Back to Journals » Clinical Epidemiology » Volume 9
Misclassification of hypertrophic cardiomyopathy: validation of diagnostic codes
Authors Magnusson P, Palm A, Branden E, Mörner S
Received 10 April 2017
Accepted for publication 13 July 2017
Published 9 August 2017 Volume 2017:9 Pages 403—410
DOI https://doi.org/10.2147/CLEP.S139300
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Irene Petersen
Peter Magnusson,1,2 Andreas Palm,2,3 Eva Branden,2,4 Stellan Mörner5
1Cardiology Research Unit, Department of Medicine, Karolinska Institutet, Stockholm, 2Centre for Research and Development, Uppsala University, Region Gävleborg, Gävle, 3Department of Medical Sciences, Respiratory, Allergy and Sleep Research, Uppsala University, Uppsala, 4Department of Medicine, Karolinska Institutet, Stockholm, 5Heart Center and Department of Public Health and Clinical Medicine, Umeå University, Umeå, Sweden
Purpose: To validate diagnostic codes for hypertrophic cardiomyopathy (HCM), analyze misclassfications, and estimate the prevalence of HCM in an unselected Swedish regional cohort.
Patients and methods: Using the hospitals’ electronic medical records (used for the Swedish National Patient Register), we identified 136 patients from 2006 to 2016 with the HCM-related codes 142.1 and 142.2 (International Classification of Diseases).
Results: Of a total of 129 residents in the catchment area, 88 patients were correctly classified as HCM (positive predictive value 68.2%) and 41 patients (31.8%) were misclassified as HCM. Among the 88 HCM patients (52.2% males), 74 were alive and 14 were dead (15.9%). This yields an HCM prevalence of 74/183,337, that is, 4.0 diagnosed cases per 10,000 in the adult population aged ≥18 years. The underlying diagnoses of misclassified cases were mainly hypertension (31.7%) and aortic stenosis (22.0%). Other types of cardiomyopathies accounted for several cases of misclassification: dilated (nonischemic or ischemic), left ventricular noncompaction, and Takotsubo. Miscellaneous diagnoses were amyloidosis, pulmonary stenosis combined with ventricular septal defect, aortic insufficiency, athelete’s heart, and atrioventricular conduction abnormality. The mean age was not significantly different between HCM and misclassified patients (65.8±15.8 vs 70.1±13.4 years; P=0.177). There were 47.8% females among HCM and 60.8% females among misclassified (P=0.118).
Conclusion: One-third of patients diagnosed as HCM are misclassified, so registry data should be interpreted with caution. A correct diagnosis is important for decision-making and implementation of optimal HCM care; efforts should be made to increase awareness of HCM and diagnostic competence throughout the health care system.
Keywords: diagnostic error, diagnosis, epidemiology, hypertrophic cardiomyopathy, International Classification of Diseases, register
Introduction
The diagnosis of hypertrophic cardiomyopathy (HCM) is based on cardiac imaging, genetics, and exclusion of phenocopies or abnormal circulation pathophysiology as an explanation for cardiac hypertrophy.1,2 Echocardiograhpy is a cornerstone among diagnostic tools, even though cardiac magnetic resonance (CMR) imaging can be useful, ie, when visualization of the whole chambers is difficult (eg, apical hypertrophy) and to rule out secondary causes.1,2 According to the European Society of Cardiology, the diagnosis of HCM requires the myocardial thickness of at least 15 mm (or 13 mm if there exists a definite diagnosis in a parent, sibling, or child), which cannot be attributed solely to increased loading conditions.1 The loading conditions resulting in hypertrophy may be an adaptation to increased volume/pressure conditions caused by hypertension and/or aortic stenosis. Hypertension affects one-fourth of the adult population,3,4 aortic stenosis affects ~2–7% of the elderly,5 and both may coexist with HCM. Thus, careful clinical judgment is required to determine if HCM solely explains the hypertrophy.1,2
The prevalence of HCM is usually stated as 1:500 (0.2%), which has been consistently reported in several population studies.6–10 In a US cohort (aged 23–35 years), 7 out of 4,111 (0.17%) unrelated persons showed signs of hypertrophy on echocardiography but only 1 had cardiac symptoms.6 This has been confirmed in later studies of cohorts (mean age 47–60 years) in other US communities, China, and Japan, which report the HCM prevalence of 0.16–0.23%.7–10 Despite advances in health care since Teare’s report on HCM >50 years ago, accurate HCM diagnosis remains challenging and delayed diagnosis is frequently encountered.11
Recently, a database search of 169 million Americans using the International Classification of Diseases, Ninth Revision (ICD-9) for HCM-related diagnosis yielded the prevalence of 1:3,195. This study had two important limitations. First, it did not validate HCM diagnoses, so the prevalence may not be correctly reported. Second, this database search excluded the somewhat large subset of patients without health care insurance, suggesting that some patients are never diagnosed.12
Patient registries are important sources of information for research and clinical decision making, but the quality of the data depends on the integrity of the diagnostic codes. The Swedish National Patient Register covers >99% of all visits in cardiology units nationwide, but even though the quality is generally high, HCM has not been validated specifically.13
The aims of this study were to validate diagnostic codes for HCM, analyze misclassifications, and estimate the prevalence of HCM in a Swedish regional cohort.
Methods
Setting
The region of Gävleborg is situated in the geographical middle part of Sweden and has 281,815 inhabitants of whom 80.7% are >18 years of age (227,304); the catchment area of the hospitals Gävle Sjukhus and Hudiksvall Sjukhus in Region Gävleborg has 183,337 persons >18 years of age.14 The main referral university hospital is Uppsala Akademiska Sjukhus, but other tertiary centers are occasionally involved in the cases of highly specialized management of HCM.
Data validation source
We searched in the Cyklop™ system for the HCM-related diagnostic codes I42.1 (obstructive HCM) and I42.2 (other HCM) from International Classification of Diseases, Tenth Revision (ICD-10) from 2006 until October 2016. The hospital’s electronic medical records system Cyklop provides complete coverage of inpatient and outpatient visits, and data are entered into the national Swedish National Patient Register.13 Thus, the records in Cyklop are identical to data from the Swedish National Patient Register. The Swedish population Census Bureau Register is connected to the medical record system Melior™ and provides a daily update on case fatalities in order to get dates of death. Validation of diagnostic codes using the medical records was performed by a cardiologist with knowledge in the field (PM) and consultation of another expert (SM) when appropriate. The validation was based on guideline definition of HCM.1 The validation work was part of a study on HCM and the current study was approved by The Regional Ethical Committee in Uppsala (protocol number 2016/280), which was conducted in compliance with the Declaration of Helsinki. The committee did not require that written informed consent be obtained from the patients as all data was de-identified.
Statistics
Data were described as frequencies and percentages, and continuous variables were expressed as a mean value with a standard deviation (SD). A Student’s t-test was used for the comparison of continuous variables, and chi-squared test was used for categorical variables. A two-sided P-value of <0.05 was considered statistically significant. The positive predictive value refers to the probability that individuals with a diagnosis of HCM truly have the condition HCM and was calculated as the number of “true positives” divided by the sum of “true positives and false positives”. The file in Excel 2010 (Microsoft Corporation, Redmond, WA, USA) was transferred into SPSS Version 22 (IBM Corporation, Armonk, NY, USA).
Results
A total of 136 patients were diagnosed (principal or secondary diagnosis) at least once with HCM (I42.1 and/or I42.2). Seven of these patients were not permanent residents but only temporarily in the region; one of them was misclassified as HCM but was, in fact, situs inversus. Of the remaining 129 patients, 88 were correctly classified as HCM (positive predictive value 68.2%), while 41 patients (31.8%) were misclassified as HCM. Among the 88 HCM patients, 74 were alive and 14 were dead (15.9%) at the time of our survey. This yields an HCM prevalence of 74/183,337, that is, 4.0 cases per 10,000 in the adult population aged ≥18 years.
HCM patients
The characteristics of HCM patients are summarized in Table 1. Slightly more than half of the cohort was male (n=46; 52.2%). The mean age was 65.8±15.7 years among the 74 living HCM patients. The mean age was different between males and females (60.3 vs 73.2 years; P<0.001). Concomitant hypertension that could not solely explain the hypertrophy that was diagnosed in 34% (n=30) of patients and these patients are discussed in more detail in Table S1. The localization of the maximal wall thickness was predominantly the left ventricular septum (n=81; 92%) with the others atypically localized to the apex, posterior, or lateral wall or showing concentric distribution. Echocardiogram was the mode of diagnosis in all cases, although CMR was an adjunctive diagnostic tool in a few cases. Genetic analysis confirmed a disease-causing mutation in a fourth of the patients.
 | Table 1 Characteristics of 88 patients with validated HCM diagnosis Notes: aAmong living patients. bIschemic heart disease requiring intervention. cAmong patients without previous myectomy/alcohol septal ablation. Abbreviations: COPD, chronic obstructive pulmonary disease; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter defibrillator; LVEF, left ventricular ejection fraction; SD, standard deviation. |
One patient had a heart transplant due to end-stage HCM, but in the vast majority (86.2%, 75/87) of the remaining patients, beta-blockade was the pharmacological treatment. In one patient, beta-blockade was combined with a calcium channel antagonist and five patients were administered isoptin monotherapy. The choice and dosage of beta-blockers varied: metoprolol (n=38; 43.7%), bisoprolol (n=29; 33.3%), carvedilol (n=4; 4.6%), atenolol (n=3; 3.4%), and propranolol (n=1; 1.1%).
The causes of deaths among HCM patients were progression to end-stage heart failure with depressed ejection fraction (n=7), sudden cardiac death due to ventricular arrhythmia (n=2) and cerebral infarction (n=1), a combination of chronic obstructive pulmonary disease and the early phase of end-stage heart failure (n=1), perioperative myectomy death (n=1), and noncardiovascular causes (n=2) (renal failure in one patient and abdominal ileus in the other). The mean age at death was 78.8 years and ranged from 43 to 90 years.
Misclassifications
The most common misclassifications were hypertension (n=13; 31.7%) and aortic stenosis (n=9; 22%). Among the 13 cases of hypertension, six patients had a history of maximal hypertrophy of ≥15 mm at any echocardiographic evaluation, while the remaining patients had less pronounced hypertrophy or no hypertrophy at all (Table S2). Other forms of cardiomyopathy without a history of hypertrophy accounted for several cases of misclassification: dilated (nonischemic or ischemic), left ventricular noncompaction, and Takotsubo. Amyloidosis was diagnosed in four cases prior to the publication of new European guidelines, which is in contrast to American guidelines, suggesting that amyloidosis may be a subgroup of HCM.1,2,15 Valvular dysfunction, such as pulmonary stenosis, combined with ventricular septal defect and aortic insufficiency was noted among the misclassfications. In addition, athelete’s heart was wrongly interpreted as HCM (n=1) and, in another case, a conduction abnormality in the absence of structural disease was misclassified. The case of athelete’s heart (the patient was a long-distance runner and weight lifter) was evaluated several times, once with the hypertrophy of 16 mm, but an expert opinion assessed it at 13–14 mm. When the patient decreased training intensity, further regression followed. The complete list of categories of misclassification is shown in Table 2.
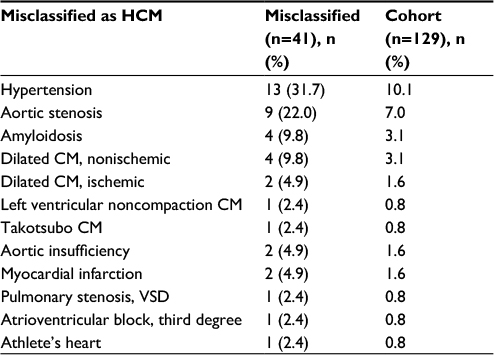 | Table 2 Patients with HCM-related diagnostic codes who were misclassified Abbreviations: CM, cardiomyopathy; HCM, hypertrophic cardiomyopathy; VSD, ventricular septal defect. |
The mean age among the living misclassified patients was 70.1 years and ranged from 46 to 96 years. There were seven fatal cases (16.7%) among the misclassified patients attributed to end-stage heart failure (n=5), stroke (n=1), and sepsis (n=1).
Comparison between HCM and misclassifications
The mean age among living HCM and misclassified patients did not differ significantly (65.8±15.8 vs 70.1±13.4 years; P=0.177). There were 47.8% (n=42) females among HCM and 60.8% (n=25) females among misclassified, but the difference was not significant (P=0.118).
Discussion
HCM remains underdiagnosed
In this Swedish regional cohort, we confirmed the diagnosis of HCM in 0.040%, which corresponds to 1:2,478. This is in line with US findings of 1:3,195 (0.031%) using diagnostic codes during year 2013 in a large sample based on insurance statistics.12 We share the limitation that the true prevalence of HCM cannot be derived from these databases and sensitivity remains unknown because false classification of true HCM is not accounted for. Nevertheless, the discrepancy from prevalence studies based on outreach programs is striking.6–10 HCM likely goes undiagnosed in some patients because they are asymptomatic, have vague symptoms, or do not get included in family screenings.
Diverse diagnoses underlie misclassifications
Validation of HCM-related diagnoses revealed a large proportion of misclassification. Approximately one-third of the sample had an incorrect HCM diagnosis from a cardiology/internal medicine facility, which only underlines the diagnostic difficulties in a real-world setting. Notably, there were cases without verified hypertrophy at any cardiac imaging evaluation, which we interpreted as unawareness of the diagnostic cutoff for hypertrophy in HCM. Moreover, slight or borderline hypertrophy in the setting of hypertension was occasionally overlooked, especially when these conditions were persistent and uncontrolled. Guidelines pinpoint that conditions such as severe aortic stenosis and hypertension should be excluded if it explains the hypertrophy, but our findings suggest that this has not been fully implemented in clinical practice.1 To address this issue, continuous educational efforts in physician training and close cooperation with highly specialized units are needed. Increased knowledge among cardiologists will improve specificity, but in order to affect sensitivity, ie, to achieve a accurate diagnoses of HCM patients, a more widespread approach is warranted. Increased awareness of HCM throughout the health care system is crucial as symptoms, such as shortness of breath, chest pain, dizziness, and syncope, are common but may relate to any number of causes. An ECG may show signs of hypertrophy and/or T-wave abnormalities in HCM.1,16 Echocardiography, although widely available, requires some clinical discernment, and the diagnosis of HCM should be made only after considering other clinical information.1,2,17–20 This also includes cases of hypertrophy regression within a year after controlled hypertension. We noted that only a fourth of HCM patients were genopositive. The finding of a genetic mutation that confirms the disease may help to identify other individuals in the family with HCM and requires cascade screening in collaboration with a molecular genetic unit capable of providing an HCM relevant panel.1,2 From our data, we could not determine what percentage of patients underwent genetic testing and why some patients did not.
Reflection on register data
A Danish effort to study the diagnostic validity of cardiovascular diagnoses included 20 HCM patients; after scrutinizing the medical records, 18 of the diagnoses were deemed correct.21 In general, the Swedish National Patient Register data are considered reliable and are used for research and the evaluation of health care quality.13 However, the findings from our study underscore that the quality of these data may vary with regard to diagnoses. Registry data should be carefully evaluated, since diagnostic errors might be introduced to cause-of-death registries if invalidated data are taken at face value. Of course, the extent of such errors depends on the nature and purpose of the registry search. Fortunately, in this limited sample, the mean age and sex distribution did not significantly vary between validated HCM patients and misclassifications, which implies less risk of differential bias in epidemiological studies.
Clinical perspectives on HCM
Few diagnoses have had so many different names as HCM. Over the decades, >75 names have been used to describe the entity nowadays known as HCM.22 Today, there are two diagnostic codes I42.1 and I42.2 to cover HCM, but in our cohort, they were often used interchangeably. Obstruction as a diagnostic criterion imposes some difficulties in that it is dynamic, may vary with conditions, such as dehydration, and can change with pharmacological regimens. Moreover, observers may vary in how they assess and measure obstruction. The same holds true for outflow gradient, which can change over time in the same patient. All of these things make it difficult to diagnose “obstructive” accurately. While obstruction is not necessary for an HCM diagnosis, it may add confusion to the diagnostic workup. In addition, disease progression can vary substantially among patients and end-stage heart failure is a pathway that often results in premature death.23
Atrial fibrillation is common in HCM and warrants anticoagulation.24 Because correct risk stratification with regard to sudden cardiac death is may improve patient survival, a correct diagnosis of HCM is crucial. Unfortunately, it still seems to be common to overlook cardiac symptoms preceding sudden death in HCM.25 Nevertheless, in general, mortality is low among HCM patients with contemporary management.26
Overall, the heterogeneous nature of HCM has to be recognized and each patient followed, according to guidelines, and managed by a multidisciplinary team, which brings expertise in heart failure, cardiac imaging, genetics, electrophysiology, implantable cardioverter defibrillators, and caring sciences in holistic patient-centered care.1,2 In addition to specialized HCM teams, we advocate the implementation of educational efforts to a wider range of health care professionals in order to increase general awareness of HCM and raise clinical suspicion for potential HCM cases. Based on the high percentage of misclassifications, it seems necessary to organize and promote multidisciplinary teams with readily available resources to accurately diagnose and optimally treat HCM.
Strength and weaknesses
This observational study on diagnostic codes with relevance to HCM covers all patients in a regional cohort without selection bias, as all patients are covered by a national insurance system. The high proportion of misclassifications in our study should to be addressed in other geographical areas and health care systems. This study is unable to estimate sensitivity and, thus, cannot address the negative predictive value of HCM. It is likely that in a heterogeneous disease, numerous patients are undiagnosed/misdiagnosed, which implies a high proportion of false negative resulting in lower sensitivity. Due to the small sample size,, statistical hypothesis testing may be subject to type II errors.
Conclusion
The number of diagnosed HCM is lower than that suggested in population studies. One-third of patients diagnosed with HCM is misclassified as miscellaneous cardiac conditions, mainly hypertension, and aortic stenosis. A correct diagnosis is not only important for individual decision-making on disease management but also important for research purposes. An implementation of improved HCM care should include efforts to increase awareness and competence throughout the health care system.
Acknowledgments
The authors acknowledge editing by Jo Ann LeQuang from LeQ Medical who reviewed the manuscript for American English use. Britt-Inger Reiterer from Region Gävleborg provided data extraction from the Cyklop™ register. Region Gävleborg funded this research project.
Author contributions
PM contributed to the design, statistics, data analysis and interpretation, writing the article, and project management. AP contributed to the design, critical revision, and project management. EB contributed to the design and critical revision. SM contributed to the design, data interpretation, and critical revision. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–2779. | ||
Sen-Chowdhry S, Jacoby D, Moon J, McKenna WJ. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat Rev Cardiol. 2016;13(11):651–675. | ||
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He Jiang J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–223. | ||
Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368(9540):1005–1011. | ||
Iung B, Baron G, Butchart EG, et al. A prospective survey of patients with valvular heart disease in Europe: the euro heart survey on valvular heart disease. Eur Heart J. 2003;24(13):1231–1243. | ||
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation. 1995;92(4):785–789. | ||
Zou Y, Song L, Wang Z, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116(1):14–18. | ||
Hada Y, Sakamoto T, Amano K, et al. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am J Cardiol. 1987;59(1):183–184. | ||
Maron BJ, Spirito P, Roman MJ, et al. Prevalence of hypertrophic cardiomyopathy in a population-based sample of American Indians aged 51 to 77 years (the Strong Heart Study). Am J Cardiol. 2004;93(12):1510–1514. | ||
Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol. 1999;33(6):1590–1595. | ||
McKenna WJ, Sen-Chowdhry S. From Teare to the present day: a fifty year odyssey in hypertrophic cardiomyopathy, a paradigm for the logic of the discovery process. Rev Esp Cardiol. 2008;61(12):1239–1244. | ||
Maron MS, Hellawell JL, Lucove JC, Farzaneh-Far R, Olivotto I. Occurrence of clinically diagnosed hypertrophic cardiomyopathy in the United States. Am J Cardiol. 2016;117(10):1651–1654. | ||
Ludvigsson JF, Andersson E, Ekbom A, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450. | ||
Statistikdatabasen. [Statistics Sweden]; 2016. Available from: http://www.statistikdatabasen.scb.se/pxweb/sv/ssd/START__BE__BE0101__ BE0101A/BefolkningNy/?rxid=67c80f1d-e68b-424b-981d-c843cf 448195. Accessed Dec 28, 2016. | ||
Gersh BJ, Maron BJ, Bonow RO, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation. 2011;124(24):2761–2796. | ||
McLeod CJ, Ackerman MJ, Nishimura RA, Tajik AJ, Gersh BJ, Ommen SR. Outcome of patients with hypertrophic cardiomyopathy and a normal electrocardiogram. J Am Coll Cardiol. 2009;54(3):229–233. | ||
Smith N, Steeds R, Masani N, et al. A systematic approach to echocardiography in hypertrophic cardiomyopathy: a guideline protocol from the British Society of Echocardiography. Echo Res Pract. 2015;2(1):G1–G7. | ||
Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2013;34(19):1448–1458. | ||
Losi MA, Nistri S, Galderisi M, et al; Working Group of Echocardiography of the Italian Society of Cardiology. Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc Ultrasound. 2010;8:7. | ||
Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26(7):1699–1708. | ||
Sundbøll J, Adelborg K, Munch T, et al. Positive predictive value of cardiovascular diagnoses in the Danish National Patient Registry: a validation study. BMJ Open. 2016;6(11):e012832. | ||
Maron BJ, Maron MS. A discussion of contemporary nomenclature, diagnosis, imaging, and management of patients with hypertrophic cardiomyopathy. Am J Cardiol. 2016;118(12):1897–1907. | ||
Pasqualucci D, Fornaro A, Castelli G, et al. Clinical spectrum, therapeutic options, and outcome of advanced heart failure in hypertrophic cardiomyopathy. Circ Heart Fail. 2015;8(6):1014–1021. | ||
Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465–472. | ||
Lynge TH, Risgaard B, Jabbari R, et al. Cardiac symptoms before sudden cardiac death caused by hypertrophic cardiomyopathy: a nationwide study among the young in Denmark. Europace. 2016;18(12):1801–1808. | ||
Maron BJ, Rowin EJ, Casey SA, et al. Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J Am Coll Cardiol. 2015;65(18):1915–1928. |
Supplementary materials
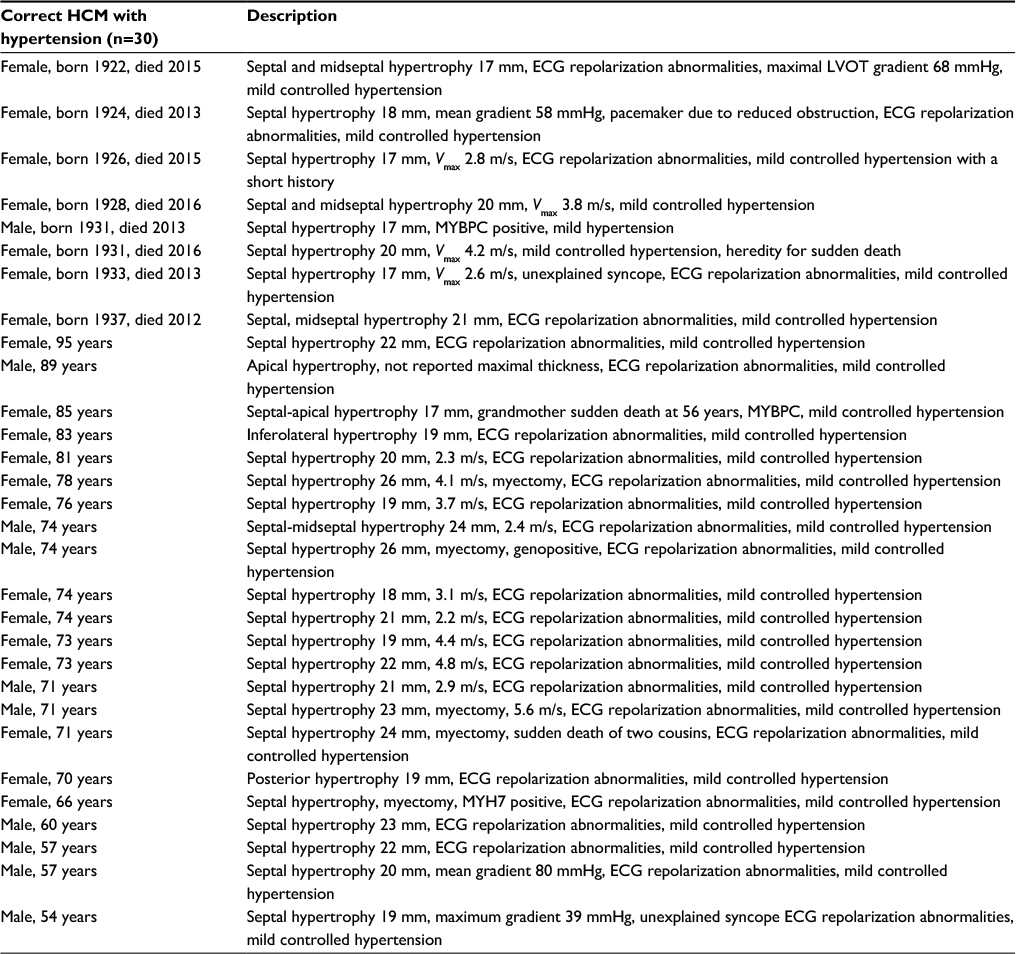 | Table S1 Patients with HCM and concomitant hypertension Abbreviations: ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; LVOT, left ventricular outflow tract; MYBPC, myosin binding protein C; Vmax, velocity maximal. |
 | Table S2 Patient with hypertension misclassified as HCM Abbreviation: HCM, hypertrophic cardiomyopathy. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.