and generating more ubiquitin chains.65–67 Ubiquitin chains on their own do not interact with isolated autophagic membranes or ATG8 family proteins attached to those membranes. As a result, when ubiquitin binds to phagosomes harboring Mtb, it attracts autophagy adaptor proteins, such as SQSTM1/p62, Optineurin (OPTN), CALCOCO2, Tax1 binding protein 1 (TAX1BP1), and NBR1 autophagy cargo receptor (NBR1), which facilitate their recognition via ubiquitin-binding domain (UBD), and then interaction with MAP1LC3 (microtubule associated protein 1 light chain 3) through an LC3 interacting region (LIR). This linkage directs autophagy substrates to ubiquitinated Mtb or Mtb-containing compartments including Mtb-containing phagosomes, autophagosomes, phagolytic lysosomes, and autophagolytic lysosomes.68,69 Impaired or depolarized mitochondria are eventually encapsulated by autophagosomes and digested by lysosomes70,71 (Figure 1B).
E3-Ub Ligase PRKN, SMURF1 and TRIM Family Confer Innate Defense Against Mtb
Parkin, an E3-Ub ligase, is found in the cytoplasm, and contains a ring finger protein 1 domain that interacts with phosphorylated Ub, thereby leading to Parkin activation and subsequent conformational changes.72 Following activation, Parkin ubiquitinates enhances E3 ligase function and affinity to promote the attraction of phosphorylated Ub, and orchestrates ubiquitination of various OMM proteins including voltage-dependent anion channel-1 (VDAC1), MFN1 and MFN2 on the surface of mitochondria, and enhancing mitophagy. During Mtb infection, Park2-/- cells were defective conversion of LC3 into its lipidated form LC3-II, indicating the necessity of Parkin for effective autophagy of mycobacteria.73 In Parkin-deficient macrophages, K63-linked ubiquitination of organelles harboring Mtb is impaired, further demonstrating that Parkin’s essential role in K63-dependent ubiquitination in Mtb-infected macrophages73 (Figure 1B).
Recent studies have revealed that Parkin modulates MFN2-mediated mitochondrial degradation, thereby impacting the intracellular survival of Mtb through apoptotic pathways.56 Beyond Parkin’s role in promoting K63-linked ubiquitination and recruiting autophagy adaptor proteins, other E3-Ub ligases drive the ubiquitination event: SMAD-specific E3 ubiquitin protein ligase 1 (Smurf1) and the tripartite motif (TRIM) proteins 16.63,74 Specifically, Smurf1, as a well-conserved HECT E3-Ub ligase, facilitates the ubiquitination of multiple substrates, marking them for proteasomal degradation.75,76 It is essential for regulating various biologically important processes such as cell growth and migration, immune response, bone morphogenesis, and embryonic development.77 Smurf1 has also been implicated in the regulation of mitophagy.78
Smurf1 mediates selective autophagy, particularly through K48-linked ubiquitination, to regulate the degradation of Mtb within host cells (Figure 1B). Additionally, this study reveals that Smurf1 works in concert with Parkin, utilizing distinct ubiquitin linkages to jointly mediate the selective autophagy of Mtb. Increased growth of Mtb is observed not only in cells doubly knockout for Smurf1 and Parkin but also in Smurf1-deficient mice model and human primary macrophages.63 In addition, similarly to Smurf1, HECT-type E3-Ub ligases NEDD4-1 also enhances autophagy and Mtb clearance.79
The tripartite motif (TRIM) proteins are a large family of ubiquitin ligases, and many of them function as E3 ubiquitin ligases that influence the autophagy process at various stages.74 Among them, TRIM16 mediates Mtb ubiquitination and autophagosome phagocytosis though attaching to Galectin-3.80 TRIM27 has been identified as a host-protective factor that enhances autophagy flux against Mtb independent of E3 ligase activity.81 TRIM32 was found to protect against Mtb by supporting the recruitment of Beclin-1 for autophagosome formation and by ubiquitination-mediated phagocytosis of Mtb autophagosomes (Figure 1B). And, its expression in THP-1 cells leads to increased Mtb growth, which was associated with impaired ubiquitination of Mtb and impaired formation of autophagosomes.82 Given their capacity to regulate protein stability and function with high substrate specificity, E3-Ub ligases represent highly attractive candidates for pharmacological targeting. In the field of oncology and diabetic nephropathy, several researches have explored the role of ubiquitination in targeted drug development and disease treatment.56,83,84 The development of selective inhibitors targeting specific E3-Ub ligases has emerged as a critical area of focus, particularly for the membrane-associated RING-CH family of E3 ligases85 and F-box only protein 38 (FBXO38), which regulates the proteasomal breakdown of programmed cell death protein 1.86,87 The role of Parkin, Smurf1 and TRIM in Mtb-induced mitophagy is now well recognized, but the specific molecular mechanisms by which these E3-Ub ligases regulate mitophagy and the specific substrates during Mtb infection deserve further investigation.
Considering the intriguing role of E3-Ub ligases in host innate defense against Mtb, the development of modulators of E3-Ub linker activity that increase ubiquitination of intracellular pathogens represents an innovative and largely unexplored approach. This strategy holds significant potential to leverage ubiquitin-mediated mitophagy as a means to promote Mtb clearance.
The Ubiquitin-Independent Mitophagy Pathways
In addition to Ub-dependent mitophagy, a number of mitophagy receptor proteins, such as BCL2 interacting protein 3 (BNIP3), BCL2 interacting protein 3 like (BNIP3L/NIX),88 FUN14 domain containing 1 (FUNDC1),89 BCL2 like 13 (BCL2L13),90 and FK506 binding protein 8 (FKBP8)91 and NOD-like receptor X1 (NLRX1)92 possess a unique capacity to interact with processed LC3. These receptors function by anchoring to the OMM and interacting directly with LC3, thereby triggering mitophagy independently of ubiquitin. Notably, NLRX1 has emerged as a new mitophagy receptor found in Listeria monocytogenes. Its virulence factor listeriolysin O (LLO) induces the oligomerization of NLRX1, facilitating the interaction between the LIR motif and LC3, thus initiating mitophagy.92 Although no ubiquitin-dependent mitophagy appears to play a key role in cellular maturation and specialization, BNIP3/NIX-mediated mitophagy is of particular important in TB. As mentioned above, the most well-recognized mechanism by which BNIP3/NIX-mediated mitophagy is through interactions with ATG8 family proteins, through which BNIP3L attracts autophagosomes to targeted mitochondria.92 However, there may be other potential mechanisms underlying BNIP3/NIX-mediated mitophagy in Mtb-infected macrophages. Due to resulting mitochondrial malfunction, BNIP3/NIX may induce ROS generation, thereby activating mitophagy,93,94 or competes with Beclin-1 for binding to BCL-xL, disrupting the BCL2–Beclin1 complex and releasing Beclin-1 to activate mitophagy95 (Figure 1C). The same seems to be true in macrophages infected with Mtb.93 Initially perceived as distinct from the ubiquitin-dependent PINK1/Parkin pathway, the BNIP3 and BNIP3L mitophagy pathways have now been shown by recent studies to be tightly regulated together. For example, NIX can participate in the Parkin-regulated mitophagy by serving as a substrate for Parkin via ubiquitination. In addition, BNIP3 also interacts with PINK1, promoting its accumulation on the OMM, which facilitates Parkin’s translocation to mitochondria and triggers mitophagy to remove destroyed mitochondria88 (Figure 1C).
BNIP3/NIX Plays a Key Role in the Regulation of Mitophagy During Mtb Infection
BNIP3 and its highly similar protein, BNIP3-like (BNIP3L), also known as NIX, are mitochondrial OMM proteins. They carry an atypical BCL2-homology 3 (BH3) domain that mediates hypoxia-induced apoptosis. BNIP3 and NIX not only involve in cell death while also triggering autophagy.95 Further investigations have indicated that the mitophagy pathways driven by distinct among BCL-2 family members, since it has been found that neither Bax/Bak, Bim, nor PUMA are required for mitophagy.96 Nevertheless, while BNIP3/NIX may operate through distinct mechanisms, they are functionally interconnected.95
Functionally, BNIP3/NIX-mediated mitophagy plays a crucial role in the regulation of mitochondrial mass. During erythrocyte maturation, NIX-mediated mitophagy is upregulated to facilitate the complete removal of mitochondria,96 and a significant decrease in mitochondrial mass is observed during embryonic retinal neuron differentiation.97 Besides, BNIP3 triggers mitochondrial swelling and increases membrane permeability via the Bax/Bak pathway. Cells overexpressing BNIP3 exhibit loss of mitochondrial electrical potential and activation of permeability transition channel.98 During Mtb infection, mitophagy is induced in macrophages, with strong upregulation of BNIP3/NIX.99 Comparative RNA sequencing analysis of peripheral blood mononuclear cells (PBMC) from TB patients reveals that Mtb infection disrupts multiple metabolic routes and upregulates the expression of BNIP3/NIX within the mitophagy pathway. Knockdown of NIX not only inhibits glycolysis and mitophagy, but also significantly impairs the secretion of inflammatory mediators such as TNF-α, IL-6 and IL-1β, which are essential for bacterial clearance. These observations imply that modulation of BNIP3/NIX-mediated mitophagy facilitates host resistance to Mtb through multiple pathways.99 The similar phenomenon has been observed in the study conducted by Lee et al. On the one hand, upregulation of BNIP3 is required to induce mitophagy, while on the other hand, downregulation of BNIP3 inhibits mitophagy, resulting in increased mROS generation and decreased Mtb survival. This manner may be achieved through activation of the HIF1α-BNIP3 axis.93 In renal fibrosis, HIF1 α-BNIP3-mediated mitophagy guards against hypoxia-triggered injury of renal epithelial cells by inhibiting mtROS and preventing activation of NLRP3 inflammasome.100 Additionally, the Chinese medicine baicalein inhibits pyroptosis induced by Mtb by promoting autophagy and downregulating AIM2 and NLRP3 protein levels.101 These findings suggest that mitophagy-directed drugs could be used as an adjunctive treatment for TB by both modulating immune function and enhancing host antimicrobial resistance.
Additional studies on various cell types indicate that BNIP3 activation leads to mitochondrial dysfunction and increased ROS generation.102 Zhu et al thought that BNIP3-induced mitophagy functions as a defense mechanism against mitochondrial malfunction in response to lactate by controlling ROS production.103 These findings suggest a close link between BNIP3 and ROS generation, with both contributing synergistically to mitophagy in response to Mtb infection. Moreover, BNIP3 has been shown to promote mtROS production in Mtb-infected macrophages and suppressed Mtb intracellular survival through the production of pro-inflammatory cytokine.93 Thus, BNIP3 emerges as a critical factor in the host’s ability to control intracellular Mtb. Meanwhile, BNIP3 represents a promising candidate for targeted therapeutic interventions in TB.
In contrast to the above studies, Song et al demonstrate that Mycobacterium bovis (M. bovis) employs mitophagy mechanism to promote its own survival.104 M. bovi-induced PINK1-dependent mitophagy demands phosphorylated TBK1 (pTBK1), that is significant for bacteriophagy.105 Thus, the competitive utilization of pTBK1 between mitophagy and phagolysis contributes to Mtb evasion of degradation mechanisms. Furthermore, M. bovis-induced mitophagy is affected by PINK1 knockdown.104 However, analysis of gene expression in TB patients revealed that PINK1/Parkin pathway genes are not significantly upregulated.99 Instead, NIX and BNIP3 expression is markedly increased in PBMCs and THP-1 cells.99 Although the differences between Mtb- and M. bovis-mediated mitophagy require further exploration, current evidence suggests that BNIP3/NIX-mediated mitophagy plays a predominant role in Mtb-infected macrophages.
Regarding drug development, an antibiotic called thiopeptide has been shown potent antibacterial activity against Mtb, and to induce mitophagy i independently of bacterial protein translation inhibition. However, this autophagy does not appear to be significant to Mtb clearance.106 Excitingly, a small-molecule agonist, imiquimod, can help clear Mtb in macrophages by enhancing BNIP3-induced mitophagy. Imiquimod also enhances mitophagy by inducing mtROS production.106 Although imidazoquinoline derivatives, known as TLR7 agonists, are effective in inducing autophagy to inhibit tumor growth,107 their antitumor and pro-inflammatory effects may be independent of TLR7 activation. While there is no direct evidence that TLR7 is required for mitophagy, autophagy triggered by TLR signaling may contribute to anti-tuberculosis activity. Findings also suggest that imiquimod enhances mitophagy through interaction between BNIP3 and Beclin-1, thereby limiting Mtb survival. This study provides crucial insights into the potential of BNIP3/NIX-mediated mitophagy as a therapeutic target for Mtb clearance.107 Presently, there exists no definitive proof of a fundamental role for BNIP3/NIX in particular disease nor are there clinical approaches targeting BNIP3/NIX. However, we believe that BNIP3/NIX-mediated mitophagy is essential for controlling Mtb replication.
Challenges and Future Perspectives
Given the rise of drug resistance, host-targeted therapies (HDTs) are emerging as promising adjuncts aimed at mitigating host pathology and enhancing immune response, particularly by restoring mitochondrial health. This study provides preliminary evidence supporting the advancement of mitochondrial function as a feasible HDT strategy. However, the crucial question remains whether targeting human mitophagy can modulate beneficial immune responses in tuberculosis? The current literature lacks consensus on this question, with conflicting perspectives regarding its implications. While mitophagy has the potential to confer protective effects by removing damaged mitochondria and contribute to innate immunity through mechanisms like ROS release, but when overactivated or improperly regulated it may cause cellular dysfunction and death, and even facilitate pathogen immune evasion.
Mitophagy is a key upstream regulator of host cell fate and cell death, with profound implications for HDT. Analogous evidence from other infectious models reinforces this concept. In influenza-infected mice, mitophagy-mediated reduction of mROS suppresses NLRP3 inflammasome activation, alleviating inflammatory lung lesions.108 Purple sweet potato anthocyanins (PSPAs) activate the NRF2 pathway to enhance PINK1/Parkin-mediated mitophagy, which attenuates Klebsiella pneumoniae-induced pyroptosis in alveolar macrophages and reduces lung injury and inflammation in infected mice.109 Conversely, dengue virus (DENV) and hepatitis B virus (HBV) both exploit mitophagy to prolong host cell survival, facilitating viral replication and persistence.110,111 In the rice gall dwarf virus (RGDV) vector, the Pns11 protein interacts with VDAC1 to activate PINK1/Parkin-mediated mitophagy, suppress excessive apoptosis, and enable ongoing infection.112 These examples demonstrate that mitophagy’s role varies dramatically across infection contexts: protecting the host from inflammatory and necrotic death in some cases, yet being hijacked by pathogens to delay apoptosis and promote their own survival in others. Future research must elucidate how mitophagy regulates distinct forms of regulated cell death including apoptosis, pyroptosis, necroptosis, and ferroptosis, to guide the development of more precise and effective HDT.
Moreover, many researches have shown that BNIP3 plays complex roles across a range of diseases, including tumor, neurodegenerative diseases, and acute brain injury. BNIP3 fulfills a twofold function in the context of cancer: on the one hand, it has been shown to support tumor cell survival; on the other hand, it may also act as an inhibitor.113–115 Research into glioma cells revealed that BNIP3L is indispensable for the cell death induced by the naturally occurring substance AT 101.116 Concurrently, another research discovered that the breakdown of endogenous BNIP3L facilitated the viability of Ewing sarcoma cells.117 On the other hand, BNIP3 has also been implicated in promoting tumor aggressiveness. In advanced tumors, such as cervical and breast cancers, BNIP3 expression is often upregulated, suggesting that it plays a role in supporting tumor progression.118,119 In contrast, high BNIP3L levels in murine pancreatic intraepithelial neoplasia organoids are associated with delayed progression of pancreatic ductal adenocarcinoma,120 indicating that BNIP3L-mediated mitophagy may promote tumorigenesis. Additionally, in acute brain injury, BNIP3 can mitigate cerebral damage by promoting protective mitophagy.121 This duality also extends to neurodegenerative disorders such as Parkinson’s disease and acromegaly, where BNIP3L accelerates disease progression.122,123 These impacts might be connected to the various diseases’ distinct metabolic settings, but the exact mechanisms remain unclear. From this understanding, several questions arise: What are the molecular determinants that modulate BNIP3’s dual role across different types of tumors and neurological conditions? How do oncogenes and specific metabolic conditions influence BNIP3-mediated mitophagy, and how can these pathways be therapeutically manipulated to yield positive outcomes? Additionally, could targeting BNIP3/NIX provide a synergistic approach when combined with other therapeutic strategies, such as immune checkpoint inhibitors or metabolic modulators? Addressing these questions will be essential to fully elucidate the potential of BNIP3/NIX as a therapeutic target and to understand how its activity can be harnessed or mitigated in different pathological contexts.
E3 ubiquitin ligases have also emerged as potential therapeutic targets for combination immunotherapy. However, their clinical utility remains limited by issues such as resistance and adverse effects in cancer treatment.124 Given the complexity and ubiquity of ubiquitin-mediated regulatory processes in cellular biology, targeting E3 ligases for immunotherapy may lead to unintended consequences on normal physiological functions. While preclinical data suggest that E3 ligases could be exploited therapeutically in TB, results from in vivo studies might be less promising, and significant challenges remain in translating these findings to clinical application.
Moreover, since mitophagy is typically studied under condition of significant mitochondrial disruption within cultured cells, it is crucial to explore these pathways under conditions that closely mimic physiological environments, with understanding their temporal and spatial regulation. To benefit the patient, it is essential to consider how host-targeted interventions impact every kind of cell and tissue, influence systemic responses, and affect the entire progression of the infection. Consequently, positive outcomes at the cellular level could be offset by harmful systemic effects affecting various responses, and the reverse may also occur. Furthermore, interventions that are beneficial at one stage of the infection, such as early in the inflammatory process, may no longer be beneficial during later stages. Therefore, careful consideration of the timing and duration of HDT administration is essential to maximize therapeutic efficacy and minimize risks. While advancements in our understanding of mitophagy in Mtb infection have been made, numerous gaps in knowledge persist.
The utilization of HDT drugs aimed at enhancing mitophagy represents a promising approach to enhancing tuberculosis treatment outcomes, potentially extending to the management of drug-resistant strains. However, comprehensive understanding of the underlying mechanisms remains limited. Research into mitophagy in the field of tuberculosis is also at an emerging stage. Consequently, more relevant studies are necessary in the future to confirm the efficacy and feasibility of current HDT drugs as adjuvants to conventional chemotherapy and to identify novel HDT drugs that target mitophagy in TB treatment.
Funding
Beijing Nova Program, Grant/Award Number: 20220484169& 20230484295 and National Key Research and Development Program of China, Grant/Award Number: 2022YFC2304802.
Disclosure
The authors declare that they have no competing interests.
References
1. Organization WH. Global tuberculosis report 2019 [1]. Geneva: World Health Organization; 2019.
2. Diacon AH, Donald PR, Pym A, et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother. 2012;56(6):3271–3276. doi:10.1128/AAC.06126-11
3. Lee M, Lee J, Carroll MW, et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med. 2012;367(16):1508–1518. doi:10.1056/NEJMoa1201964
4. Keam SJ. Pretomanid: first Approval. Drugs. 2019;79(16):1797–1803. doi:10.1007/s40265-019-01207-9
5. Kadura S, King N, Nakhoul M, et al. Systematic review of mutations associated with resistance to the new and repurposed Mycobacterium tuberculosis drugs bedaquiline, clofazimine, linezolid, delamanid and pretomanid. J Antimicrob Chemother. 2020;75(8):2031–2043. doi:10.1093/jac/dkaa136
6. Zhuang L, Yang L, Li L, et al. Mycobacterium tuberculosis: immune response, biomarkers, and therapeutic intervention. MedComm. 2024;5(1):e419. doi:10.1002/mco2.419
7. Chai Q, Wang L, Liu CH, et al. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol Immunol. 2020;17(9):901–913. doi:10.1038/s41423-020-0502-z
8. Patrick KL, Watson RO. Mitochondria: powering the innate immune response to Mycobacterium tuberculosis infection. Infect Immun. 2021;89(4). doi:10.1128/IAI.00687-20
9. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO j. 2017;36(13):1811–1836. doi:10.15252/embj.201796697
10. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5. doi:10.1089/rej.2005.8.3
11. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406–417. doi:10.1016/j.immuni.2015.02.002
12. West AP. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology. 2017;391:54–63. doi:10.1016/j.tox.2017.07.016
13. Lu Y, Li Z, Zhang S, et al. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13(2):736–766. doi:10.7150/thno.79876
14. Huang T, Pu Q, Zhou C, et al. MicroRNA-302/367 cluster impacts host antimicrobial defense via regulation of mitophagic response against Pseudomonas aeruginosa infection. Front Immunol. 2020;11:569173. doi:10.3389/fimmu.2020.569173
15. Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy. 2018;14(10):1665–1673. doi:10.1080/15548627.2018.1466014
16. Zhu L, Mou C, Yang X, et al. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget. 2016;7(19):27122–27141. doi:10.18632/oncotarget.8345
17. Cavalcante GC, Marinho ANR, Anaissi AK, et al. Whole mitochondrial genome sequencing highlights mitochondrial impact in gastric cancer. Sci Rep. 2019;9(1):15716. doi:10.1038/s41598-019-51951-x
18. Bahat A, MacVicar T, Langer T. Metabolism and innate immunity meet at the mitochondria. Front Cell Dev Biol. 2021;9:720490. doi:10.3389/fcell.2021.720490
19. Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science. 2004;304(5668):253–257. doi:10.1126/science.1094884
20. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. doi:10.1038/nri.2017.21
21. Harrington JS, Choi AMK, Nakahira K. Mitochondrial DNA in Sepsis. Curr Opin Crit Care. 2017;23(4):284–290. doi:10.1097/MCC.0000000000000427
22. Lin MM, Liu N, Qin Z-H, et al. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. 2022;43(10):2439–2447. doi:10.1038/s41401-022-00879-6
23. Yao CH, Wang R, Wang Y, et al. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife. 2019;8.
24. Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6(8):657–663. doi:10.1038/nrm1697
25. Kim IS, Silwal P, Jo EK. Mitofusin 2, a key coordinator between mitochondrial dynamics and innate immunity. Virulence. 2021;12(1):2273–2284. doi:10.1080/21505594.2021.1965829
26. Tiku V, Tan MW, Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020;30(4):263–275. doi:10.1016/j.tcb.2020.01.006
27. Song Y, Zhou Y, Zhou X. The role of mitophagy in innate immune responses triggered by mitochondrial stress. Cell Commun Signal. 2020;18(1):186. doi:10.1186/s12964-020-00659-x
28. Suliman HB, Kraft B, Bartz R, et al. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2017;313(4):L699–l709. doi:10.1152/ajplung.00197.2017
29. Kim MJ, Bae SH, Ryu J-C, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12(8):1272–1291. doi:10.1080/15548627.2016.1183081
30. Cataldo AM, McPhie DL, Lange NT, et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol. 2010;177(2):575–585. doi:10.2353/ajpath.2010.081068
31. Asalla S, Mohareer K, Banerjee S. Small molecule mediated restoration of mitochondrial function augments anti-mycobacterial activity of human macrophages subjected to cholesterol induced asymptomatic dyslipidemia. Front Cell Infect Microbiol. 2017;7:439. doi:10.3389/fcimb.2017.00439
32. Jamwal S, Midha MK, Verma HN, et al. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci Rep. 2013;3:1328. doi:10.1038/srep01328
33. Mohareer K, Asalla S, Banerjee S. Cell death at the cross roads of host-pathogen interaction in Mycobacterium tuberculosis infection. Tuberculosis. 2018;113:99–121. doi:10.1016/j.tube.2018.09.007
34. Zhao D, Lin D, Xu C. A protein fragment of Rv3194c located on mycobacterial cell surface efficiently prevents adhesion of recombinant Mycobacterium smegmatis, and promises a new anti-adhesive drug. Microb Pathog. 2020;149:104498. doi:10.1016/j.micpath.2020.104498
35. Jayaraman P, Sada-Ovalle I, Nishimura T, et al. IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. 2013;190(8):4196–4204. doi:10.4049/jimmunol.1202688
36. Kundu M, Basu J. The role of microRNAs and long non-coding RNAs in the regulation of the immune response to Mycobacterium tuberculosis infection. Front Immunol. 2021;12:687962. doi:10.3389/fimmu.2021.687962
37. Ge P, Lei Z, Yu Y, et al. M. tuberculosis PknG manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy. 2022;18(3):576–594. doi:10.1080/15548627.2021.1938912
38. Bach H, Papavinasasundaram KG, Wong D, et al. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe. 2008;3(5):316–322. doi:10.1016/j.chom.2008.03.008
39. Miller JL, Velmurugan K, Cowan MJ, et al. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 2010;6(4):e1000864. doi:10.1371/journal.ppat.1000864
40. Billingham LK, Stoolman JS, Vasan K, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. 2022;23(5):692–704. doi:10.1038/s41590-022-01185-3
41. Ellzey LM, Patrick KL, Watson RO. Mitochondrial reactive oxygen species: double agents in Mycobacterium tuberculosis infection. Curr Opin Immunol. 2023;84:102366. doi:10.1016/j.coi.2023.102366
42. Chao WC, Yen C-L, Hsieh C-Y, et al. Mycobacterial infection induces higher interleukin-1β and dysregulated lung inflammation in mice with defective leukocyte NADPH oxidase. PLoS One. 2017;12(12):e0189453. doi:10.1371/journal.pone.0189453
43. Smulan LJ, Martinez N, Kiritsy MC, et al. Sirtuin 3 downregulation in mycobacterium tuberculosis-Infected macrophages reprograms mitochondrial metabolism and promotes cell death. mBio. 2021;12(1). doi:10.1128/mBio.03140-20
44. Kim YJ, Lee S-H, Jeon SM, et al. Sirtuin 3 is essential for host defense against Mycobacterium abscessus infection through regulation of mitochondrial homeostasis. Virulence. 2020;11(1):1225–1239. doi:10.1080/21505594.2020.1809961
45. Pajuelo D, Gonzalez-Juarbe N, Tak U, et al. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep. 2018;24(2):429–440. doi:10.1016/j.celrep.2018.06.042
46. Choi HH, Shin D-M, Kang G, et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 2010;584(11):2445–2454. doi:10.1016/j.febslet.2010.04.050
47. Lim YJ, Choi J-A, Lee J-H, et al. Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis. 2015;20(3):358–370. doi:10.1007/s10495-014-1080-2
48. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, et al. Cutting edge: Mycobacterium tuberculosis Induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol. 2016;196(6):2444–2449. doi:10.4049/jimmunol.1501612
49. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi:10.1084/jem.20151570
50. Cumming BM, Addicott KW, Adamson JH, et al. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife. 2018;7.
51. Huang L, Nazarova EV, Tan S, et al. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. 2018;215(4):1135–1152. doi:10.1084/jem.20172020
52. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. 2015;6:164. doi:10.3389/fimmu.2015.00164
53. Howard NC, Marin ND, Ahmed M, et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat Microbiol. 2018;3(10):1099–1108. doi:10.1038/s41564-018-0245-0
54. Howard NC, Khader SA. Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. 2020;28(10):832–850. doi:10.1016/j.tim.2020.04.010
55. Ning Y, Cai Y, Dai Y, et al. Mitochondrial fusion mediated by Mitofusin 1 regulates macrophage mycobactericidal activity by enhancing autophagy. Infect Immun. 2021;89(11):e0030621. doi:10.1128/IAI.00306-21
56. Lee J, Choi J-A, Cho S-N, et al. Mitofusin 2-deficiency suppresses Mycobacterium tuberculosis survival in macrophages. Cells. 2019;8(11):1355. doi:10.3390/cells8111355
57. Aguilar-López BA, Correa F, Moreno‐ Altamirano MMB, et al. LprG and PE _ PGRS 33 Mycobacterium tuberculosis virulence factors induce differential mitochondrial dynamics in macrophages. Scand J Immunol. 2019;89(1):e12728. doi:10.1111/sji.12728
58. Okatsu K, Kimura M, Oka T, et al. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci. 2015;128(5):964–978. doi:10.1242/jcs.161000
59. Xu Y, Tang Y, Lu J, et al. PINK1-mediated mitophagy protects against hepatic ischemia/reperfusion injury by restraining NLRP3 inflammasome activation. Free Radic Biol Med. 2020;160:871–886. doi:10.1016/j.freeradbiomed.2020.09.015
60. Tiwari S, Casey R, Goulding CW, et al. Infect and inject: how Mycobacterium tuberculosis exploits its major virulence-associated Type VII secretion system, ESX-1. Microbiol Spectr. 2019;7(3). doi:10.1128/microbiolspec.BAI-0024-2019
61. Kishi-Itakura C, Ktistakis NT, Buss F. Ultrastructural insights into pathogen clearance by autophagy. Traffic. 2020;21(4):310–323. doi:10.1111/tra.12723
62. Perrin AJ, Jiang X, Birmingham CL, et al. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol. 2004;14(9):806–811. doi:10.1016/j.cub.2004.04.033
63. Franco LH, Nair VR, Scharn CR, et al. The Ubiquitin Ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017;21(1):59–72. doi:10.1016/j.chom.2016.11.002
64. Chai Q, Wang X, Qiang L, et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun. 2019;10(1):1973. doi:10.1038/s41467-019-09955-8
65. Ryter SW, Bhatia D, Choi ME. Autophagy: a lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid Redox Signal. 2019;30(1):138–159. doi:10.1089/ars.2018.7518
66. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. doi:10.1038/ncb2012
67. Chen Y, Dorn GW. 2nd, PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–475. doi:10.1126/science.1231031
68. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi:10.1038/nature14893
69. Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol. 2018;9:1283. doi:10.3389/fimmu.2018.01283
70. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19(2):93–108. doi:10.1038/nrm.2017.129
71. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–r185. doi:10.1016/j.cub.2018.01.004
72. Tang MY, Vranas M, Krahn AI, et al. Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat Commun. 2017;8:14697. doi:10.1038/ncomms14697
73. Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501(7468):512–516. doi:10.1038/nature12566
74. Di Rienzo M, Romagnoli A, Antonioli M, et al. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020;27(3):887–902. doi:10.1038/s41418-020-0495-2
75. Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004;23(11):1972–1984. doi:10.1038/sj.onc.1207436
76. Wei X, Wang X, Zhan J, et al. Smurf1 inhibits integrin activation by controlling Kindlin-2 ubiquitination and degradation. J Cell Biol. 2017;216(5):1455–1471. doi:10.1083/jcb.201609073
77. Cao Y, Zhang L. A Smurf1 tale: function and regulation of an ubiquitin ligase in multiple cellular networks. Cell Mol Life Sci. 2013;70(13):2305–2317. doi:10.1007/s00018-012-1170-7
78. Orvedahl A, Sumpter R, Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. doi:10.1038/nature10546
79. Pei G, Buijze H, Liu H, et al. The E3 ubiquitin ligase NEDD4 enhances killing of membrane-perturbing intracellular bacteria by promoting autophagy. Autophagy. 2017;13(12):2041–2055. doi:10.1080/15548627.2017.1376160
80. Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and Galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39(1):13–27. doi:10.1016/j.devcel.2016.08.003
81. Zhao D, Qiang L, Lei Z, et al. TRIM27 elicits protective immunity against tuberculosis by activating TFEB-mediated autophagy flux. Autophagy. 2024;20(7):1483–1504. doi:10.1080/15548627.2024.2321831
82. Romagnoli A, Di Rienzo M, Petruccioli E, et al. The ubiquitin ligase TRIM32 promotes the autophagic response to Mycobacterium tuberculosis infection in macrophages. Cell Death Dis. 2023;14(8):505. doi:10.1038/s41419-023-06026-1
83. Deng L, Meng T, Chen L, et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5(1):11. doi:10.1038/s41392-020-0107-0
84. Akhouri V, Majumder S, Gaikwad AB. The emerging insight into E3 ligases as the potential therapeutic target for diabetic kidney disease. Life Sci. 2023;321:121643. doi:10.1016/j.lfs.2023.121643
85. Lin H, Li S, Shu HB. The membrane-associated MARCH E3 ligase family: emerging roles in immune regulation. Front Immunol. 2019;10:1751. doi:10.3389/fimmu.2019.01751
86. Serman TM, Gack MU. FBXO38 drives PD-1 to destruction. Trends Immunol. 2019;40(2):81–83. doi:10.1016/j.it.2018.12.005
87. Meng X, Liu X, Guo X, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564(7734):130–135. doi:10.1038/s41586-018-0756-0
88. Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi:10.1038/nature07006
89. Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi:10.1038/ncb2422
90. Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi:10.1038/ncomms8527
91. Bhujabal Z, Birgisdottir ÅB, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–961. doi:10.15252/embr.201643147
92. Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446. doi:10.1038/s41590-019-0324-2
93. Lee J, Lee S-A, Son S-H, et al. Impaired mitophagy induces antimicrobial responses in macrophages infected with Mycobacterium tuberculosis. Cell Biosci. 2023;13(1):158. doi:10.1186/s13578-023-01107-2
94. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30–38. doi:10.1016/j.tibs.2010.07.007
95. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. doi:10.1038/cdd.2009.16
96. Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–19505. doi:10.1073/pnas.0708818104
97. Esteban-Martínez L, Sierra‐Filardi E, McGreal RS, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO j. 2017;36(12):1688–1706. doi:10.15252/embj.201695916
98. Dhingra A, Jayas R, Afshar P, et al. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med. 2017;112:411–422. doi:10.1016/j.freeradbiomed.2017.08.010
99. Mahla RS, Kumar A, Tutill HJ, et al. NIX-mediated mitophagy regulate metabolic reprogramming in phagocytic cells during mycobacterial infection. Tuberculosis. 2021;126:102046. doi:10.1016/j.tube.2020.102046
100. Li J, Lin Q, Shao X, et al. HIF1α-BNIP3-mediated mitophagy protects against renal fibrosis by decreasing ROS and inhibiting activation of the NLRP3 inflammasome. Cell Death Dis. 2023;14(3):200. doi:10.1038/s41419-023-05587-5
101. Ning B, Shen J, Liu F, et al. Baicalein suppresses NLRP3 and AIM2 inflammasome-mediated pyroptosis in macrophages infected by Mycobacterium tuberculosis via induced autophagy. Microbiol Spectr. 2023;11(3):e0471122. doi:10.1128/spectrum.04711-22
102. Gao A, Jiang J, Xie F, et al. Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin Chim Acta. 2020;506:72–83. doi:10.1016/j.cca.2020.02.024
103. Zhu Y, Ji -J-J, Yang R, et al. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal. 2019;58:53–64. doi:10.1016/j.cellsig.2019.03.006
104. Song Y, Ge X, Chen Y, et al. Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy. 2022;18(6):1401–1415. doi:10.1080/15548627.2021.1987671
105. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 2016;113(24):E3349–58. doi:10.1073/pnas.1523810113
106. Bird KE, Xander C, Murcia S, et al. Thiopeptides induce proteasome-independent activation of cellular mitophagy. ACS Chem Biol. 2020;15(8):2164–2174. doi:10.1021/acschembio.0c00364
107. Cho JH, Lee H-J, Ko H-J, et al. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget. 2017;8(15):24932–24948. doi:10.18632/oncotarget.15326
108. Liu H, You L, Wu J, et al. Berberine suppresses influenza virus-triggered NLRP3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS. J Leukoc Biol. 2020;108(1):253–266. doi:10.1002/JLB.3MA0320-358RR
109. Dong G, Xu N, Wang M, et al. Anthocyanin extract from purple sweet potato exacerbate mitophagy to ameliorate pyroptosis in Klebsiella pneumoniae infection. Int J Mol Sci. 2021;22(21):11422. doi:10.3390/ijms222111422
110. Barbier V, Lang D, Valois S, et al. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology. 2017;500:149–160. doi:10.1016/j.virol.2016.10.022
111. Kim SJ, Syed GH, Khan M, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA. 2014;111(17):6413–6418. doi:10.1073/pnas.1321114111
112. Chen Q, Jia D, Ren J, et al. VDAC1 balances mitophagy and apoptosis in leafhopper upon arbovirus infection. Autophagy. 2023;19(6):1678–1692. doi:10.1080/15548627.2022.2150001
113. Lazarini M, Machado-Neto JA, Duarte ADSS, et al. BNIP3L in myelodysplastic syndromes and acute myeloid leukemia: impact on disease outcome and cellular response to decitabine. Haematologica. 2016;101(11):e445–e448. doi:10.3324/haematol.2016.142521
114. Moussay E, Kaoma T, Baginska J, et al. The acquisition of resistance to TNFα in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011;7(7):760–770. doi:10.4161/auto.7.7.15454
115. Jung J, Zhang Y, Celiku O, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 2019;79(20):5218–5232. doi:10.1158/0008-5472.CAN-19-0198
116. Meyer N, Zielke S, Michaelis JB, et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy. 2018;14(10):1693–1709. doi:10.1080/15548627.2018.1476812
117. Gallegos ZR, Taus P, Gibbs ZA, et al. EWSR1-FLI1 activation of the cancer/Testis Antigen FATE1 promotes Ewing Sarcoma survival. Mol Cell Biol. 2019;39(14). doi:10.1128/MCB.00138-19
118. Leo C, Horn LC, Höckel M. Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int J Gynecol Cancer. 2006;16(3):1314–1320. doi:10.1136/ijgc-00009577-200605000-00055
119. Sowter HM, Ferguson M, Pym C, et al. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol. 2003;201(4):573–580. doi:10.1002/path.1486
120. Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019;9(9):1268–1287. doi:10.1158/2159-8290.CD-18-1409
121. Yuan Y, Zheng Y, Zhang X, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13(10):1754–1766. doi:10.1080/15548627.2017.1357792
122. Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci Rep. 2017;7:44373. doi:10.1038/srep44373
123. Duval N, Sumner WA, Andrianakos AG, et al. The Bcl-2 homology-3 domain (BH3)-only proteins, Bid, DP5/Hrk, and BNip3L, are upregulated in reactive astrocytes of end-stage mutant SOD1 mouse spinal cord. Front Cell Neurosci. 2018;12:15. doi:10.3389/fncel.2018.00015
124. Ye P, Chi X, Cha J-H, et al. Potential of E3 ubiquitin ligases in cancer immunity: opportunities and challenges. Cells. 2021;10(12):3309. doi:10.3390/cells10123309
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.