Back to Journals » OncoTargets and Therapy » Volume 13
miR-4999-5p Predicts Colorectal Cancer Survival Outcome and Reprograms Glucose Metabolism by Targeting PRKAA2
Authors Zhang Q, Hong Z, Zhu J, Zeng C, Tang Z, Wang W, Huang H
Received 15 October 2019
Accepted for publication 10 January 2020
Published 11 February 2020 Volume 2020:13 Pages 1199—1210
DOI https://doi.org/10.2147/OTT.S234666
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Qiwei Zhang,1 Zhi Hong,1 Jieyao Zhu,2 Chao Zeng,1 Zhen Tang,1 Weiqiang Wang,1 He Huang1
1Department of Gastrointestinal Surgery, The First Affiliated Hospital, Yijishan Hospital of Wannan Medical College, Wuhu 241000, Anhui, People’s Republic of China; 2Department of General Surgery, Lujiang County People’s Hospital, Hefei 231500, Anhui, People’s Republic of China
Correspondence: He Huang
Department of Gastrointestinal Surgery, The First Affiliated Hospital, Yijishan Hospital of Wannan Medical College, 2 Zheshan West Road, Jinhu District, Wuhu 241000, Anhui, People’s Republic of China
Tel +86 553 5739558
Fax +86 553 5738279
Email [email protected]
Purpose: Colorectal cancer (CRC) is the third most common cancer, and the second leading cause of cancer death worldwide. Dysregulation of microRNAs has been shown to modulate glucose metabolic reprogramming in CRC. However, the functional role of miR-4999-5p in the CRC glucose metabolic shift has not been characterized.
Patients and Methods: The levels of miR-4999-5p and PRKAA2 were evaluated by RT-qPCR. Univariate and multivariate survival analyses were conducted to evaluate the prognostic value of miR-4999-5p. Cell proliferation was assessed using the CCK-8 and colony formation assays. Extracellular acidification rate, glucose uptake, cellular glucose-6-phosphate level, and lactate production were evaluated to assess the effects of miR-4999-5p on CRC glycolysis. Dual-luciferase reporter assay was conducted to investigate the direct interaction between miR-4999-5p and PRKAA2. Mouse xenograft models were established to assess the functions of miR-4999-5p in vivo.
Results: miR-4999-5p was highly expressed in CRC tissues and cell lines. In addition, miR-4999-5p was associated with tumor differentiation and TNM stage, and elevated expression of miR-4999-5p was an independent predictor of poorer overall survival. Furthermore, miR-4999-5p promoted cell proliferation and glycolysis in CRC. miR-4999-5p targeted PRKAA2 to exert its tumor-promoting functions, and PRKAA2 knockdown rescued decreased cell proliferation and glycolysis in miR-4999-5p-silenced CRC cells. In vivo experiments showed that miR-4999-5p promoted CRC growth.
Conclusion: miR-4999-5p facilitated cell growth and glucose metabolic reprogramming through direct targeting of PRKAA2. Our results showed that miR-4999-5p may be a novel prognostic marker and therapeutic target for CRC.
Keywords: miR-4999-5p, PRKAA2, colorectal cancer, glycolysis, prognosis
Introduction
Colorectal cancer (CRC) is the third most common cancer, and is the second leading cause of cancer-related death, with 1.8 million new cases and 881,000 deaths worldwide in 2018.1 Characterization of the molecular mechanisms of CRC is essential for development of effective preventive strategies, early-stage detection approaches, and identification of novel therapeutic targets. Reprogramming of energy metabolism is a hallmark of cancer.2,3 Dysregulated glucose metabolism enables sustained tumor cell growth and division. Under normoxic conditions, cancer cells convert glucose to lactate, a phenomenon known as the Warburg effect.4,5 This metabolic shift toward aerobic glycolysis provides adequate macromolecules for biosynthesis, and minimizes production of reactive oxygen species.6 Therefore, elucidating the role of glucose metabolic reprogramming in CRC progression is of great importance.
MicroRNAs (miRNAs), a class of noncoding RNAs 21–23 nucleotides in length, inhibit target mRNA translation and expression via binding to the 3ʹ-untranslated region (3ʹ-UTR).7 A previous study showed that miR-143-3p inhibited CRC tumorigenesis and progression by targeting CTNND1.8 miR-125a-5p was shown to suppress CRC cell migration, invasion, and epithelial-mesenchymal transition by targeting TAZ.9 MicroRNAs have been shown to be crucial modulators of glucose metabolic reprogramming and cancer progression.10 A previous study showed that miR-3662 regulated the Warburg effect and suppressed hepatocellular carcinoma cell growth by decreasing HIF-1α levels.11 Overexpression of miR-140-5p significantly suppressed chronic myeloid leukemia cell proliferation and glucose metabolic reprogramming by targeting SIX1.12
The role of miR-4999-5p in malignancies has not been characterized. In the present study, we showed that miR-4999-5p was highly expressed in CRC tissues, and was associated with unfavorable prognosis. miR-4999-5p enhanced CRC cell proliferation and glucose metabolic reprogramming through suppression of PRKAA2. miR-4999-5p may be a prognostic indicator for CRC, and may be a novel target for CRC therapy.
Materials and Methods
Patients and Tissue Specimens
Fifty matched CRC tissues and adjacent non-cancer tissues were obtained from patients undergoing surgical resections at the First Affiliated Hospital of Wannan Medical College. None of the patients had received preoperative chemotherapy. Written informed consent was obtained from the enrolled patients. The ethics committee of the First Affiliated Hospital of Wannan Medical College reviewed and approved this study.
Cell Culture
CRC cell lines (LoVo, HCT116, Caco2, and HCT15) and a normal human colon epithelial cell line (FHC) were acquired from American Type Culture Collection (Manassas, VA, USA). All cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were incubated in a humid atmosphere containing 5% CO2 at 37°C.
Isolation of RNA and Real-Time Quantitative PCR (RT-qPCR)
Total RNA was extracted from tissues and cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Complementary DNA was synthesized using the PrimeScript RT reagent kit (TaKaRa, Dalian, China), and TB Green Premix Ex Taq Ⅱ (TaKaRa) was used for qPCR analysis. Bulge-loop miRNA RT-qPCR primer sets specific for miR-4999-5p and U6 were purchased from RiboBio (Guangzhou, China). The primers used in this study were as follows: PRKAA2 (forward: 5ʹ-GTGAAGATCGGACACTACGTG-3ʹ, reverse: 5ʹ-CTGCCACTTTATGGCCTGTTA-3ʹ); β-actin (forward: 5ʹ-TGACGTGGACATCCGCAAAG-3ʹ, reverse: 5ʹ- CTGGAAGGTGGACAGCGAGG-3ʹ). The relative expression levels of miRNAs or mRNAs were calculated using the 2−△△Ct method with U6 or β-actin as the internal control. These experiments were repeated three times independently.
Lentivirus Transfection
miR-4999-5p overexpressing or knockdown lentiviruses, PRKAA2 knockdown lentivirus, and control lentiviruses were purchased from GenePharma (Shanghai, China). CRC cells were transfected with the lentiviruses with 10 μg/mL polybrene. Puromycin (Sigma-Aldrich, St. Louis, MO, USA) was used for one week to select the stably transfected cells.
Cell Proliferation Assay
Cell proliferation was evaluated using the Cell Counting Kit-8 (CCK-8) assay. The cells were seeded into 96-well plates at a density of 1000 cells/well. After incubation for 24, 48, 72, 96, or 120 h, the cells were treated with 10 μL of CCK-8 (Dojindo, Kumamoto, Japan) for 2 h. After incubation, absorbance was measured at 450 nm using a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). For colony formation assay, the cells were seeded into 6-well plates at a density of 500 cells per well. After two weeks, the plates were fixed using paraformaldehyde and stained with crystal violet. Three independent experiments were performed.
Extracellular Acidification Rate (ECAR) Assay
Extracellular acidification rate was determined using a Seahorse XFe 24 extracellular flux analyzer (Agilent, Santa Clara, CA, USA). A total of 70,000 cells was seeded into the microplates and incubated overnight. The culture medium was then replaced with assay medium, and the cells were incubated for 1 h at 37 °C. Then, 100 mM glucose, 10 μM oligomycin, and 1 M 2-deoxyglucose (2-DG) was added to each well. The assays were repeated three times independently.
Measurement of Glucose Uptake, Cellular Glucose-6-Phosphate (G6P) Levels, and Lactate Production
The Glucose Uptake-GloTM Assay (Promega) was used to measure glucose uptake. Cellular levels of G6P were detected using a Glucose-6-phosphate Fluorometric Assay kit (Cayman, Ann Arbor, MI, USA). We quantified lactate production using the Lactate Assay kit (BioVision, Mountain View, CA, USA). The experiments were repeated three times independently.
Dual-Luciferase Reporter Assay
The wildtype (WT) 3ʹ-UTR sequence of human PRKAA2 containing the putative miR-4999-5p binding site was cloned into the pMIR-REPORT plasmid (Ambion, Austin, TX, USA). A plasmid with a mutant (MUT) binding site was also constructed. Cells were transfected with PRKAA2 3ʹ-UTR WT or MUT plasmid with miR-4999-5p or negative control (NC) using Lipofectamine 2000 (Invitrogen). Two days after transfection, the cells were harvested. Luciferase activity was determined using the dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Three independent assays were performed.
Animal Models
Nude male BALB/c mice were purchased from the Department of Experimental Animals, Yangzhou University (Yangzhou, China). A total of 2 × 106 HCT15 cells were suspended in 100 μL of PBS, then subcutaneously injected into the nude mice. The xenografts were regularly monitored and harvested after one month. Xenograft volume was calculated using the following equation: tumor volume = 1/2 × length × width2. Xenograft weight was recorded. Tumor samples were analyzed using RT-qPCR. All animal experiments were approved by the Animal Ethics Committee of the First Affiliated Hospital of Wannan Medical College (No. WNMC1808) prior commencement of the study. All experiments were performed according to the NC3Rs ARRIVE guidelines
Immunohistochemistry Staining
Tissues were fixed in 4% paraformaldehyde and cut into 4-μm sections. Heat-mediated antigen retrieval was performed. The sections were blocked with 2% bovine serum albumin for 1 h, and incubated with primary antibodies against AMPKα2 (ab3760, Abcam, Cambridge, MA, USA; 1:100) or Ki-67 (#9027, Cell Signaling Technology, Beverly, MA, USA; 1:400) at 4°C overnight. After the sections were incubated with the secondary antibody, color was developed using diaminobenzidine tetrahydrochloride.
Statistical Analysis
The data are presented as the mean ± SEM. The expression levels of miR-4999-5p or PRKAA2 between cancer tissues and normal tissues were compared using paired t-tests. Student’s t-test and chi-squared test were performed to compare differences between groups. The correlation between miR-4999-5p and PRKAA2 expression levels was analyzed using Pearson’s correlation. Data were analyzed using SPSS 22 and GraphPad Prism 8. P values less than 0.05 were considered as statistically significant.
Results
miR-4999-5p Was Highly Expressed in CRC Tissues and Cells, and Independently Predicted Patient Survival
To characterize the role of miR-4999-5p in CRC, we evaluated the expression level of miR-4999-5p in CRC tissues and adjacent normal tissues. We analyzed TCGA data and found that miR-4999-5p was more highly expressed in CRC tissues than in normal specimens (logFC = 3.025, P = 0.002). We then evaluated the expression of miR-4999-5p in 50 paired CRC tissues and adjacent non-cancer tissues. The expression of miR-4999-5p was markedly higher in the CRC samples than that in the adjacent normal tissues (Figure 1A). Furthermore, the expression of miR-4999-5p was significantly higher in CRC cell lines than that in normal colon epithelial cells (FHC) (Figure 1B).
 |
Figure 1 miR-4999-5p was highly expressed in CRC tissues and cells, and was an independent predictor of patient survival. (A) The expression level of miR-4999-5p in CRC tissues was determined using RT-qPCR. The data are shown as the mean ± SEM, n = 50. (B) miR-4999-5p expression was evaluated in normal human colon epithelial cell line (FHC) and CRC cell lines (LoVo, HCT116, Caco2, and HCT15) using RT-qPCR. The data are shown as the mean ± SEM, n = 3. (C) The effect of miR-4999-5p on overall survival of patients with CRC was evaluated using TCGA database. (D) A Kaplan-Meier curve was plotted to compare the overall survival of patients with high miR-4999-5p expression levels and those with low miR-4999-5p expression levels. **P < 0.01, ***P < 0.001. |
To assess the clinical significance of miR-4999-5p in CRC, we divided the enrolled patients into two groups based on the median expression value of miR-4999-5p. As shown in Table 1, high miR-4999-5p expression level was associated with unfavorable clinicopathological features, including differentiation and TNM stage. Data from TCGA suggested that high miR-4999-5p expression levels were correlated with poor overall survival of patients with CRC (Figure 1C; P = 0.002817). We used follow-up data from our 50-patient cohort to evaluate the prognostic value of miR-4999-5p for CRC. We generated a Kaplan-Meier curve, which showed that higher expression levels of miR-4999-5p were significantly associated with decreased overall survival (Figure 1D, P = 0.001). Furthermore, we conducted multivariate survival analysis using the Cox proportional hazard regression model (Table 2). The results showed that miR-4999-5p was an independent predictor of overall survival [hazard ratio (HR) = 4.094, 95% confidence interval (CI) 1.203–13.935, P = 0.024]. These results showed that miR-4999-5p was upregulated in CRC tissues and cell lines, and high level of miR-4999-5p could independently predict poor overall survival of patients with CRC.
 |
Table 1 Correlation Between miR-4999-5p Expression and Clinicopathological Features of Colorectal Cancer Patients (n = 50) |
 |
Table 2 Univariate and Multivariable Analysis of Overall Survival After Surgery |
miR-4999-5p Promoted Cell Proliferation and Induced Glucose Metabolic Reprogramming in CRC
To evaluate the functional roles of miR-49999-5p in CRC, we performed various in vitro experiments. We knocked down the expression of miR-4999-5p in HCT15 cells, and overexpressed miR-4999-5p in LoVo cells. Knockdown or overexpression efficiency of miR-4999-5p was confirmed using RT-qPCR (Figure 2A and B). We performed CCK-8 assays to estimate the effects of miR-4999-5p on CRC cell proliferation. As shown in Figure 2C, miR-4999-5p knockdown suppressed cell proliferation in HCT15 cells. In contrast, overexpression of miR-4999-5p resulted in increased proliferation in LoVo cells (Figure 2D). Colony formation assay further demonstrated that proliferation was decreased in miR-4999-5p-knockdown HCT15 cells (Figure 2E). In addition, miR-4999-5p overexpression significantly increased proliferation of LoVo cells (Figure 2F).
 |
Figure 2 miR-4999-5p promoted cell proliferation in CRC. (A) The knockdown efficiency of miR-4999-5p in HCT15 cells was evaluated using RT-qPCR. The data are shown as the mean ± SEM, n = 3. (B) The overexpression efficiency of miR-4999-5p in LoVo cells was evaluated using RT-qPCR. The data are shown as the mean ± SEM, n = 3. (C) Cell Counting Kit-8 was used to evaluate the effect of miR-4999-5p knockdown on HCT15 cell proliferation. The data are shown as the mean ± SEM, n = 3. (D) Cell Counting Kit-8 was used to evaluate the effect of miR-4999-5p overexpression on LoVo cell proliferation. The data are shown as the mean ± SEM, n = 3. (E) Colony formation assay was performed to estimate the proliferative capability of sh-miR-4999-5p and sh-NC HCT15 cell lines. The data are shown as the mean ± SEM, n = 3. (F) Colony formation assay was performed to estimate the proliferative capability of miR-4999-5p and NC LoVo cell lines. The data are shown as the mean ± SEM, n = 3. **P < 0.01, ***P < 0.001. |
Glucose metabolic reprogramming is a hallmark of cancer cells. To evaluate the influence of miR-4999-5p on glycolysis in CRC, we measured ECAR in miR-4999-5p knockdown or overexpressing CRC cells using a Seahorse XFe 24 extracellular flux analyzer. Knockdown of miR-4999-5p resulted in increased glycolysis rate and glycolytic capacity in HCT15 cells (Figure 3A). Conversely, overexpression of miR-4999-5p resulted in reduced glycolysis rate and glycolytic capacity in LoVo cells (Figure 3B). We also investigated glucose uptake, cellular G6P levels, and lactate production in miR-4999-5p knockdown or overexpressing CRC cells. As indicated in Figure 3C–E, miR-4999-5p knockdown significantly reduced glucose uptake, cellular G6P levels, and lactate production in HCT15 cells. In contrast, miR-4999-5p overexpression resulted in increased glucose uptake, cellular G6P levels, and lactate production in LoVo cells.
 |
Figure 3 miR-4999-5p induced glucose metabolic reprogramming in CRC. (A) Extracellular acidification rate was used to evaluate the effects of miR-4999-5p knockdown on glycolysis in HCT15 cells. The data are shown as the mean ± SEM, n = 3. (B) Extracellular acidification rate was used to evaluate the effects of miR-4999-5p overexpression on glycolysis in LoVo cells. The data are shown as the mean ± SEM, n = 3. (C) The effects of miR-4999-5p knockdown or overexpression on glucose uptake were examined. The data are shown as the mean ± SEM, n = 3. (D) The effects of miR-4999-5p knockdown or overexpression on cellular glucose-6-phosphate (G6P) level were examined. The data are shown as the mean ± SEM, n = 3. (E) The effects of miR-4999-5p knockdown or overexpression on lactate production were examined. The data are shown as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001. |
miR-4999-5p Directly Targeted PRKAA2 in CRC
To elucidate the downstream molecular mechanisms of miR-4999-5p, we performed bioinformatics prediction using three different algorithms. As shown in Figure 4A, TargetScan, miRDB, and DIANA databases were used, and PRKAA2 was predicted as a target of miR-4999-5p by all three databases. The complementary sequence of PRKAA2 and miR-4999-5p is shown in Figure 4B. PRKAA2 encodes the α2 subunit of 5′-AMP activated protein kinase (AMPK) and plays an important role in glucose metabolism.13,14 Data from TCGA showed that PRKAA2 expression was significantly decreased in CRC tissues compared to that in non-tumor samples (Figure 4C). We also examined the expression levels of PRKAA2 in 50 paired CRC tumor samples and non-tumor tissues, and showed that PRKAA2 was significantly downregulated in tumor samples (Figure 4D). Correlation analysis showed that the expression of miR-4999-5p was negatively correlated with PRKAA2 expression (Figure 4E). As indicated in Figure 4F, higher expression levels of PRKAA2 were detected in miR-4999-5p knockdown HCT15 than in control cells. In miR-4999-5p overexpressing LoVo cells, the expression of PRKAA2 was significantly downregulated (Figure 4G). Dual-luciferase reporter assay showed that PRKAA2 was a direct target of miR-4999-5p (Figure 4H).
 |
Figure 4 miR-4999-5p directly targeted PRKAA2 in CRC. (A) TargetScan, miRDB, and DIANA databases were used to predict the downstream target of miR-4999-5p, and PRKAA2 was predicted by each of the algorithms. (B) The complementary sequence of PRKAA2 and miR-4999-5p. (C) Data from TCGA were used to determine the expression levels of PRKAA2 in CRC tissues and non-tumor samples. (D) Real-time qPCR was used to determine the expression levels of PRKAA2 in 50 paired CRC tumor samples and non-tumor tissues. (E) The expression levels of miR-4999-5p and PRKAA2 were negatively correlated. n = 50, P < 0.001. (F) The expression levels of PRKAA2 in miR-4999-5p knockdown HCT15 cells. The data are shown as the mean ± SEM, n = 3. (G) The expression levels of PRKAA2 in miR-4999-5p overexpressing LoVo cells. The data are shown as the mean ± SEM, n = 3. (H) Dual-luciferase reporter assay was used to show that PRKAA2 was a direct target of miR-4999-5p. The data are shown as the mean ± SEM, n = 3. **P < 0.01, ***P < 0.001. |
To confirm that miR-4999-5p promoted CRC progression by targeting PRKAA2, we knocked down PRKAA2 in miR-4999-5p-silenced HCT15 cells. The knockdown efficiency of PRKAA2 was confirmed using RT-qPCR (Figure 5A). Analysis using the CCK-8 assay showed that PRKAA2 knockdown rescued blunted cell proliferation observed in miR-4999-5p-silenced HCT15 cells (Figure 5B). Extracellular acidification rate analysis showed that PRKAA2 knockdown rescued reduced glycolysis observed in miR-4999-5p-silenced HCT15 cells (Figure 5C). In addition, glucose uptake, cellular G6P level, and lactate production were restored by PRKAA2 knockdown in miR-4999-5p-silenced HCT15 cells (Figure 5D–F). These results indicated that miR-4999-5p promoted CRC progression via targeting PRKAA2.
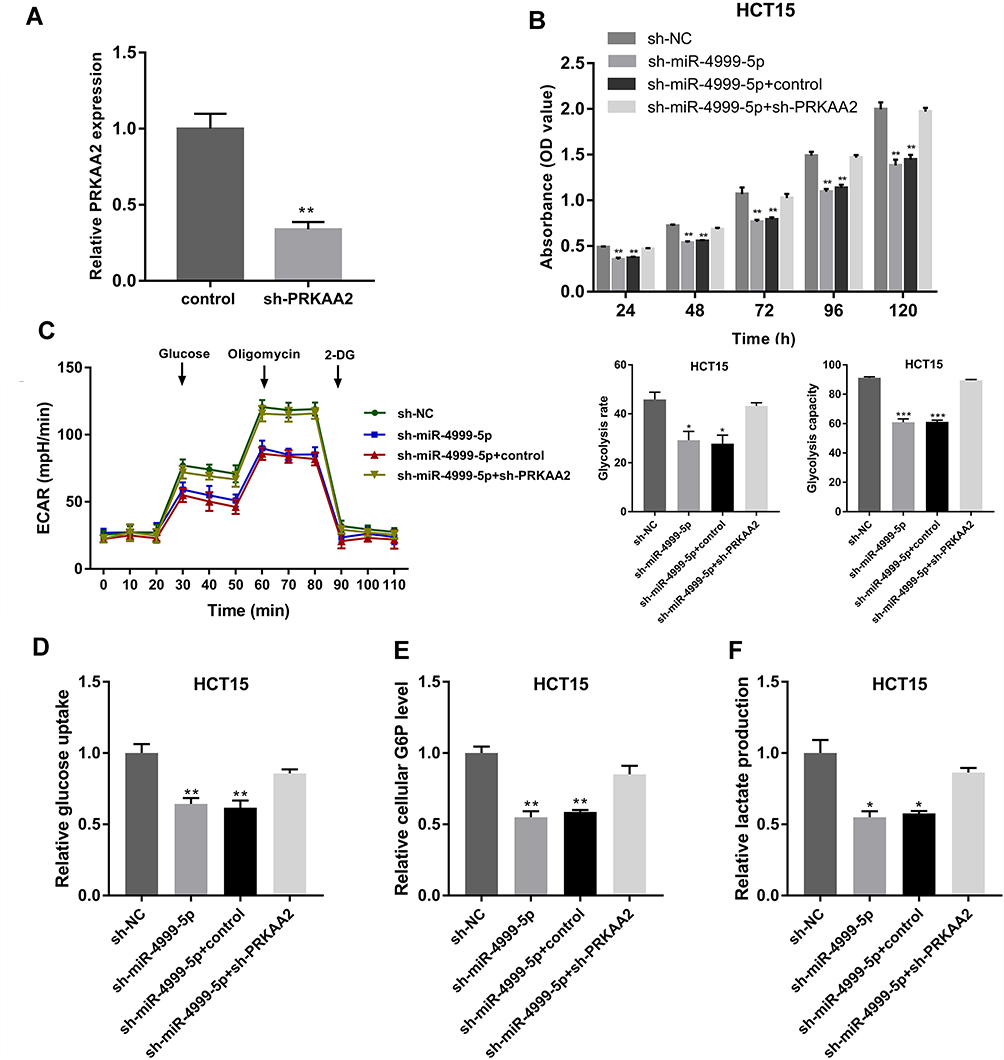 |
Figure 5 miR-4999-5p promoted CRC progression by targeting PRKAA2. (A) The knockdown efficiency of PRKAA2 was verified using RT-qPCR. The data are shown as the mean ± SEM, n = 3. (B) Cell Counting Kit-8 assay was used to evaluate the cell proliferation of miR-4999-5p-silenced HCT15 cells with or without PRKAA2 knockdown. The data are shown as the mean ± SEM, n = 3. (C) Extracellular acidification rate was used to determine the effect of PRKAA2 knockdown on glycolysis in miR-4999-5p-silenced HCT15 cells. The data are shown as the mean ± SEM, n = 3. (D) Glucose uptake was measured in miR-4999-5p-silenced HCT15 cells with or without PRKAA2 knockdown. The data are shown as the mean ± SEM, n = 3. (E) Cellular glucose-6-phosphate (G6P) levels were detected in miR-4999-5p-silenced HCT15 cells with or without PRKAA2 knockdown. The data are shown as the mean ± SEM, n = 3. (F) Lactate production was detected in miR-4999-5p-silenced HCT15 cells with or without PRKAA2 knockdown. The data are shown as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001. |
miR-4999-5p Promoted CRC Growth in vivo
To further characterize the tumor-promoting role of miR-4999-5p in CRC, we constructed an in vivo xenograft mouse model. Smaller xenografts were observed in animals injected with miR-4999-5p knockdown HCT15 cells (Figure 6A). The miR-4999-5p knockdown xenografts grew at a slower rate than the control xenografts (Figure 6B). Xenograft weights were markedly lower in mice inoculated with sh-miR-4999-5p HCT15 cells (Figure 6C). Real-time qPCR results confirmed that miR-4999-5p knockdown was stable in the xenografts (Figure 6D). Immunohistochemistry analysis showed that the protein level of PRKAA2 was higher in the sh-miR-4999-5p group than that in the control group (Figure 6E), which was consistent with our in vitro data. Furthermore, Ki-67 staining showed that miR-4999-5p knockdown resulted in reduced proliferation in vivo.
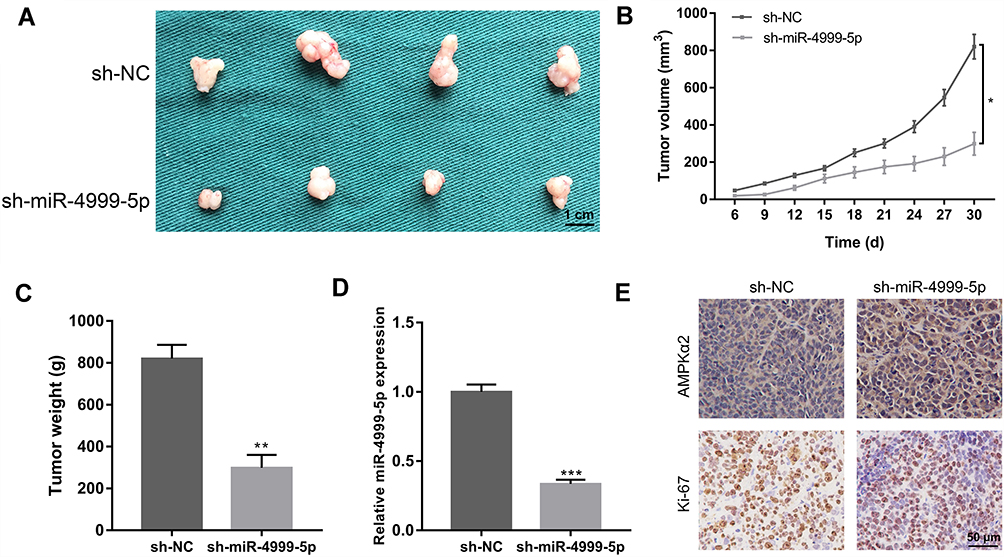 |
Figure 6 miR-4999-5p promoted CRC growth in vivo. (A) Xenograft mouse models were used to assess the effect of miR-4999-5p knockdown on CRC growth. (B) The growth curve of xenograft volume was plotted. The data are shown as the mean ± SEM, n = 3. (C) Xenograft weights. The data are shown as the mean ± SEM, n = 3. (D) The expression level of miR-4999-5p in xenografts was evaluated using RT-qPCR. The data are shown as the mean ± SEM, n = 3. (E) Immunohistochemistry results for AMPKα2 and Ki-67 in the xenograft tissues. *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
In the present study, we mined TCGA data and found that miR-4999-5p was significantly overexpressed in CRC. We then analyzed CRC tissue samples and cell lines to confirm that miR-4999-5p was upregulated in CRC. Further analyses showed that high miR-4999-5p expression levels were associated with malignant clinicopathological features, including differentiation and TNM stage. miR-4999-5p was also an independent predictor of worse overall survival of patients with CRC.
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of tumor cells. In cancer cells, glucose is preferably converted to lactate under aerobic conditions, and aerobic glycolysis plays a crucial role in regulation of cell progression.15–17 Increasing numbers of studies have focused on the regulatory roles of non-coding RNAs in cancer metabolism.18,19 A study showed that circRNA_100290 acted as a ceRNA to sponge miR-378a, which resulted in decreased expression of GLUT1, and increased cell proliferation in oral squamous cell carcinoma.20 In hepatocellular carcinoma, the long non-coding RNA MALAT1 accelerated cancer cell progression through enhanced translation of the transcription factor TCF7L2, which promoted the Warburg effect.21 Another showed that miR-31-5p promoted the Warburg effect and played an important role in metabolic reprogramming by directly targeting FIH in lung cancer.22
In CRC, many miRNAs have been shown to be involved in regulation of glucose metabolism reprogramming. miR-493-5p has been shown to interact with DDK2, which resulted in stimulation of CRC glycolysis.23 miR-328 was shown to target SLC2A1 and to mediate the glucose metabolic shift in CRC.24 miR-98 inhibited glycolysis by targeting HK2 in CRC cells.25 To understand the functional roles of miR-4999-5p in CRC, we performed loss-of-function and gain-of-function assays, and showed that miR-4999-5p facilitated proliferation and glycolysis in CRC cells. The results of in vivo experiments verified the tumor-promoting effects of miR-4999-5p in CRC.
AMPK functions as a cellular energy sensor and is a crucial regulator in various cancers.26 AMPK exists as heterotrimeric complexes with α1 and α2 as the catalytic subunits. The gene that encodes AMPKα2, PRKAA2, has been reported to modulate the expression levels of multiple genes during the late stages of differentiation and myogenesis.27 Furthermore, PRKAA2 has been shown to be involved in cell energy metabolism in various cancers. In bladder cancer, loss of PRKAA2 resulted in increased proliferation and larger xenografts.28 In liver cancer, PRKAA2 knockout enhanced tumor inflammation.29 In addition, PRKAA2 has been shown to act as a tumor repressor in CRC.30,31 As a subunit of AMPK, PRKAA2 has been shown to inhibit cell proliferation through the p53/p21 pathway, and through modulation of the expression of p27.32,33 Previous studies have shown that PRKAA2/mTOR signaling was crucial to cancer cell proliferation.34,35 Our mechanistic studies showed that PRKAA2 was a direct target of miR-4999-5p. Rescue experiments suggested that miR-4999-5p promoted cancer progression via regulation of PRKAA2 in CRC.
In summary, our study showed that the expression of miR-4999-5p was increased in CRC tissues and cell lines. miR-4999-5p was associated with CRC clinicopathological features and was an independent predictor of patient overall survival. miR-4999-5p promoted CRC cell proliferation and glycolysis by directly targeting PRKAA2. These findings suggested that miR-4999-5p might be a promising prognostic indictor for CRC, and a potential target for treatment of CRC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.v68.6
2. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12(11):829–846. doi:10.1038/nrd4145
3. Rodriguez-Enriquez S, Marin-Hernandez A, Gallardo-Perez JC, et al. Transcriptional regulation of energy metabolism in cancer cells. Cells. 2019;8(10):1225. doi:10.3390/cells8101225
4. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001
5. Unterlass JE, Curtin NJ. Warburg and Krebs and related effects in cancer. Expert Rev Mol Med. 2019;21:e4. doi:10.1017/erm.2019.4
6. Orang AV, Petersen J, McKinnon RA, Michael MZ. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol Metab. 2019;23:98–126. doi:10.1016/j.molmet.2019.01.014
7. Li D, Zhang J, Li J. Role of miRNA sponges in hepatocellular carcinoma. Clin Chim Acta. 2019.
8. Ding X, Du J, Mao K, Wang X, Ding Y, Wang F. MicroRNA-143-3p suppresses tumorigenesis by targeting catenin-delta1 in colorectal cancer. Onco Targets Ther. 2019;12:3255–3265. doi:10.2147/OTT.S184118
9. Tang L, Zhou L, Wu S, et al. miR-125a-5p inhibits colorectal cancer cell epithelial-mesenchymal transition, invasion and migration by targeting TAZ. Onco Targets Ther. 2019;12:3481–3489. doi:10.2147/OTT.S191247
10. Subramaniam S, Jeet V, Clements JA, Gunter JH, Batra J. Emergence of MicroRNAs as key players in cancer cell metabolism. Clin Chem. 2019;65(9):1090–1101. doi:10.1373/clinchem.2018.299651
11. Chen Z, Zuo X, Zhang Y, et al. MiR-3662 suppresses hepatocellular carcinoma growth through inhibition of HIF-1alpha-mediated Warburg effect. Cell Death Dis. 2018;9(5):549. doi:10.1038/s41419-018-0616-8
12. Nie ZY, Liu XJ, Zhan Y, et al. miR-140-5p induces cell apoptosis and decreases Warburg effect in chronic myeloid leukemia by targeting SIX1. Biosci Rep. 2019;39:4. doi:10.1042/BSR20190150
13. Vila IK, Park MK, Setijono SR, et al. A muscle-specific UBE2O/AMPKalpha2 axis promotes insulin resistance and metabolic syndrome in obesity. JCI Insight. 2019;4:13. doi:10.1172/jci.insight.128269
14. Randrianarisoa E, Lehn-Stefan A, Krier J, et al. AMPK subunits harbor largely non-overlapping genetic determinants for body fat mass, glucose- and cholesterol metabolism. J Clin Endocrinol Metab. 2019. doi:10.1210/jcem/dgz020
15. Meng Y, Xu X, Luan H, et al. The progress and development of GLUT1 inhibitors targeting cancer energy metabolism. Future Med Chem. 2019;11(17):2333–2352. doi:10.4155/fmc-2019-0052
16. Yan L, Raj P, Yao W, Ying H. Glucose metabolism in pancreatic cancer. Cancers. 2019;11:10. doi:10.3390/cancers11101460
17. Shen Y, Chen G, Zhuang L, Xu L, Lin J, Liu L. ARHGAP4 mediates the Warburg effect in pancreatic cancer through the mTOR and HIF-1alpha signaling pathways. Onco Targets Ther. 2019;12:5003–5012. doi:10.2147/OTT.S207560
18. Yang D, Sun L, Li Z, Gao P. Noncoding RNAs in regulation of cancer metabolic reprogramming. Adv Exp Med Biol. 2016;927:191–215.
19. Shankaraiah RC, Veronese A, Sabbioni S, Negrini M. Non-coding RNAs in the reprogramming of glucose metabolism in cancer. Cancer Lett. 2018;419:167–174. doi:10.1016/j.canlet.2018.01.048
20. Chen X, Yu J, Tian H, et al. Circle RNA hsa_circRNA_100290 serves as a ceRNA for miR-378a to regulate oral squamous cell carcinoma cells growth via glucose transporter-1 (GLUT1) and glycolysis. J Cell Physiol. 2019;234(11):19130–19140. doi:10.1002/jcp.v234.11
21. Malakar P, Stein I, Saragovi A, et al. Long noncoding RNA MALAT1 regulates cancer glucose metabolism by enhancing mTOR-Mediated translation of TCF7L2. Cancer Res. 2019;79(10):2480–2493. doi:10.1158/0008-5472.CAN-18-1432
22. Zhu B, Cao X, Zhang W, et al. MicroRNA-31-5p enhances the Warburg effect via targeting FIH. FASEB J. 2019;33(1):545–556. doi:10.1096/fj.201800803R
23. Deng F, Zhou R, Lin C, et al. Tumor-secreted dickkopf2 accelerates aerobic glycolysis and promotes angiogenesis in colorectal cancer. Theranostics. 2019;9(4):1001–1014. doi:10.7150/thno.30056
24. Santasusagna S, Moreno I, Navarro A, et al. miR-328 mediates a metabolic shift in colon cancer cells by targeting SLC2A1/GLUT1. Clin Transl Oncol. 2018;20(9):1161–1167. doi:10.1007/s12094-018-1836-1
25. Zhu W, Huang Y, Pan Q, Xiang P, Xie N, Yu H. MicroRNA-98 suppress warburg effect by targeting HK2 in colon cancer cells. Dig Dis Sci. 2017;62(3):660–668. doi:10.1007/s10620-016-4418-5
26. Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019;9(7):190099. doi:10.1098/rsob.190099
27. Okamoto S, Asgar NF, Yokota S, Saito K, Minokoshi Y. Role of the alpha2 subunit of AMP-activated protein kinase and its nuclear localization in mitochondria and energy metabolism-related gene expressions in C2C12 cells. Metabolism. 2019;90:52–68.
28. Kopsiaftis S, Sullivan KL, Garg I, Taylor JA
29. Qiu S, Liu T, Piao C, et al. AMPKalpha2 knockout enhances tumour inflammation through exacerbated liver injury and energy deprivation-associated AMPKalpha1 activation. J Cell Mol Med. 2019;23(3):1687–1697. doi:10.1111/jcmm.13978
30. Ji S, Tang S, Li K, et al. Licoricidin inhibits the growth of SW480 human colorectal adenocarcinoma cells in vitro and in vivo by inducing cycle arrest, apoptosis and autophagy. Toxicol Appl Pharmacol. 2017;326:25–33. doi:10.1016/j.taap.2017.04.015
31. Das B, Neilsen BK, Fisher KW, et al. A functional signature ontology (FUSION) screen detects an AMPK inhibitor with selective toxicity toward human colon tumor cells. Sci Rep. 2018;8(1):3770.
32. Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5ʹ-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287(2):562–567. doi:10.1006/bbrc.2001.5627
33. Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–293. doi:10.1016/j.molcel.2005.03.027
34. Vila IK, Yao Y, Kim G, et al. A UBE2O-AMPKα2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell. 2017;31(2):208–224. doi:10.1016/j.ccell.2017.01.003
35. Xu Q, Wu N, Li X, et al. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019;10(12):874. doi:10.1038/s41419-019-2073-4
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.