")
Back to Journals » OncoTargets and Therapy » Volume 7
miR-7 inhibits colorectal cancer cell proliferation and induces apoptosis by targeting XRCC2
Authors Xu K , Chen Z, Qin C , Song X
Received 18 December 2013
Accepted for publication 16 January 2014
Published 20 February 2014 Volume 2014:7 Pages 325—332
DOI https://doi.org/10.2147/OTT.S59364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Kaiwu Xu, Zhihui Chen, Changjiang Qin, Xinming Song
Gastrointestinal and Pancreatic Surgery Department, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, People’s Republic of China
Background: Analysis using publicly available algorithms predicts that X-ray repair complementing defective repair in Chinese hamster cells 2 (XRCC2), a key component in the homologous recombination repair pathway, is a potential target of micro-ribonucleic acid-7 (miR-7). Some studies have shown that both miR-7 and XRCC2 are associated with cancer development. For this purpose, we searched for the possible relationship between miR-7 and XRCC2 in the development of colorectal cancer (CRC).
Methods: miR-7 expression was assessed in CRC specimens and cell lines using real-time polymerase chain reaction (PCR). Luciferase reporter assay was used to confirm the target associations. The effect of miR-7 on cell proliferation and apoptosis was confirmed in vitro by the methylthiazol tetrazolium (MTT) assay, colony formation assay, and flow cytometry. Gene and protein expression were examined using real time PCR and western blotting, respectively.
Results: miR-7 was downregulated in CRC specimens and cell lines, and targeted the 3' untranslated region of XRCC2. miR-7 overexpression reduced cyclin D1 expression and increased p21, caspase-3, and BAX expression, which subsequently inhibited CRC cell proliferation and induced CRC cell apoptosis. However, XRCC2 can repress the inhibitory effects of miR-7 on proliferation.
Conclusion: Our findings suggest that miR-7 plays a protective role by inhibiting proliferation and increasing apoptosis of CRC cells. It may identify new targets for anti-cancer treatment.
Keywords: MiRNA, DNA, overexpression, downregulated
Introduction
Colorectal cancer (CRC) is one of the most malignant types of cancers. In the United States and Europe, it is the second most frequent cancer (after lung cancer) that leads to death.1 In the People’s Republic of China, it has been reported that the incidence of CRC increases annually and will continue to rise in the next few years.2 Currently, approximately 1.25 million people are diagnosed with CRC, and more than 600,000 patients will die from it every year around the world.3 In recent years, significant advances have been made in the integration of targeted therapies in CRC treatment. However, better targeted drugs are required because the effects of existing drugs are not satisfactory.
X-ray repair complementing defective repair in Chinese hamster cells 2 (XRCC2) is located at 7q36.1 and encodes a member of the Rad51-related protein family that participates in homologous recombination to maintain chromosome stability and repairs deoxyribonucleic acid (DNA) damage. This gene is involved in the homologous recombination repair (HRR) pathway of double-stranded DNA; it is believed that it repairs chromosomal fragmentation, translocations, and deletions.4–6 Some studies have shown that XRCC2 is associated with the development of CRC. Curtin et al found that XRCC2 single-nucleotide polymorphisms can increase the risk of CRC.7,8 Haines et al found that XRCC2 haploinsufficiency reduced spontaneous tumor incidence in a multiple intestinal neoplasia (min/+) mice background but increased the tumorigenic response to radiation.9
Micro-ribonucleic acids (miRNAs) are a class of small noncoding ribonucleic acids (RNAs) that can negatively regulate posttranscriptional gene expression, mainly by binding to the 3′ untranslated region (UTR) of a target gene, and can act as tumor suppressors or oncogenes. Recent advances in miRNA research have provided new insights into the mechanisms driving CRC.10,11 Numerous studies have demonstrated that aberrant miRNA expression is closely associated with cellular proliferation, differentiation, apoptosis, and tissue development.10,11 An increasing amount of evidence finds that miRNAs are involved in the development of different human cancers, and they have been widely proposed as potential targets for anticancer therapies.12–15 Recently, multiple miRNAs were implicated in CRC progression, such as miR-622,16 miR-34b,17 miR-1226,18 miR-150,19 miR-561,20 miR-608,21 and miR-153.22
Publicly available algorithms (TargetScan, PicTar, miRanda) indicate that mirR-7 (micro-ribonucleic acid-7) may directly target the 3′ UTR of XRCC2. In our study, we demonstrated that miR-7 inhibits CRC cell proliferation and induces apoptosis by targeting the 3′ UTR of XRCC2 mRNA. Our results suggest that miR-7 may play an important role in CRC tumorigenesis.
Materials and methods
Patients and ethics statement
The eight malignant CRC tissues and adjacent noncancerous tissues used in this study were obtained from patients who underwent surgery at the First Affiliated Hospital of Sun Yat-Sen University, People’s Republic of China. The Sun Yat-Sen University and First Affiliated Hospital Institutional Ethical Board approved the use of clinical materials for research purposes in this study, and written informed consent was obtained from all patients.
Cell culture
Normal colonic mucosa epithelial cells were isolated and purified from the adjacent noncancerous tissues of the patient who underwent surgery at our hospital. CRC cell lines SW620, SW480, LoVo, RKO, and LS174T were obtained from American Type Culture Collection (Manassas, VA, USA). Cell lines were cultured in Dulbecco’s Modified Eagle Medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin. Colorectal normal tissue was used as the normal control. All cells were maintained in a humidified atmosphere at 37°C with 5% CO2.
RNA extraction, reverse transcription, and real-time PCR
Total RNA was extracted from the cultured cells, CRC tumors, and the adjacent noncancerous tissues using TRIzol® (Life Technologies) according to the manufacturer’s instructions. Real-time quantitative reverse transcription–polymerase chain reaction was performed using SYBR® Green I (Life Technologies) with an ABI 7500 system (Life Technologies). Gene expression data were normalized to the geometric mean of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene to control for variability in expression levels and calculated as:
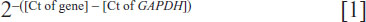
where Ct represents the threshold cycle for each transcript. Quantitation of miRNA was based on the Ct, and relative expression was calculated as:
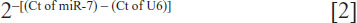
after normalization to U6 small nuclear RNA. The following primers were used:
- p21 forward: 5′-CGATGCCAACCTCCTCAACGA-3′, p21 reverse: 5′-TCGCAGACCTCCAGCATCCA-3′;
- cyclin D1 forward: 5′-AACTACCTGGA CCGCTTCCT-3′, cyclin D1 reverse: 5′-CCACTTGAGCTTGTTCACCA-3′;
- Bcl-xL forward: 5′-TTGGACAATGGACTGGTTG-3′, Bcl-xL reverse: 5′-GTAGAGTGGATGGTCAGTG-3′;
- caspase-3 forward: 5′-ATGGGAGCAAGTCAG TGGAC-3′, caspase-3 reverse: 5′-CGTACCAGAGCG AGATGACA-3′;
- GAPDH forward: 5′-GACTCATGACCACAGTCC ATGC-3′, GAPDH reverse: 5′-AGAGGCAGGGATGAT GTTCTG-3′;
- XRCC2 forward: 5′-TCACCTGTGCATGGTG ATATT-3′, XRCC2 reverse: 5′-TTCCAGGCCACCTTC TGATT-3′.
Western blotting
Western blotting was performed according to standard methods as previously described.23 Membranes were probed with polyclonal rabbit antibodies against XRCC2 (1:1,500; Abcam, Cambridge, UK), cyclin D1, p21, retinoblastoma protein, phosphorylated retinoblastoma protein, caspase-3, BAX, and Bcl-xL (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Then, the membranes were stripped and reprobed with anti-β-actin mouse monoclonal antibody (1:1,000; Sigma-Aldrich, St Louis, MO, USA) as a loading control.
Plasmids and transfection
The 100–900 base pair region of the human XRCC2 3′ UTR was PCR-amplified from genomic DNA from SW480 cells and cloned into pGL3 vectors (Promega Corporation, Fitchburg, WI, USA). Transfection of miR-7 mimic, miR-7 inhibitor, negative control (NC), XRCC2-small interfering RNA (siRNA; RiboBio Co, Ltd, Guangzhou, People’s Republic of China), and plasmids were performed using Lipofectamine® 2000 (Life Technologies) according to the manufacturer’s instruction.
Methylthiazol tetrazolium assay
Methylthiazol tetrazolium (MTT) assay was performed to quantify CRC cell viability following transfection with miR-7 mimic, miR-7-inhibitor, and NC. Cells were stained with 100 μL sterile 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (0.5 mg/mL; Sigma-Aldrich) for 4 hours at 37°C. Culture medium was then removed and formazan crystals in the cells were solubilized using dimethyl sulfoxide (Sigma-Aldrich) with plate shaking for 30 minutes. Cells were incubated for 36 hours at 37°C in a humidified 5% CO2 atmosphere. Absorbance was measured at 570 nm with a reference wavelength of 655 nm. All experiments were performed in triplicate.
Colony formation and apoptosis assays
Cells were seeded on a 6-well plate (0.5 × 103 cells per well) and cultured for 10 days. Colonies were fixed for 5 minutes with 10% formaldehyde and stained with 1.0% crystal violet for 1 minute. The experiment was performed independently three times for each cell line. For the apoptosis assay, cultured cells were harvested 72 hours following transfection by trypsinization and washed with phosphate-buffered saline. Then,1 × 106 cells from each sample were processed for Annexin V FITC/PI apoptosis detection (BD, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. The result was assessed by FACSCalibur flow cytometry (BD).
Luciferase reporter assay
Cells (5 × 105 cells per well) were seeded in triplicate wells in 6-well plates and allowed to settle for 12 hours. XRCC2 3′ UTR luciferase plasmid (100 ng) or control luciferase plasmid (100 ng) plus 10 ng pRL-TK Renilla plasmid (Promega) were transfected into CRC cells using Lipofectamine® 2000 (Life Technologies). Culture medium was replaced after 6 hours. Cells were harvested 48 hours after transfection, and luciferase activity was measured using a Dual-Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s protocol. Three independent experiments were performed and the data are presented as the mean ± standard deviation.
Statistical analysis
The Student’s t-test was used to evaluate significant differences between two groups of data in all relevant experiments. All results are presented as means ± standard deviation; P<0.05 (two-tailed paired t-test) was considered statistically significant.
Results
miR-7 was downregulated in CRC cell lines and clinical tissues
Real-time PCR showed that miR-7 was markedly downregulated in all five CRC cell lines compared with expression in normal colonic mucosa epithelial cells. (Figure 1A). miR-7 expression was examined in eight CRC tumors and in the matched adjacent noncancerous tissues from the same patient using real-time PCR. miR-7 was significantly downregulated in tumors compared with normal tissues (Figure 1B).
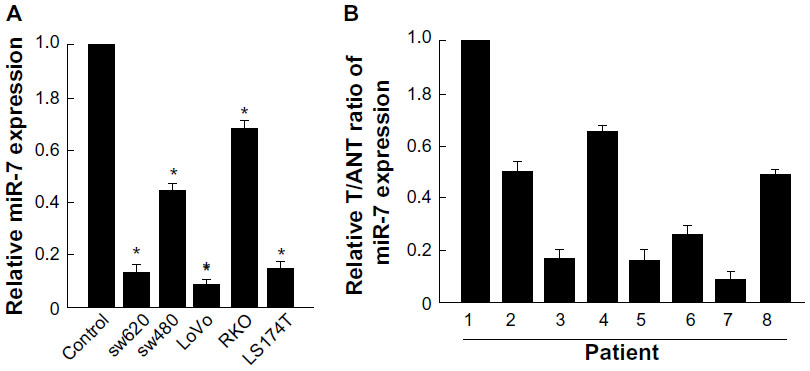 | Figure 1 miR-7 is downregulated in CRC cell lines and tissues. |
miR-7 overexpression inhibited proliferation and induced apoptosis in CRC cells
To investigate whether miR-7 plays a role in the development and progression of CRC, we transfected SW480 cells with miR-7 mimic, miR-7 inhibitor, and NC RNA and examined the effect of miR-7 overexpression on CRC cell proliferation by MTT assay and colony formation assay. MiR-7 upregulation significantly decreased the growth rate of SW480 cells following 48-hour transduction with miR-7 mimic compared with miR-7 inhibitor (Figure 2A and C). The rate of apoptosis in SW480 cells transfected with miR-7 mimic was significantly higher that than in NC or miR-7 inhibitor transfected cells (Figure 2B). As expected, miR-7 overexpression increased p21 expression while significantly decreasing cyclin D1 expression (Figure 3A), and significantly increased caspase-3 and BAX expression while decreasing Bcl-xL expression (Figure 3B). The real-time PCR analysis results were consistent with those of the western blotting assays (Figure 3C). These findings indicate that miR-7 may play an important role in the proliferation and apoptosis of CRC cells.
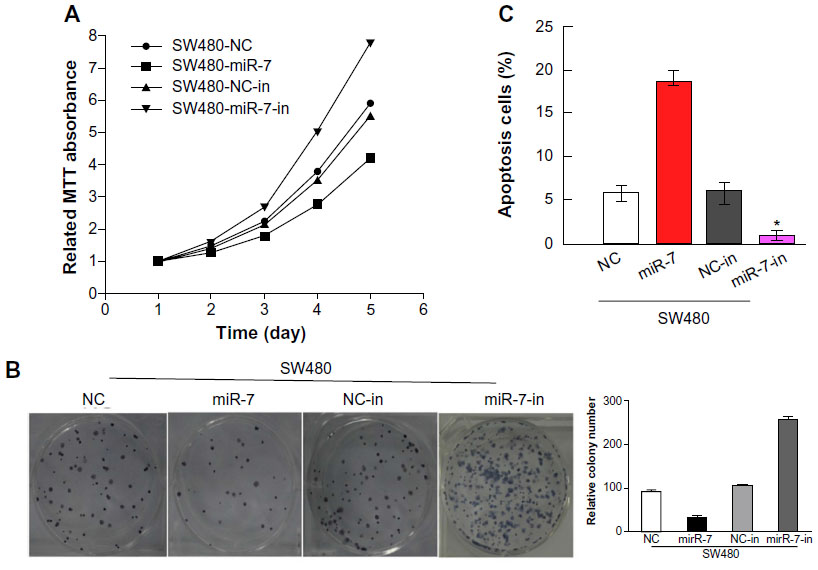 | Figure 2 Upregulation of miR-7 inhibits CRC cell proliferation. |
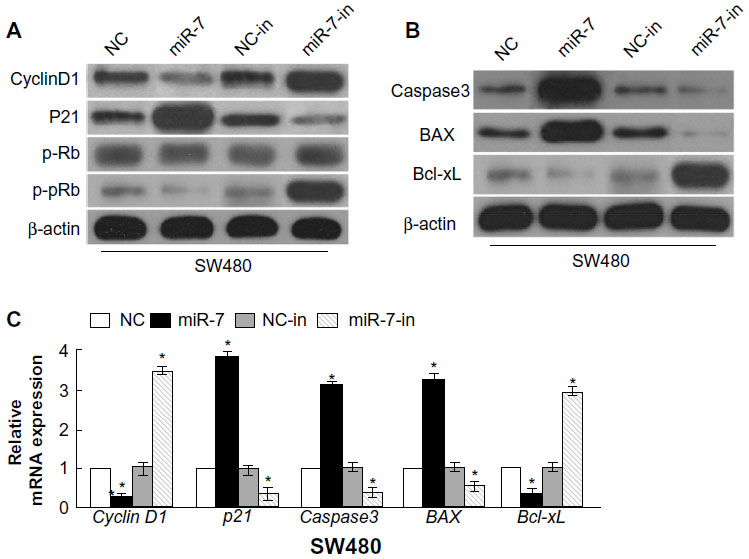 | Figure 3 Gene and protein expression in transfected cells. |
MiR-7 targeted XRCC2 in CRC cells directly
We used the prediction programs TargetScan, PicTar, and miRanda to analyze the potential interaction between miR-7 and XRCC2, which is a potential target of miR-7 and a key member of the HRR pathway (Figure 4A). To examine whether XRCC2 is a direct target of miR-7, the human XRCC2 wild-type 3′ UTR containing the miR-7-binding site and mutated XRCC2 3′ UTR sequence were cloned into modified pGL-3 luciferase reporter vectors, which were cotransfected into SW480 cells with NC, miR-7 mimic, or miR-7 inhibitor. miR-7 overexpression reduced the luciferase reporter activity of the XRCC2 3′ UTR in a consistent and dose-dependent manner; inhibition of miR-7 had the opposite effect. However, XRCC2 3′ UTR luciferase reporter activity was unaffected by point mutations in the miR-7-binding seed region (Figure 4B). Collectively, these data suggest that miR-7 may inhibit post-transcriptional XRCC2 expression by targeting its 3′ UTR. These results demonstrate that XRCC2 is a bona fide target of miR-7. As predicted, western blotting revealed that XRCC2 expression was decreased in SW480 cells transfected with miR-7 mimic and was increased in cells transfected with miR-7 inhibitor (Figure 4C).
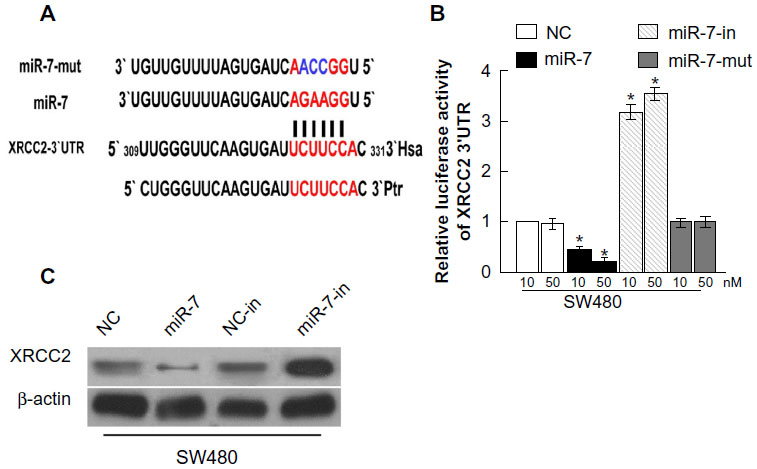 | Figure 4 miR-7 directly targets XRCC2. |
XRCC2 repressed the function of miR-7 in SW480 cells
miR-7 was downregulated in CRC cell lines and clinical tissues (Figure 1), while the expression of XRCC2 in tumor tissue was significantly higher than the matched adjacent noncancerous tissues (Figure 5A). Meanwhile, the results of the western blotting showed that XRCC2 expression in SW480 cells transfected with the miR-7 inhibitor and XRCC2-siRNA were significantly lower than in the cells transfected with miR-7 inhibitor alone (Figure 5B). SW480 cells transfected with a miR-7 inhibitor were more viable than controls (Figure 2C). However, those transfected with both the miR-7 inhibitor and XRCC2-siRNA had significantly decreased growth rates compared to treatment with the miR-7 inhibitor alone (Figure 5C). These findings suggested that XRCC2 may repress the inhibitory effects of miR-7 on proliferation.
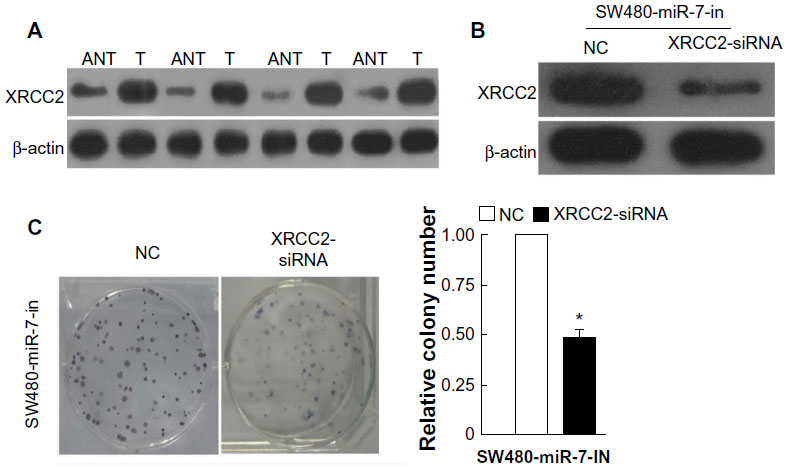 | Figure 5 The expression of XRCC2 and colony formation assays for the function of XRCC2 on miR-7. |
Discussion
miR-7 has been characterized as a tumor suppressor in several human cancers that targets epidermal growth factor receptor, p21-activated kinase 1, and other genes.24 Liu et al demonstrated that miR-7 suppressed cell growth and induced apoptosis in cervical cancer cells.25 miR-7 downregulation was reported to be associated with epithelial-to-mesenchymal transition and metastasis in breast cancer.26 Kong et al found that miR-7 expression was significantly decreased in gastric tumors and that miR-7 downregulation was related to the severity of inflammatory response.27 Xiong et al observed miR-7 reduction in non-small cell lung cancer cell lines, finding that miR-7 overexpression induced non-small cell lung cancer cell apoptosis and suppressed cell proliferation, migration, and tumorigenicity by targeting Bcl-2.28 Skalsky and Cullen, and Kefas et al reported that miR-7 expression was downregulated in glioblastoma, potently suppressing epidermal growth factor receptor expression and independently inhibiting the protein kinase B pathway by targeting upstream regulators.29,30 These data are consistent with our findings that miR-7 acts as a tumor suppressor. However, a study reported that miR-7 was upregulated in renal cancer cells compared with normal tissues and that miR-7 downregulation inhibited cell proliferation and induced apoptosis, suggesting that miR-7 could be characterized as an oncogene in renal cancer cells.31
Bioinformatics analysis predicted that XRCC2 was a potential direct target of miR-7. We deduced that miR-7 may function by interacting with the XRCC2 gene. To validate this hypothesis experimentally, we searched for a correlation between miR-7 and XRCC2 expression levels. We demonstrated that XRCC2 is a bona fide miR-7-targeted gene. Furthermore, these results showed that miR-7 was downregulated in CRC cell lines and clinical tissue samples, while the expression of XRCC2 in tumor tissue was significantly higher than in the matched adjacent noncancerous tissues. Moreover, we determined that XRCC2 may repress the inhibitory effects of miR-7 on cell proliferation. However, the function of XRCC2 in the ability of miR-7 to induce apoptosis was not studied. Furthermore, how the level of XRCC2 affects the expression of miR-7, and whether miR-7 functions through interactions with the XRCC2-dependent HRR signaling pathway, remains unclear. Multiple signaling pathways are thought to make important contributions to CRC progression. Further studies are necessary to clarify these complex mechanisms, and much remains to be done before miR-7 can be used for downregulating XRCC2 in CRC patients.
CRC is a highly malignant tumor that leads to more than 600,000 deaths worldwide each year.3 In the present study, miR-7 expression was markedly downregulated in CRC cells and clinical tissues compared with adjacent noncancerous tissues from the same patient. Furthermore, we demonstrated that miR-7 overexpression in CRC cells targeted XRCC2 to inhibit cell proliferation, while miR-7 downregulation had the opposite effect. Although the precise mechanisms are not fully understood, these findings nevertheless suggest that miR-7 may play an important role in the proliferation of CRC.
In conclusion, we found that miR-7 can reduce cyclin D1 expression and increase p21, caspase-3, and BAX expression, which subsequently inhibits CRC cell proliferation and induces apoptosis by targeting the 3′ UTR of XRCC2. Our findings suggest that miR-7 may play an important role in the development and progression of CRC. Further exploration of the molecular link between miR-7 and XRCC2 will advance our understanding of the intricate molecular regulation of CRC cells and may provide new targets for anticancer treatment.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (NSFC-2011-81172339) and Guangdong Natural Science Foundation (2011B031800118).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. | |
Dai Z, Zheng RS, Zou XN, et al. [Analysis and prediction of colorectal cancer incidence trend in China]. Zhonghua Yu Fang Yi Xue Za Zhi. 2012;46(7):598–603. Chinese. | |
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | |
Jones NJ, Zhao Y, Siciliano MJ, Thompson LH. Assignment of the XRCC2 human DNA repair gene to chromosome 7q36 by complementation analysis. Genomics. 1995;26(3):619–622. | |
Thacker J, Tambini CE, Simpson PJ, Tsui LC, Scherer SW. Localization to chromosome 7q36.1 of the human XRCC2 gene, determining sensitivity to DNA-damaging agents. Hum Mol Genet. 1995;4(1):113–120. | |
Liu N, Lamerdin JE, Tebbs RS, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1(6):783–793. | |
Curtin K, Lin WY, George R, et al. Genetic variants in XRCC2: new insights into colorectal cancer tumorigenesis. Cancer Epidemiol Biomarkers Prev. 2009;18(9):2476–2484. | |
Krupa R, Sliwinski T, Wisniewska-Jarosinska M, et al. Polymorphisms in RAD51, XRCC2 and XRCC3 genes of the homologous recombination repair in colorectal cancer – a case controlstudy. Mol Biol Rep. 2011;38(4):2849–2854. | |
Haines JW, Coster MR, Adam J, et al. Xrcc2 Modulates Spontaneous and Radiation-Induced Tumorigenesis in Apcmin/+ Mice. Mol Cancer Res. 2010;8(9):1227–1233. | |
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. | |
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | |
Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–436. | |
Petri A, Lindow M, Kauppinen S. MicroRNA silencing in primates: towards development of novel therapeutics. Cancer Res. 2009;69(2):393–395. | |
Stamatopoulos B, Meuleman N, Haibe-Kains B, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113(21):5237–5245. | |
Landi MT, Zhao Y, Rotunno M, et al. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin Cancer Res. 2010;16(2):430–441. | |
Della Vittoria Scarpati G, Falcetta F, Carlomagno C, et al. A specific miRNA signature correlates with complete pathological response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer. Int J Radiat Oncol Biol Phys. 2012;83(4):1113–1119. | |
Siemens H, Neumann J, Jackstadt R, et al. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and β-catenin predicts distant metastasis of colon cancer. Clin Cancer Res. 2013;19(3):710–720. | |
Ragusa M, Statello L, Maugeri M, et al. Specific alterations of the microRNA transcriptome and global network structure in colorectal cancer after treatment with MAPK/ERK inhibitors. J Mol Med (Berl). 2012,90(12):1421–1438. | |
Pizzini S, Bisognin A, Mandruzzato S, et al. Impact of microRNAs on regulatory networks and pathways in human colorectal carcinogenesis and development of metastasis. BMC Genomics. 2013;14(1):589. | |
Yu XF, Zou J, Bao ZJ, Dong J. miR-93 suppresses proliferation and colony formation of human colon cancer stem cells. World J Gastroenterol. 2011;17(42):4711–4717. | |
Ryan BM, McClary AC, Valeri N, et al. rs4919510 in hsa-miR-608 is associated with outcome but not risk of colorectal cancer. PLoS One. 2012;7(5):e36306. | |
Zhang L, Pickard K, Jenei V, et al. miR-153 supports colorectal cancer progression via pleiotropic effects that enhance invasion and chemotherapeutic resistance. Cancer Res. 2013,73(21):6435–6447. | |
Li J, Zhang N, Song LB, et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin Cancer Res. 2008;14(11):3319–3326. | |
Saydam O, Senol O, Würdinger T, et al. miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways. Cancer Res. 2011;71(3):852–861. | |
Liu S, Zhang P, Chen Z, Liu M, Li X, Tang H. MicroRNA-7 downregulates XIAP expression to suppress cell growth and promote apoptosis in cervical cancer cells. FEBS Lett. 2013;587(14):2247–2253. | |
Kong X, Li G, Yuan Y, et al. MicroRNA-7 inhibits epithelial-to-mesenchymal transition and metastasis of breast cancer cells via targeting FAK expression. PLoS One. 2012;7(8):e41523. | |
Kong D, Piao YS, Yamashita S, et al. Inflammation-induced repression of tumor suppressor miR-7 in gastric tumor cells. Oncogene. 2012;31(35):3949–3960. | |
Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Chu Y. MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. Int J Biol Sci. 2011;7(6):805–814. | |
Skalsky RL, Cullen BR. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS One. 2011;6(9):e24248. | |
Kefas B, Godlewski J, Comeau L, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68(10):3566–3572. | |
Yu Z, Ni L, Chen D, et al. Identification of miR-7 as an oncogene in renal cell carcinoma. J Mol Histol. 2013;44(6):669–677. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.