")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
miR-455 inhibits neuronal cell death by targeting TRAF3 in cerebral ischemic stroke
Authors Yao ST, Tang B, Li G, Fan RM, Cao F
Received 31 August 2016
Accepted for publication 28 September 2016
Published 5 December 2016 Volume 2016:12 Pages 3083—3092
DOI https://doi.org/10.2147/NDT.S121183
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Shengtao Yao,* Bo Tang,* Gang Li, Ruiming Fan, Fang Cao
Department of Cerebrovascular Disease, The First Affiliated Hospital of Zunyi Medical College, Zunyi, People’s Republic of China
*These authors contributed equally to this work
Abstract: Ischemic stroke is one of the leading causes of brain disease, with high morbidity, disability, and mortality. MicroRNAs (miRNAs) have been identified as vital gene regulators in various types of human diseases. Accumulating evidence has suggested that aberrant expression of miRNAs play critical roles in the pathologies of ischemic stroke. Yet, the precise mechanism by which miRNAs control cerebral ischemic stroke remains unclear. In the present study, we explored whether miR-455 suppresses neuronal death by targeting TRAF3 in cerebral ischemic stroke. The expression levels of miR-455 and TRAF3 were detected by quantitative real-time polymerase chain reaction and Western blot. The role of miR-455 in cell death caused by oxygen–glucose deprivation (OGD) was assessed using Cell Counting Kit-8 (CCK-8) assay. The influence of miR-455 on infarct volume was evaluated in mouse brain after middle cerebral artery occlusion (MCAO). Bioinformatics softwares and luciferase analysis were used to find and confirm the targets of miR-455. The results showed that the expression levels of miR-455 significantly decreased in primary neuronal cells subjected to OGD and mouse brain subjected to MCAO. In addition, forced expression of miR-455 inhibited neuronal death and weakened ischemic brain infarction in focal ischemia-stroked mice. Furthermore, TRAF3 was proved to be a direct target of miR-455, and miR-455 could negatively suppress TRAF3 expression. Biological function analysis showed that TRAF3 silencing displayed the neuroprotective effect in ischemic stroke and could enhance miR-455-induced positive impact on ischemic injury both in vitro and in vivo. Taken together, miR-455 played a vital role in protecting neuronal cells from death by downregulating TRAF3 protein expression. These findings may represent a novel latent therapeutic target for cerebral ischemic stroke.
Keywords: miR-455, neuronal cell, TRAF3, cerebral ischemic stroke, MCAO
Introduction
Ischemic stroke is one of the principal diseases of brain, with a high rate of morbidity, disability, and mortality.1 Extensive research has reported that cerebral ischemia could induce a series of pathological events and finally contribute to apoptotic and necrotic neuron death in the ischemic regions.2–4 Neuronal damage following cerebral ischemia could trigger an intricate series of signaling cascades that result in neuronal cell death.5 Despite the advances in current therapeutic strategies for decreasing ischemic brain injury, pathophysiological mechanisms of this deadly disease are not completely clear. Hence, elaborating the molecular mechanism of ischemic injury is necessary in order to develop therapeutic strategies for patients with stroke.
MicroRNAs (miRNAs) are a group of endogenous 18- to 22-nucleotide (nt)-long non-protein-coding short RNA molecules, which regulate gene expression by recognizing binding sites situated in the 3′-untranslated region (3′-UTR) of mRNA targets.6,7 It is well understood that miRNAs are capable of regulating the expression of at least one-third of all human genes and play a vital role in many biological processes, such as cell differentiation, apoptosis, and development.8 Yin et al8 first demonstrated the functional importance of unparalleled miRNAs in ischemic brain injury. Data reported showed that miR-497 was induced after ischemic injury and confirmed that miR-497 increased ischemic neuronal death by downregulating Bcl-2 and Bcl-w.9 Additionally, Ziu et al10 verified that the levels of miRNAs were changed in numerous central nervous system (CNS) injuries, indicating the underlying function of miRNAs in the cellular response to damage. Recently, miR-455 was reported to be downregulated in ischemic stroke.11,12 However, the definite role and underlying mechanism of miR-455 in ischemic stroke remain unclear.
TRAF3 is a member of the TRAF families and ubiquitously expressed in most tissues and cell types, including brain, lung, heart, spleen, and liver.13,14 TRAF3 experiences interactions with various biological molecules, such as receptors, kinases, adaptors, and regulatory proteins, to exert its different biological functions.15 For example, Jiang et al15 demonstrated that TRAF3 acted as a regulator of pathological cardiac hypertrophy and heart injury. Other investigators reported that TRAF3 was significantly upregulated in neurons to respond to ischemia/reperfusion (I/R) injury and TRAF3 deletion can be neuroprotective.16 Gu et al17 also found that inhibiting TRAF3 expression can protect neuronal cells from high glucose-induced apoptosis. Thus, it is essential to illustrate the delicate regulation of TRAF3 as an important modulator in neuronal death in ischemic brain damage.
In the present study, miR-455 expression levels were detected in ischemic stroke in vitro and in vivo. Then, the effect of miR-455 in protecting neuronal cells from death was investigated. Additionally, a potential target by which miR-455 exerted its effect on ischemic stroke was identified.
Materials and methods
Reagents
The miR-455 mimics, miR-455 inhibitor, lentivirus encoding shRNA for TRAF3 (sh-TRAF3), and the corresponding controls were purchased from Ambion (Austin, TX, USA). The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). The Trizol reagent and the M-MLV Reverse Transcriptase kit were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies against TRAF3 (Cell Signaling Technology, Beverly, MA, USA) and β-actin (Sigma, St Louis, MO, USA) were used. All other chemical reagents were purchased from Sigma unless indicated otherwise.
Cortical neuron culture and oxygen–glucose deprivation (OGD)
Neurons were detached from the cerebral cortex of ~17-day-old C57BL/6 mice as in previously described protocols.10 Neurons were plated onto plates at a density of 1×106 cells/mL. They were cultured in neurobasal medium (Gibco, Grand Island, NY, USA) with 2% B27 supplement (Gibco). The fresh medium was a substitute for the cultured medium at an interval of 2 days. Cell cultures were kept at 37°C in a humidified incubator with a mixture of 95% air and 5% CO2. All other related experiments were performed 24 hours after seeding.
Cultured neurons exposed to OGD were transferred to glucose-free Dulbecco’s Modified Eagle’s Medium (DMEM) in a humidified incubator with 94% N2, 5% CO2, and 1% O2 at 37°C for 1–2 hours. Neurons treated with OGD were kept in DMEM containing glucose under normoxic conditions for reoxygenation for 24 hours. Control neuron cultures were grown in glucose-containing DMEM (25 mM) and cultured under normal culture conditions simultaneously.8 Control neuronal cells were not subjected to OGD.
Mouse transient middle cerebral artery occlusion (MCAO) model of ischemic stroke
Adult male C57BL/6J mice (20±5 g) were purchased from the Experimental Animal Center of the Chinese Academy of Medical Sciences, People’s Republic of China. They were bred and maintained in a pathogen-free facility. The animal care and procedures for the experiments were approved by Institutional Animal Care and Use Committee of The First Affiliated Hospital of Zunyi Medical College. To guarantee longer survival time of the mice after MCAO, the mice MCAO model of ischemic stroke was constructed as described previously,18 with minor alterations. Briefly, mice were anesthetized with sodium pentobarbital (40 mg/kg). The left common artery and the left external carotid artery were exposed. A piece of 6-0 monofilament nylon suture with its cusp slightly rounded by heat was plugged via the right internal carotid artery to the base of the middle cerebral artery. Following 1 hour of MCAO, the mice were permitted to recover for 24 hours. Mice were analgesia sacrificed to obtain the brains for biochemical assays. In the sham group, the sham control animals underwent similar operations to expose the carotid arteries without occlusion of the middle cerebral artery. Following 24-hour reperfusion, the brains were rapidly removed for the subsequent analysis. The lesion area was used to determine the mRNA and protein expression. Mortality rate was ~21%, resulting from the individual resistance to the surgery and vascular variation. The data were analyzed by two pathologists.
Treatment of neurons with miR-455 mimics or inhibitor
Cortical neuronal cells were seeded in 24-well or 6-well plates in antibiotic-free medium for 24 hours, and miR-455 mimics, miR-455 inhibitor, or sh-TRAF3 (20 nM) was transfected using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. Samples were collected for quantitative real-time polymerase chain reaction (qRT-PCR), Western blot analysis, or cell death assay at the indicated time points.
Cortical injection of miR-455 mimics, miR-455 inhibitor, and lentiviral sh-TRAF3
Cortical injections of miR-455 mimics, miR-455 inhibitor, sh-TRAF3, and the corresponding controls were administered as reported previously.19,20 Briefly, pentobarbital sodium was used to anesthetize mice deeply, and mice were fixed in a stereotaxic apparatus. Lentiviral sh-TRAF3 (109 TU/mL) or its control (109 TU/mL) was mixed homogeneously with the cationic lipid polybrene (4 μg/μL, GenePharma) and incubated at 37°C for 15 minutes, and cortical injection of 7 μL mixture was administered for 10 minutes. miR-455 mimics (100 μM), miR-455 inhibitor (100 μM), or the controls (100 μM) were mixed with the siRNA-Mate (GenePharma), incubated at room temperature for 20 minutes, and injected for 10 minutes. Mice were exposed to MCAO after injection.
Measurement of infarct volume
Mice were sacrificed 24 hours after MCAO, and brains were quickly removed and cut into 1.0 mm-thick coronal sections. The slices were incubated for 15 minutes in a solution of 0.5% 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C, and then the slices were scanned into a computer. The images of stained slices were analyzed in Image Pro Plus1 6.0 (Media Cybernetics, Silver Spring, MD, USA).
RNA isolation and qRT-PCR
Total RNA was extracted from tissues or cultured cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. The reverse transcription reactions were performed using a TaqMan™ microRNA assay kit (Applied Biosystems, Foster City, CA, USA) and a Prime Script™ RT reagent kit (Takara, Shiga, Japan) following the instructions from the respective manufacturers. Real-time PCR was performed using a standard SYBR Green PCR kit (Toyobo, Osaka, Japan) and a Rotor-Gene RG-3000A (Corbett Life Science, New South Wales, Australia) following the instructions from the respective manufacturers. The expression levels of miRNA were normalized against the endogenous U6 small nuclear RNA (U6-snRNA). Each sample was analyzed in triplicate. The relative expression levels of miR-455 in brain tissue and neurons were calculated by the 2−ΔΔCt method.
Luciferase reporter assay
The wild-type TRAF3-3′-UTR (WT) and mutant TRAF3-3′-UTR (MUT) containing the putative binding site of miR-455 were constructed and cloned in pmirGLO dual luciferase reporter vectors (Promega, Madison, WI, USA). The reporter vectors containing the wild type or mutant of TRAF3 3′-UTR and miR-455 mimics, miR-455 inhibitor, miR control, or inhibitor control were co-transfected into cortical neurons (ATCC, Manassas, VA, USA) using Lipofectamine 2000 (Invitrogen). After 48 hours of incubation, cells were lysed. Firefly and Renilla luciferase activities were measured using the dual-luciferase assay system (Promega).
Western blot analysis
All samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Then, the membranes were blocked for 1 hour in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST) and 5% BSA and incubated overnight with anti-TRAF3 or anti-β-actin antibodies in PBST containing 5% BSA, at 4°C. PVDF membranes containing the proteins of interest were washed in Tris buffered saline with Tween-20 and then incubated with secondary antibodies labeled with horseradish peroxidase (HRP). Finally, the proteins of interest were detected by enhanced chemiluminescence (Pierce, Rockford, IL, USA).
Assessment of neuron death
Cell death was measured using CCK-8 (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Cells were seeded in 96-well plates at a density of 2×104 cells/well, after pre-culture for 24 hours. After treatment, CCK-8 solution was added (10 μM/well), and the plates were incubated at 37°C for 4 hours. Then, the numbers of cells per well were detected by the absorbance at 450 nm of reduced WST-8 following 2 hours OGD.
Statistical analysis
All numerical data were presented as mean ± SD and analyzed using Student’s t-test or one-way analysis of variance (ANOVA). Experiments were independently repeated at least three times. Differences were considered significant when P<0.05.
Results
miR-455 level was significantly downregulated after MCAO and OGD
To evaluate the effect of miR-455 on modulating ischemic brain damage, experimental model of stroke was used. To begin with, the level of miR-455 in cortical neurons after OGD and in mouse brain after MCAO was detected by qRT-PCR. The data indicated that miR-455 in the neurons was downregulated at 24-hour post-OGD compared with that in control neuronal cultures (Figure 1A). Similarly, the level of miR-455 was significantly downregulated in the mouse brain after MCAO in comparison with the sham control (Figure 1B). These data suggest that downregulation of miR-455 may be connected with ischemic brain injury.
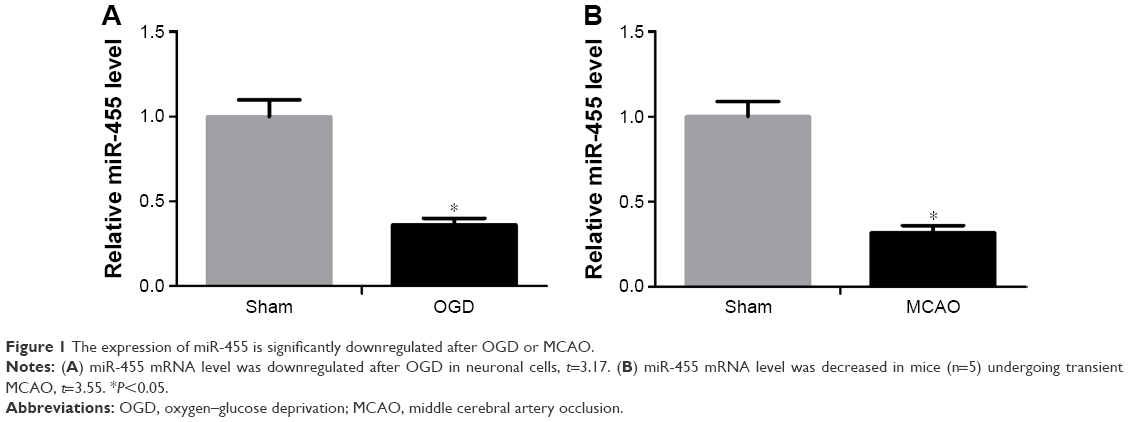 | Figure 1 The expression of miR-455 is significantly downregulated after OGD or MCAO. |
Upregulated miR-455 suppressed neuronal death
In order to investigate the biological function of miR-455 in neuronal cell survival, the neurons transfected with miR-455 mimics or their inhibitor were analyzed. The miR-455 level was significantly increased in neuronal cells that were treated with miR-455 mimics, and miR-455 was downregulated by miR-455 inhibitor (Figure 2A). The neuronal cell death rate was increased after OGD for 24 hours (Figure 2B). Furthermore, upregulated miR-455 inhibited neuronal cell death (Figure 2C), whereas downregulated miR-455 promoted neuronal cell death (Figure 2D). These results suggest that upregulated miR-455 positively regulates neuronal cell survival after ischemic brain injury.
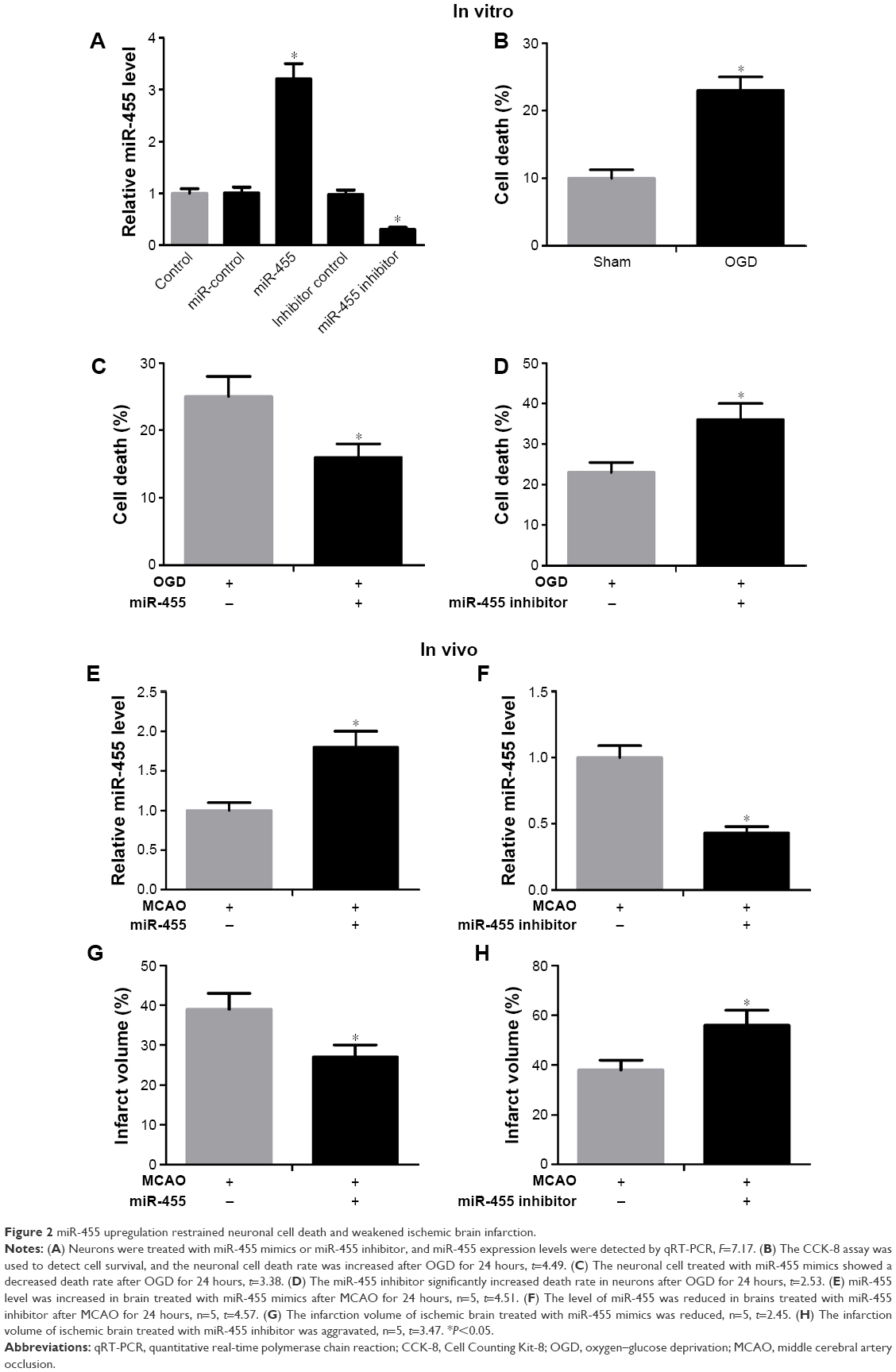 | Figure 2 miR-455 upregulation restrained neuronal cell death and weakened ischemic brain infarction. |
Upregulated miR-455 attenuated ischemic brain infarction
In order to further investigate the function of miR-455 in ischemic brain injury, miR-455 mimics and miR-455 inhibitor were injected into the cerebral cortex. The efficiency levels of upregulation and downregulation of miR-455 were confirmed by qRT-PCR. Our research showed that injection of miR-455 mimics and miR-455 inhibitor could effectively upregulate and downregulate miR-455 expression in MCAO mice, respectively (Figure 2E and F). Moreover, overexpression of miR-455 reduced the infarct volume, whereas downregulation of miR-455 effectively augmented ischemic injury after MCAO for 24 hours (Figure 2G and H).
TRAF3 was a functional target of miR-455
To define the target genes by which miR-455 regulates neuronal cell death, miRBase (http://www.mirbase.org/) and TargetScan 5.1 (http://www.targetscan.org/) miRNA databases were applied. The results demonstrated that TRAF3 was a target of miR-455 (Figure 3A). Next, firefly luciferase reporter containing wild 3′-UTR of TRAF3 or mutant 3′-UTR of TRAF3 was constructed and the luciferase reporter assay was performed. The reporter assay indicated that miR-455 was capable of significantly inhibiting luciferase expression of the reporter containing wild-type 3′-UTR of TRAF3 but not the mutant reporter gene (Figure 3B). Then neurons were co-transfected simultaneously with the reporter and miR-455 inhibitor or the inhibitor control. The reporter assay showed that miR-455 inhibitor was able to significantly promote luciferase expression of the reporter containing intact 3′-UTR of TRAF3 but not the mutant reporter gene (Figure 3C). In addition, the data revealed that miR-455 upregulation significantly reduced TRAF3 mRNA and protein levels, and miR-455 inhibitor significantly increased the mRNA and protein levels of TRAF3 in neuronal cells (Figure 3D–G). These results suggested that TRAF3 was a target of miR-455 and negatively regulated TRAF3 expression by binding to the 3′-UTR of TRAF3.
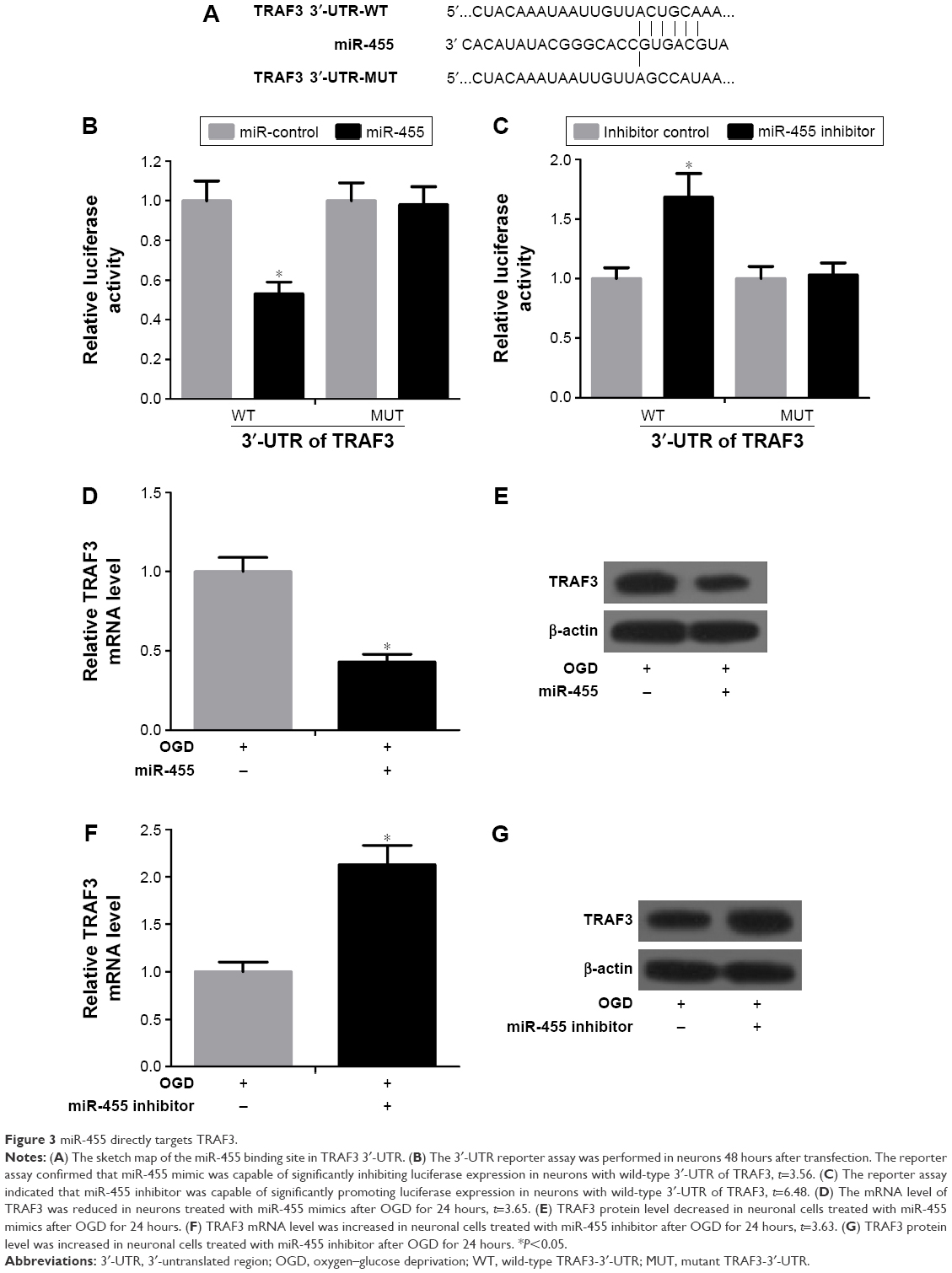 | Figure 3 miR-455 directly targets TRAF3. |
Downregulated TRAF3 expression suppressed neuronal cell death
To further confirm whether miR-455 inhibited neuronal cell death by regulating TRAF3 expression, knockdown of TRAF3 was performed in neuronal cells and mouse brains. As shown in Figure 4A and B, TRAF3 silencing significantly reduced TRAF3 expression in mRNA and protein levels in neuronal cells. Moreover, neuronal cell death was prevented when TRAF3 was downregulated (Figure 4C). In addition, death rate was determined in neuronal cells treated with miR-455 mimics and sh-TRAF3 or miR-455 inhibitor and sh-TRAF3. Our research showed that neuronal cell death was prevented by miR-455 in neuronal cells, and the addition of sh-TRAF3 strengthened protective effect of miR-455 on neuronal cells. On the other hand, miR-455 inhibitor significantly promoted neuronal cell death; however, this effect was reversed by sh-TRAF3 (Figure 4D).
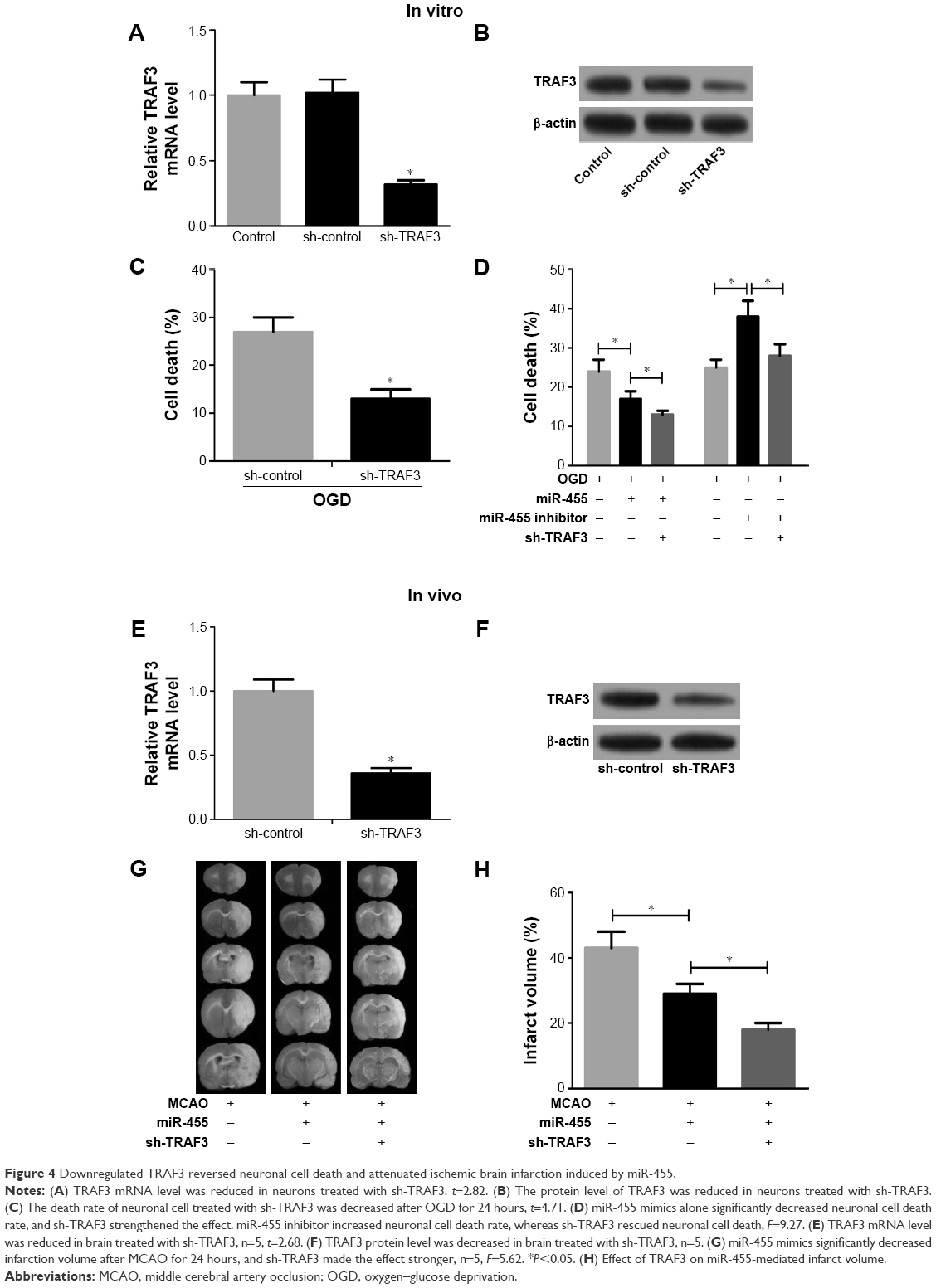 | Figure 4 Downregulated TRAF3 reversed neuronal cell death and attenuated ischemic brain infarction induced by miR-455. |
Downregulated TRAF3 attenuated ischemic brain infarction
sh-TRAF3 lentivirus was further injected into the cerebral cortex of wild-type C57BL/6J mice to investigate the effect of TRAF3 on ischemic brain infarction. The results showed that sh-TRAF3 effectively downregulated TRAF3 expression in both mRNA and protein levels in ischemic brain (Figure 4E and F). Then the infract volume of ischemic brain treated with miR-455 mimics and sh-TRAF3 lentivirus was detected. The data showed that miR-455 significantly reduced cerebral infarction volume, and this effect was enhanced by TRAF3 deletion (Figure 4G and H). These data demonstrated that miR-455 inhibited partly neuron death, via suppressing TRAF3 expression.
Discussion
Cerebral ischemia triggers frequently various pathological pathways of the ischemic cascade and finally results in irreversible neuronal injury in the ischemic region.21 Increasing evidence indicated that neuron death is a striking characteristic observed after stroke.22,23 Thrombolytic therapy using tissue-type plasminogen activator is still the only globally approved treatment for ischemic stroke, which is limited by a short treatment time window and low recanalization rates. These limitations indicated that protection from I/R injury is vital for patients with stroke. In our study, the effects of miR-455 on neuronal death and ischemic brain injury were investigated.
Identification of the miRNAs modulating the neuronal death and apoptosis networks associated with stroke could provide insight into new therapeutic ways. Xiao et al11 found that miRNAs were involved in various biological events, especially inflammatory response, resulting in cerebral damage. Peng et al24 showed that downregulated miR-181b can weaken the ischemic damage by upregulating HSPA5 and UCHL1 expressions. Similarly, previous reports indicated that miR-30a downregulation could induce neuroprotection from ischemic damage via upregulating the expression of HSPA5 in vitro and in vivo,1 and miR-29b could partly promote neuronal death by downregulating the expression of Bcl2L2.25 Also, knockdown of miR-29b at the infarct region induced stroke lesion caused by activation of the 12-lipoxygenase pathway, providing for the first time evidence linking arachidonic acid metabolism to miRNA stroke biology.26 He et al12 reported that miR-455 was markedly deregulated in ischemic stroke. Consistent with this, our research showed that the level of miR-455 decreased in neuronal cells after OGD and mouse brains after MCAO. Importantly, miR-455 upregulation in mice significantly alleviated ischemic brain injury and inhibited neuronal cell death, suggesting that miR-455 may be a promising therapeutic target for cerebral infarction.
The viability of miRNAs as therapeutic targets is confirmed by the fact that a Phase I clinical trial of an anti-miRNA was successfully concluded.27 In this case, a locked nucleic acid-based anti-miRNA targeting a liver-specific miRNA, miR-122 (miravirsen or SPC3649), is being developed as a hepatitis C therapy. miRNA overexpression strategies also exist and may be helpful in certain settings. Systemic delivery of miR-34a in a lipid-based delivery vehicle has been shown to block lung tumor growth in vivo.28 Overexpressing miR-210 can improve cardiac function in a murine model of ischemic heart disease.29 Because of the intricacy of pathophysiological molecular signaling in ischemic stroke, targeting single genes for therapeutic intervention has not yet succeeded in the clinic.
TRAF3 has been confirmed as a central modulator of ischemic signaling cascades, including neuronal death, neuroapoptosis, inflammation, and oxidative stress.16 TRAF3 serves in anti-inflammatory signaling, and its deletion in myeloid cells leads to inflammatory diseases and cancer in mice.30 Recently, Gong et al16 showed that neurons are the main target of TRAF3 in brain tissue, and TRAF3 contributes to c-Jun kinase-, nuclear factor κB-, and Rac-1-induced neuronal death via activation of transforming growth factor-β-activated kinase 1 (TAK1). In this study, using online softwares, miRBase and TargetScan, and Luciferase reporter assay, it was found that TRAF3 was a functional target of miR-455. Moreover, TRAF3 downregulation rescued neuronal cell death and attenuated ischemic brain infarction. Moreover, TRAF3 silencing strengthened the protective effect of miR-455 mimics on neurons and ischemic brain. Thus, our study confirmed that miR-455 prevented neuronal cell death by inhibiting TRAF3.
Conclusion
Our results revealed that miR-455 protected neuronal cells from death after OGD and reduced infarct volume in mouse brain after MCAO brain injury, by suppressing TRAF3 expression. Therefore, miR-455 functions as a new death regulator and may develop into a neuroprotective medicine for therapeutics of stroke. In light of this, the elucidation of miRNA mechanisms involved in neuronal death and potential pathogenesis of cerebral ischemia should be further investigated.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang P, Zhang N, Liang J, Li J, Han S, Li J. Micro-RNA-30a regulates ischemia-induced cell death by targeting heat shock protein HSPA5 in primary cultured cortical neurons and mouse brain after stroke. J Neurosci Res. 2015;93(11):1756–1768. | ||
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. | ||
An YT, Zhu P, Zhong Y, et al. A neuroprotective mechanism of YGY-E in cerebral ischemic injury in rats. CNS Neurosci Ther. 2012;18(1):14–20. | ||
Luker KE, Lewin SA, Mihalko LA, et al. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene. 2012;31(45):4750–4758. | ||
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–414. | ||
Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. | ||
Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149(3):515–524. | ||
Yin KJ, Deng Z, Huang H, et al. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 2010;38(1):17–26. | ||
Saugstad JA. MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. 2010;30(9):1564–1576. | ||
Ziu M, Fletcher L, Rana S, Jimenez DF, Digicaylioglu M. Temporal differences in microRNA expression patterns in astrocytes and neurons after ischemic injury. PLoS One. 2011;6(2):e14724. | ||
Xiao S, Ma Y, Zhu H, Sun H, Yin Y, Feng G. miRNA functional synergistic network analysis of mice with ischemic stroke. Neurol Sci. 2015;36(1):143–148. | ||
He W, Chen S, Chen X, Li S, Chen W. Bioinformatic analysis of potential microRNAs in ischemic stroke. J Stroke Cerebrovasc Dis. 2016;25(7):1753–1759. | ||
Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11(7):457–468. | ||
Yi Z, Lin WW, Stunz LL, Bishop GA. Roles for TNF-receptor associated factor 3 (TRAF3) in lymphocyte functions. Cytokine Growth Factor Rev. 2014;25(2):147–156. | ||
Jiang X, Deng KQ, Luo Y, et al. Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension. 2015;66(2):356–367. | ||
Gong J, Li ZZ, Guo S, et al. Neuron-specific tumor necrosis factor receptor-associated factor 3 is a central regulator of neuronal death in acute ischemic stroke. Hypertension. 2015;66(3):604–616. | ||
Gu H, Yu J, Dong D, Zhou Q, Wang JY, Yang P. The miR-322-TRAF3 circuit mediates the pro-apoptotic effect of high glucose on neural stem cells. Toxicol Sci. 2015;144(1):186–196. | ||
Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W, Jia LS. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun. 2011;404(4):941–945. | ||
Papadakis M, Hadley G, Xilouri M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19(3):351–357. | ||
Zhou L, Li F, Xu HB, et al. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med. 2010;16(12):1439–1443. | ||
Choi IY, Ju C, Anthony Jalin AM, Lee DI, Prather PL, Kim WK. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol. 2013;182(3): 928–939. | ||
Kim DH, Lee HE, Kwon KJ, et al. Early immature neuronal death initiates cerebral ischemia-induced neurogenesis in the dentate gyrus. Neuroscience. 2015;284:42–45. | ||
Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14(4):469–477. | ||
Peng Z, Li J, Li Y, et al. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J Neurosci Res. 2013;91(10):1349–1362. | ||
Shi G, Liu Y, Liu T, et al. Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res. 2012;216(2):225–230. | ||
Khanna S, Rink C, Ghoorkhanian R, et al. Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J Cereb Blood Flow Metab. 2013;33(8):1197–1206. | ||
Ouyang YB, Stary CM, Yang GY, et al. microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets. 2013; 14(1):90–101. | ||
Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70(14):5923–5930. | ||
Hu S, Huang M, Li Z, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122(11 suppl):S124–S131. | ||
Lalani AI, Luo C, Han Y, Xie P. TRAF3: a novel tumor suppressor gene in macrophages. Macrophage. 2015;2:e1009. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.