")
Back to Journals » Cancer Management and Research » Volume 12
MiR-26a-5p Serves as an Oncogenic MicroRNA in Non-Small Cell Lung Cancer by Targeting FAF1
Authors Ye M, Lin D, Li W, Xu H , Zhang J
Received 4 May 2020
Accepted for publication 14 July 2020
Published 11 August 2020 Volume 2020:12 Pages 7131—7142
DOI https://doi.org/10.2147/CMAR.S261131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Ming-fan Ye,1,* Dong Lin,2,* Wu-jin Li,1 Hai-peng Xu,2 Jing Zhang2
1Department of Chest Surgery, Fujian Provincial Hospital, Fuzhou, Fujian Province, People’s Republic of China; 2Department of Thoracic Oncology, Fujian Provincial Cancer Hospital, Fuzhou, Fujian Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Zhang Tel +86 13809557405
Email [email protected]
Purpose: Non-small cell lung cancer (NSCLC) accounts for approximately 80– 85% of all lung cancers, with the FAS-associated factor 1 (FAF1) acting as a tumor suppressor. MicroRNAs (miRNAs) can influence cancer progression by targeting oncogenes or anti-oncogenes. In this study, we aimed to reveal the influence of miR-26a-5p on the regulation of FAF1 expression and NSCLC progression, with the motivation of identifying a potential therapeutic target for NSCLC treatment.
Methods: A dual-luciferase reporter assay was used to check for the direct targeting of FAF1 by miR-26a-3p. The miR-26a-5p inhibitor or FAF1 shRNA plasmid was transfected into A549 and H1299 cells to modulate FAF1 expression. Then, the effect of miR-26a-5p/FAF1 on cellular functions was investigated. MTT assay was used to evaluate cell viability. EdU proliferation assay and cell cycle assay were performed to analyze the effect of miR-26a-5p on cell replication and cell cycle. We used annexin V-FITC and PI to stain apoptotic cells, followed by flow cytometric analysis. Transwell and wound healing assays were performed to investigate metastasis. Moreover, the effect of miR-26a-5p/FAF1 on cancer progression was examined in vivo. Lastly, the underlying mechanism was uncovered using RT-qPCR, Western blotting, and TOP/FOP flash assay.
Results: miR-26a-5p was found to directly target FAF1 and downregulate its expression. Blocking miR-26a-5p inhibited the cell growth, migration, and invasion, but promoted cell apoptosis. In addition, this inhibited the growth of tumor in mice. FAF1 knockdown reversed the functions of miR-26a-5p. Further, miR-26a-5p/FAF1 was observed to play an important role in the Wnt signaling pathway, regulating the expression of genes such as AXIN, c-Myc, and cyclin-D1.
Conclusion: Taken together, we show that miR-26a-5p functions as an oncogenic microRNA in NSCLC by targeting FAF1 and may serve as a potential target for NSCLC treatment.
Keywords: miR-26a-5p, FAF1, NSCLC, proliferation, apoptosis, metastasis
Introduction
Although the incidence of lung cancer is decreasing, it remains the major cause of cancer-related deaths worldwide.1,2 There are two main types of lung cancer: small cell lung cancer and non-small cell lung cancer (NSCLC). NSCLC is further classified into three subtypes, including adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. These are categorized in the same group due to similarities in the approach to treatment and prognosis.3,4 NSCLC accounts for approximately 80–85% of all lung cancers.5–7 Only a small portion of NSCLC patients are diagnosed at an early stage (stage I or II), where the tumor could be surgically resected.8 More than 60% of patients are diagnosed at an advanced or metastatic stage (stage III or IV), when surgical resection may not represent an effective choice.9 Until recently, radiotherapy and chemotherapy were the main modalities available for lung cancer treatments.10 Although the last decade had witnessed the rapid development of targeted therapy with improved clinical outcomes the 5-year survival rate for NSCLC is still less than 20%.11,12 It is, therefore, necessary to identify and utilize novel biomarkers or targets for NSCLC therapy.
FAF1, a member of the death-inducing signaling complex, is encoded by the FAF1 gene in humans.13,14 Interaction of the Fas ligand with the corresponding FAS antigen could mediate programmed cell death in a number of organs. The binding of FAF1 to FAS antigen leads to the initiation or enhancement of apoptosis mediated by the antigen.15 Overexpression of FAF1 has been shown to induce cell death in some cell types even without any extrinsic death signal.16 In addition, a low expression of FAF1 has been reported in various cancers.17 This has led to the identification of FAF1 as a tumor suppressor in several types of cancers, including NSCLC. Therefore, certain microRNAs (miRNAs) could possibly influence NSCLC progression by regulating FAF1 expression levels.
miRNAs are a class of endogenous non-coding RNAs, that consist of one single-strand RNA containing 19–22 nucleotides. These can act as either tumor suppressors or oncogenes, depending on the function of the genes they target.18 In recent years, microRNA-targeted therapeutics have emerged as an important anti-cancer strategy.19 In the present study, we have demonstrated that miR-26a-5p functions as an oncogenic miRNA in NSCLC cells by targeting FAF1, in both in vitro and in vivo conditions. Inhibition of miR-26a-5p could inhibit the proliferation, migration, and invasion of NSCLC cells, in addition to promoting their apoptosis. Moreover, FAF1 knockdown reversed these effects, suggesting that FAF1 functions as a tumor suppressor. Lastly, we investigated the regulation of downstream molecules by the miR-26a-5p/FAF1 axis.
Materials and Methods
Cell Culture
A549, H1299, and 293T cell lines were purchased from ATCC. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Detroit, MI, USA), supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin (Gibco), 100 U/mL streptomycin (Gibco), and 2 mM L-glutamine (Gibco), at 37°C in an atmosphere containing 5% CO2 and 21% O2.
Plasmid Constructs and Lentiviral Transduction
The 3ʹUTR of human FAF1 gene was amplified from genomic DNA by PCR using the primers shown in Table 1. The FAF1 3ʹUTR fragment was cloned downstream to the firefly luciferase reporter gene in the pmirGLO vector (Promega, Madison, WI, USA). The mutant FAF1-3ʹUTR was generated with the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, CA, USA).
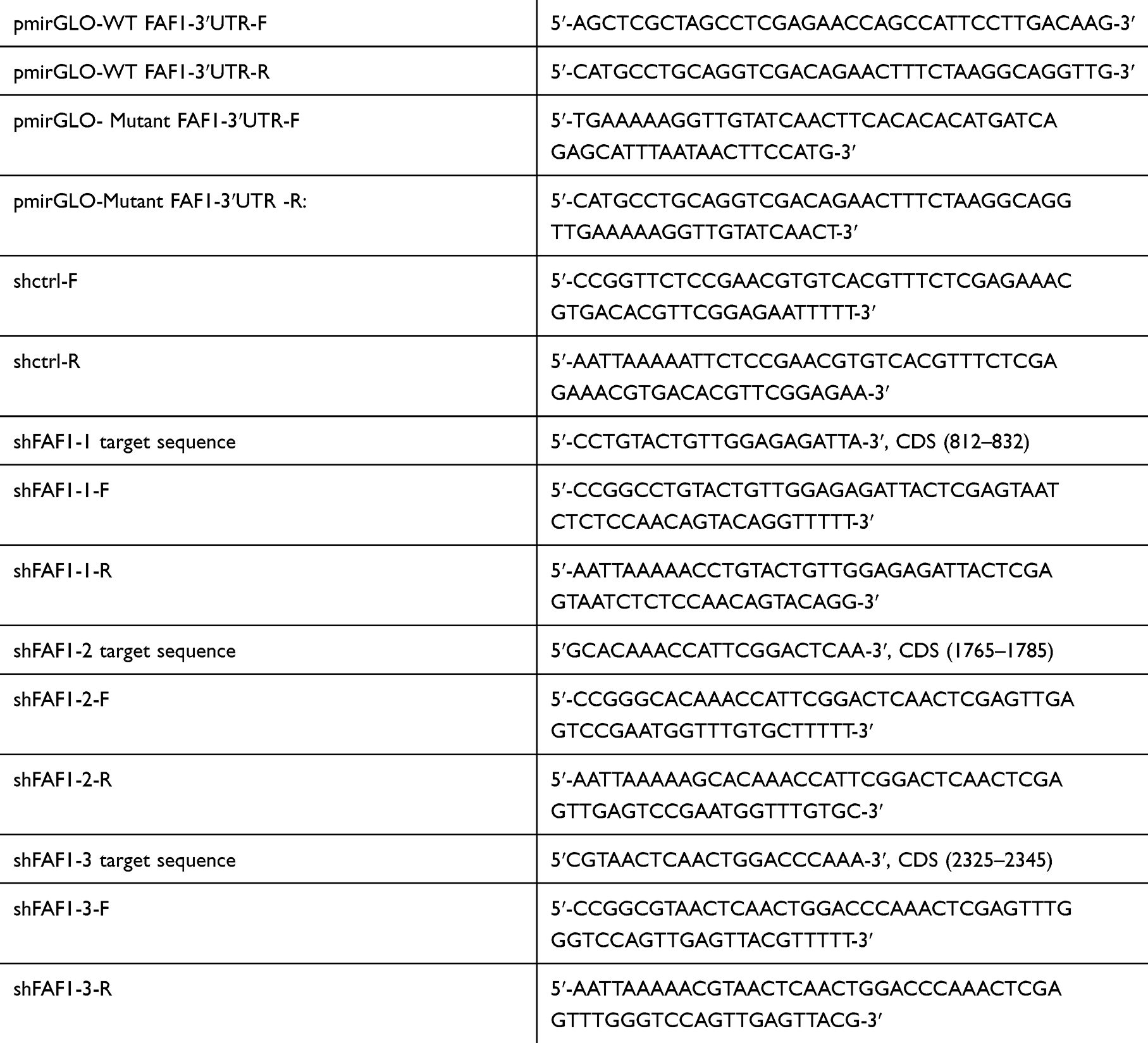 |
Table 1 Primers for Plasmid Constructs |
The miRNA sponge technology was used to block miRNAs. The oligosaccharide sequence (5′-AGCCUAUCACAAUUACUUGAA-3′), complementary to miR-26a-5p with mismatches at positions 9–12, was cloned into the lentivirus-based pCDH-EF1-MCS-T2A-Blasticidin vector (Anithela, Xiamen, Fujian, China). 293T cells were used to package the lentivirus. Two days after lentiviral infection at a multiplicity of infection (MOI) of 30, A549 cells were maintained in the presence of 4.0 µg/mL of blasticidin S (Sigma, San Francisco, CA, USA) for 10 days.
The lentivirus-based PLKO.1-TRC-Puro vector was used for FAF1 knockdown. The target sequences for FAF1 and the corresponding primers used for plasmid construction are shown in Table 1. These genetically engineered A549 cells (miR-26a-5p-sp) were infected with lentivirus at 30 MOI. Two days after infection, cells were maintained in 1.0 ug/mL of puromycin (Sigma) for 7 days.
Dual-Luciferase Reporter Assay
The miRNA and negative control (NC) mimics were obtained from GenePharma (Shanghai, China). A549 or H1299 cells were plated in a 24-well plate. When the cells reached 60–80% confluence, these were co-transfected with miR-26a-5p (or NC) mimic and pmirGLO-WT FAF1-3ʹUTR (or pmirGLO-mutant FAF1-3ʹUTR) plasmid. Thirty-six h after transfection, the cells were lysed. Then, the luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) and normalized to Renilla luciferase activity. Data acquisition for each group was performed in triplicate.
Transfection
The miR-26a-5p inhibitor and appropriate inhibitor NC were obtained from GenePharma. A549 and H1299 cells were placed into 60-mm cell culture dishes. When the cells grew to 60–80% confluence, these were transfected with miR-26a-5p inhibitor or inhibitor NC (200 pmol/well) using the Exfect Transfection Reagent (Vazyme, Nanjing, China). Four-µg aliquots of the pLKO.1-shFAF1 plasmid or pLKO.1-shctrl plasmid were used per well during co-transfection of the plasmid with inhibitor NC or miR-26a-5p inhibitor.
qRT-PCR Assay
RNA Isolater Total RNA Extraction Reagent (Vazyme) was used to isolate total RNA. cDNAs of FAF1, AXIN, C-MYC, and cyclin-D1 were synthesized using a Script High Fidelity One Step RT-PCR Kit (Vazyme). An RNA to cDNA EcoDry Premix Kit (Vazyme) was used for reverse transcription of miR-26a-5p. qPCR was conducted using the iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) with a ChamQ Universal SYBR qPCR Master Mix Kit (Vazyme) under the following conditions: 94°C for 3 min, followed by 94°C for 15 sec, 60°C for 20 sec, and 72°C for 20 sec, for 40 cycles. Each detection step was carried out in triplicate. The primers used in reverse transcription and qPCR are shown in Table 2. Expression levels were normalized to U6 or 18S rRNA levels.
 |
Table 2 Primers for RT-qPCR |
Western Blotting
Cells were lysed using the RIPA buffer (Vazyme). Lysates (30 μg/sample) were loaded on 10% denaturing SDS-PAGE gels and transferred onto PVDF membranes (Roche, Basel, Switzerland). The latter were blocked for 2 h in Tris-HCl buffer containing 5% bovine serum albumin, followed by incubation with primary antibodies overnight at 4°C. The membranes were then labeled with secondary antibodies at 28°C for 1 h. The antibodies used included those against FAF1 (Cat: 10271-1-AP, Proteintech, Chicago, IL, USA), β-catenin (Cat: 51067-2-AP, Proteintech), and GAPDH (Cat: 5174, Cell Signaling, Boston, MA, USA), and the anti-rabbit IgG, HRP-linked antibody (Cat: 7074, Cell Signaling).
EdU Proliferation Assay
The EdU proliferation assay was performed according to the manufacturer’s instructions (RiboBio, Guangzhou, China). After transfection for 24 h, A549 and H1299 cells were cultured in DMEM containing EdU at 37°C for 6 h. Then, cells were fixed with 4% formaldehyde for 20 min, followed by treatment with glycine for 5 min. We used treatment with 0.5% Triton X-100 at 28°C for 10 min to permeabilize the cell membranes. After washing twice, each well was treated with 200 μL of 1x Apollo reaction cocktail for 20 min. Subsequently, nuclear DNA was stained with Hoechst and imaged using fluorescence-based microscopy (Motic, Hongkong, China).
Cell Proliferation Assay
After transfection for 24 h, A549 and H1299 cells were seeded into 96-well plates (104 cells/well). An MTT assay kit (Cat: QF0025, Qiancheng Biotech, Shanghai, China) was used to evaluate cell viability at each time point. Absorbance was measured at a wavelength of 490 nm in a SpectraMax Absorbance Reader (Molecular Devices, San Francisco, CA, USA).
Cell Cycle Assay
After transfection for 24 h, A549 and H1299 cells were plated in 6-well plates at a density of 3 × 105 cells per well. These were collected the next day and fixed in 70% ethanol at 4°C overnight. Then, the cells were washed twice and incubated with PBS containing 10 μg/mL RNase and 0.2% Triton X-100 for 30 min at 37°C. Finally, the cells were stained at 28°C with propidium iodide (20 μg/mL) for 10 min in the dark, and subjected to flow cytometric analysis using the NovoCyte setup (Cat: 1300, ACEA, San Diego, CA, USA).
Analysis of Apoptosis
The Apoptosis Detection Kit (Cat: A211-02, Vazyme, Nanjing, China) were used to stain apoptotic cells. A549 and H1299 cells were collected after a 48-h transfection period, and stained with 200 µg/mL annexin V-fluorescein isothiocyanate and 30 µg/mL propidium iodide in the dark for 10 min at 28°C. Immediately afterwards, the samples were analyzed using the flow cytometer.
Wound Healing Assay
After transfection for 24 h, A549 and H1299 cells were seeded into 6-well plates. When the cells reached 100% confluence, wounds were induced by scratching with a 10-μL pipette tip. The cells were maintained in DMEM without serum and imaged under a microscope (MOTIC, Hongkong, China).
Transwell Assay
Transwell plates (Corning, Corning, NY, USA) were used to perform migration and invasion assays. After transfection for 24 h, 3 × 105 A549 and H1299 cells were seeded into the upper chambers of the transwell plates. After a 24-h incubation, 0.5% toluidine blue was used to stain the migrated cells and the number of migrated cells were counted in three random fields. The membranes of the upper chambers were pre-coated with 8-fold diluted Matrigel (BD Biosciences, Sparks, MD) before use in the Matrigel invasion assay.
In vivo Experiments
Six-to-eight-week-old female nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). Stably constructed A549 cells or control A549 cells (5 x 106) were subcutaneously injected into mice. The length (a) and width (b) of tumors were recorded every 4 days. The tumor volume was calculated using the formula V = ab2/2 (cm3). The tumors were also imaged 38 days after injection.
TOP/FOP Flash Assay
H1299 and A549 cells in 12-well palates were transfected with 1.0 ug of either TOP flash or FOP flash plasmid, 0.1 ug of TK-Renilla control vector (Promega), and 100 pM of miR-26a-5p (or NC) mimic per well. Twenty-four h after transfection, the cells were lysed and luciferase activities were measured using the Dual-Glo Luciferase Assay System (Promega).
Statistical Analysis
All data were analyzed using the SPSS software 24.0 (IBM SPSS, Armonk, NY, USA). Unpaired Student’s t-test was used to compare between two groups of data. A value of p < 0.05 was statistically significant.
Results
miR-26a-5p Binds to 3′UTR of FAF1 and Downregulates FAF1 Expression
The TargetScan database was used to search for FAF1-targeting miRNAs and the information extracted was used to choose several miRNAs as candidate miRNAs. As shown in Figure 1A, after transfection with only the miR-26a-5p mimic for 24 h, the mRNA expression level of FAF1 decreased appreciably in both A549 and H1299 cells. Then, we performed a dual-luciferase reporter assay. As shown in Figure 1B, transient co-transfection of miR-26a-5p mimic and pmirGLO-FAF1-3′UTR plasmid resulted in a marked decrease in the luciferase activity, compared with the NC group. This suppressive effect could be eliminated by introducing a mutation at the miR-26a-5p binding site in FAF1-3ʹUTR. These results indicated that miR-26a-5p directly targets FAF1 and downregulates its expression.
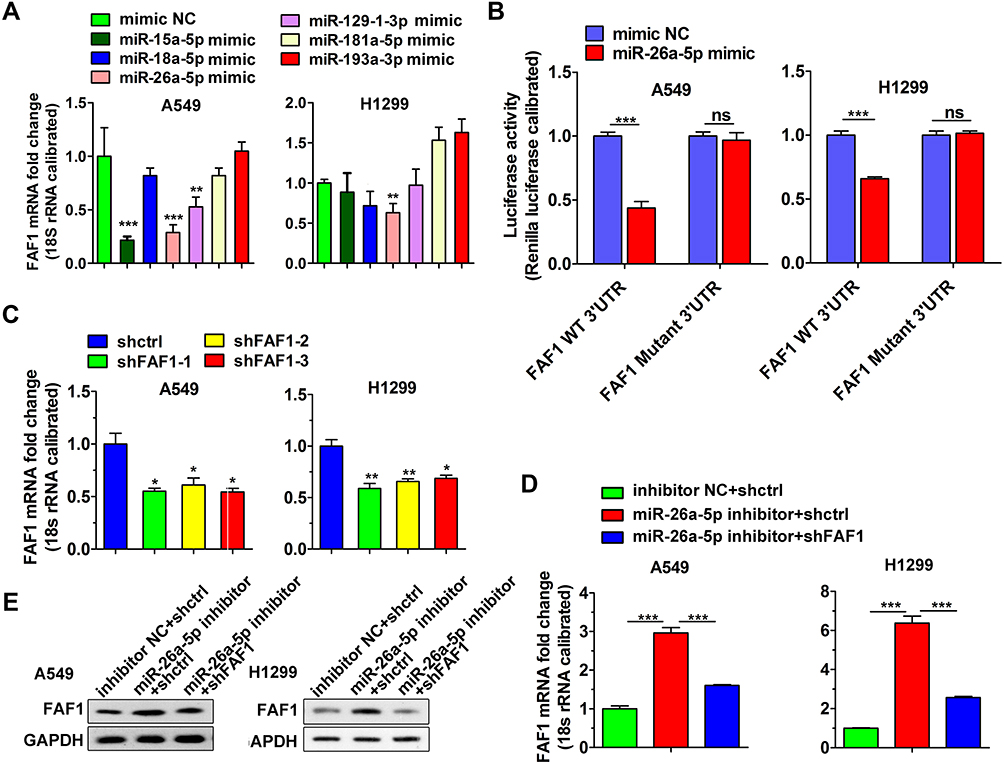 |
Figure 1 MiR-26a-5p binds to 3′UTR of FAF1 and downregulates FAF1 expression. (A) Analysis of the effect of candidate miRNAs on regulating FAF1 mRNA level by qRT-PCR. The data represent mean ± s.d. values from triplicate wells. **p < 0.01, ***p < 0.001. (B) Dual-luciferase reporter assay indicating that miR-26a-5p directly targets FAF1 3′UTR. The data represent mean ± s.d. values from triplicate wells. ***p < 0.001. (C) Analysis of knockdown efficiency of FAF1 shRNAs. Data shown represent mean ± s.d. values from triplicate wells. *p < 0.05, **p < 0.01. (D) Increase in FAF1 mRNA levels by miR-26a-5p inhibitor in both A549 and H1299 cells. Knockdown of FAF1 reversed this increase. The data represent mean ± s.d. values from triplicate wells. ***p < 0.001. (E) Increase in FAF1 protein levels by miR-26a-5p inhibitor in both A549 and H1299 cells. FAF1 knockdown reversed this effect. Representative data from two independent experiments is shown. GAPDH was used as the loading control. Abbreviation: ns, not significant. |
We used shRNA for FAF1 knockdown. As shown in Figure 1C, all three shRNAs targeting FAF1 mRNA reduced the expression of the latter in both A549 and H1299 cells. After transfection with the miR-26a-5p inhibitor, both mRNA and protein levels of FAF1 were significantly increased, and this increase could be reduced by co-transfection with the pCDH-shFAF1 plasmid (Figure 1D and E). The above data, therefore, also indicated that miR-26a-5p downregulates the expression of FAF1.
miR-26a-5p Promotes the Growth of NSCLC Cells and Inhibits Cell Apoptosis
To confirm the effects of miR-26a-5p and FAF1 on the growth of NSCLC cells, MTT assay to evaluate cellular viability was performed. The results revealed that the miR-26a-5p inhibitor suppressed the proliferation of A549 and H1299 cells. Co-transfection of the pCDH-shFAF1 plasmid with miR-26a-5p reversed this suppressive effect (Figure 2A). Moreover, the EdU proliferation assay showed that the miR-26a-5p inhibitor reduced the percentage of EdU+ cells, which represent cells in the S phase of the cell cycle (Figure 2B and C). In addition, the cell cycle assay revealed that miR-26a-5p inhibitor increased the percentage of cells in the G0/G1 phase (Figure 2D and E). Therefore, this data indicated that miR-26a-5p promotes the proliferation of NSCLC cells. We analyzed the effect of miR-26a-5p on cell apoptosis by flow cytometry. As shown in Figure 2F and G, the miR-26a-5p inhibitor promoted cell apoptosis, and FAF1 knockdown reduced the percentage of apoptotic cells. These data indicated that miR-26a-5p inhibits cell apoptosis.
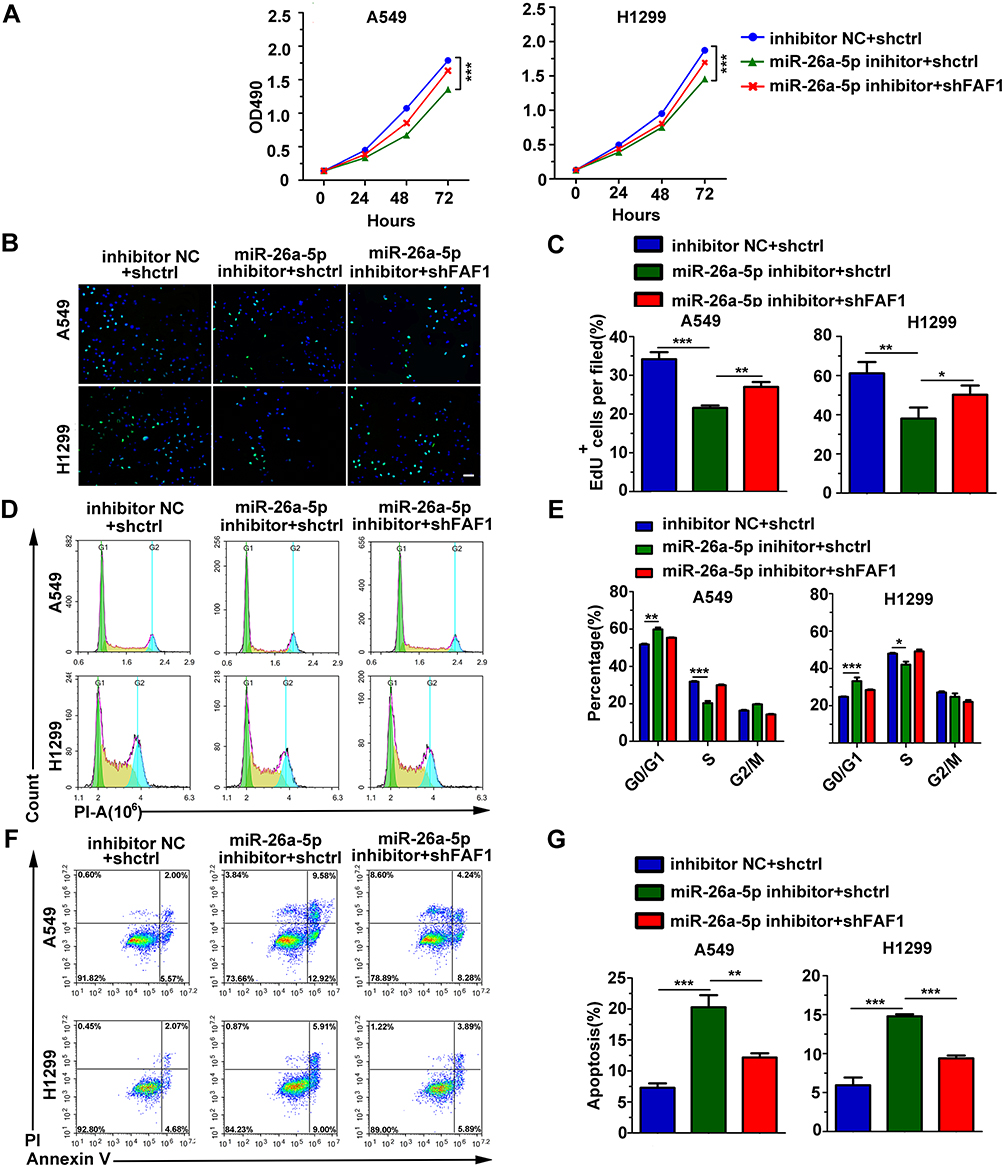 |
Figure 2 miR-26a-5p promotes cell growth and inhibits apoptosis. (A) MTT assay of NSCLC cells demonstrated that miR-25-3p promoted cell growth, with FAF1 reversing the effect. (B) Representative images from EdU proliferation assay. Scale bars, 200 μm. (C) Quantification of EdU-positive cells (n = 3). (D) Representative images from cell cycle analysis. (E) miR-26a-5p inhibition suppressed cell proliferation by inducing cell cycle arrest at G1/G0 phase and preventing entry into the S phase. Data shown represent mean ± s.d. values from triplicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (F) Representative images of apoptotic cells. (G) Blocking miR-25-3p promoted the apoptosis of NSCLC cells. The data represent mean ± s.d. values from triplicate wells. **p < 0.01, ***p < 0.001. |
miR-26a-5p Improves Cell Migration and Invasion
We used both wound healing and transwell migration assays to evaluate the function of miR-26a-5p in cell migration. As shown in Figure 3A–C, the miR-26a-5p inhibitor suppressed the migration of A549 and H1299 cells. Moreover, FAF1 knockdown reversed the miR-26a-5p-induced reduction in number of migrated cells. Consistent with these findings, the Matrigel invasion assay showed that the miR-26a-5p inhibitor also reduced the number of invasive cells (Figure 3D and E). These findings suggested that miR-26a-5p plays an important role in cell metastasis.
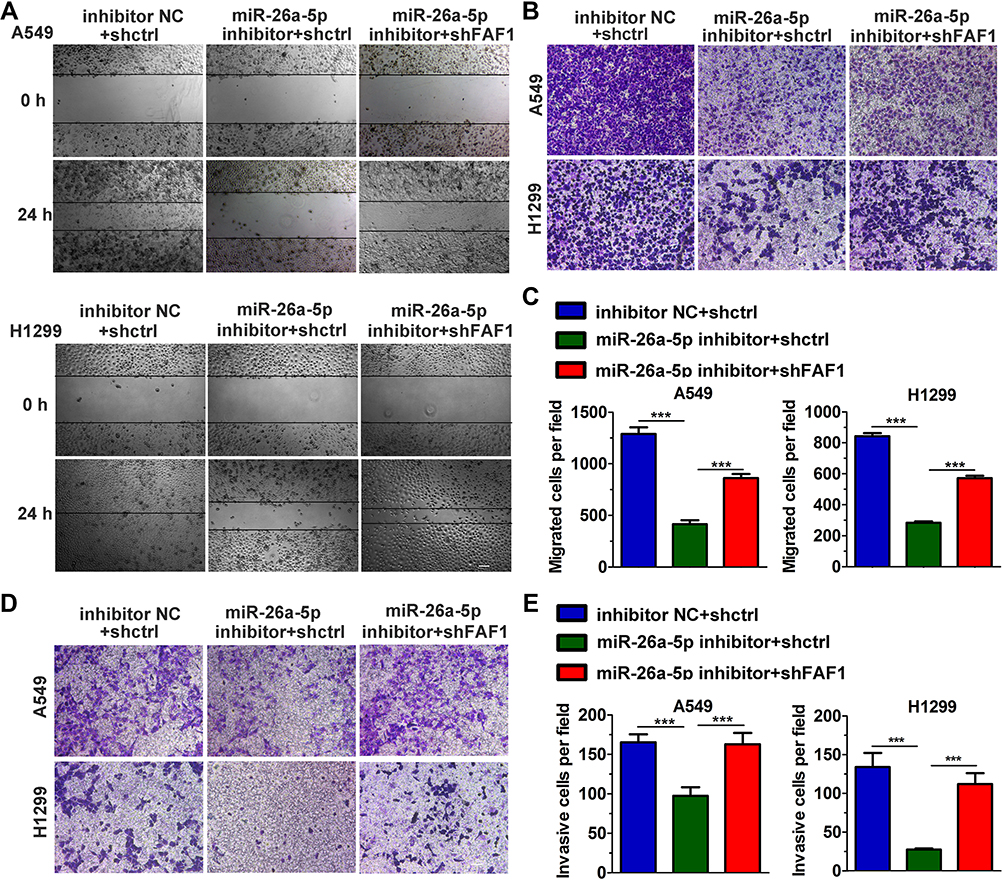 |
Figure 3 miR-26a-5p promotes cell migration and invasion. (A) Wound healing assay. Scale bars, 200 μm. (B) Representative images from the transwell migration assay. (C) Histogram of migrated cells per field. The data represent mean ± s.d. values from triplicate wells. ***p < 0.001. (D) Representative images from the Matrigel invasion assay. Scale bars, 100 μm. (E) Histogram of invasive cells per field. The data represent mean ± s.d. values from triplicate wells. ***p < 0.001. |
Blocking miR-26a-5p Inhibits NSCLC Cell Growth in Mouse Xenograft Models
The miRNA sponge method is usually used to generate long-term inhibition of miRNA activity. As shown in Figure 4A, transient co-transfection of miR-26a sponge (miR-26a-sp) and pmirGLO-FAF1-3′UTR plasmid into A549 cells led to a marked increase in luciferase activity, indicating that miR-26a-sp successfully blocked the activity of miR-26a-5p. Then, we constructed an A549 cell line with long-term inhibition of miR-26a-5p activity by miR-26a-sp, which was overexpressed in a lentiviral system. To study the effect of miR-26a-5p on cancer progression in vivo, A549 cells were subcutaneously injected into nude mice. Our results showed that miR-26a-sp suppressed tumorigenesis (Figure 4B). The volume and weight of tumor were significantly reduced in the miR-26a-sp group, compared with those in the control group (vector). (Figure 4C and D). Moreover, FAF1 knockdown reversed the effect of miR-26a-sp. These data suggested that miR-26a-5p promotes tumor growth.
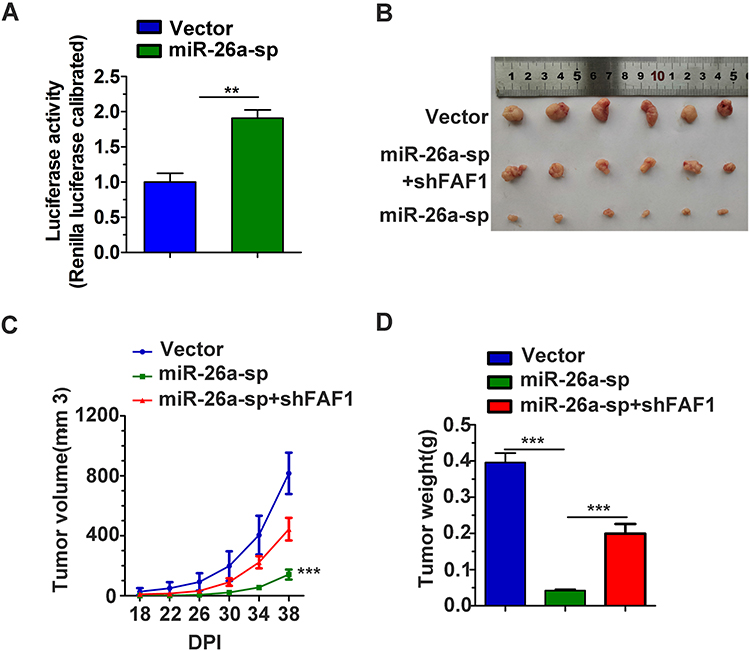 |
Figure 4 Blocking miR-26a-5p inhibits NSCLC cell growth in mouse xenograft models. (A) Efficacy of miR-26a-5p sponge (miR-26a-sp). pCDH-vector or pCDH-miR-26a-sp plasmid was co-transfected with pmirGLO-WT FAF1-3ʹUTR in A549 cells. Luciferase activity was measured 24 h post transfection. (B–D) Blocking miR-26a-5p activity suppressed tumorigenesis. Stable A549 cell lines were subcutaneously injected into nude mice (4 x 106 per mouse). The tumor size was recorded every 4 days from the 18th day after injection. The tumor volume was calculated using the formula V = ab2/2 (cm3). Tumor weight was recorded immediately after the tumors were harvested. **p < 0.01, ***p < 0.001. |
miR-26a-5p/FAF1 Plays Important Role in the Wnt Signaling Pathway
The Wnt/β-catenin signaling pathway plays a crucial role in cell proliferation and dysregulation of this pathway usually occurs during tumor progression. Therefore, we investigated the role of the miR-26a-5p/FAF1 axis in regulation of the Wnt/β-catenin signaling pathway. As shown in Figure 5A, the miR-26a-5p mimic markedly increased the luciferase activity in comparison to the NC mimic, indicating that miR-26a-5p promotes transcriptional activity of β-catenin. Western blot assays revealed that the miR-26a-5p inhibitor reduced the protein levels of β-catenin in both A549 and H1299 cells, demonstrating that miR-26a-5p upregulates the expression of β-catenin and consequently promotes the activation of Wnt/β-catenin signaling pathway (Figure 5B). As expected, the expression of several components of the canonical Wnt pathway, including AXIN, c-MYC, and cyclin-D1, was regulated in this process. As shown in Figure 5C, after transfection of the miR-26a-5p inhibitor in A549 and H1299 cells, the mRNA levels of AXIN were upregulated, but that of c-MYC and cyclin-D1 were downregulated (Figure 5C). Moreover, the increase or decrease in mRNA expression levels induced by miR-26a-5p could be reversed by FAF1 knockdown. In addition, we analyzed the expression levels of these components in harvested tumors, and the results were consistent with the findings above (Figure 5D–F). Taken together, miR-26a-5p/FAF1 axis plays important role in the Wnt signaling pathway.
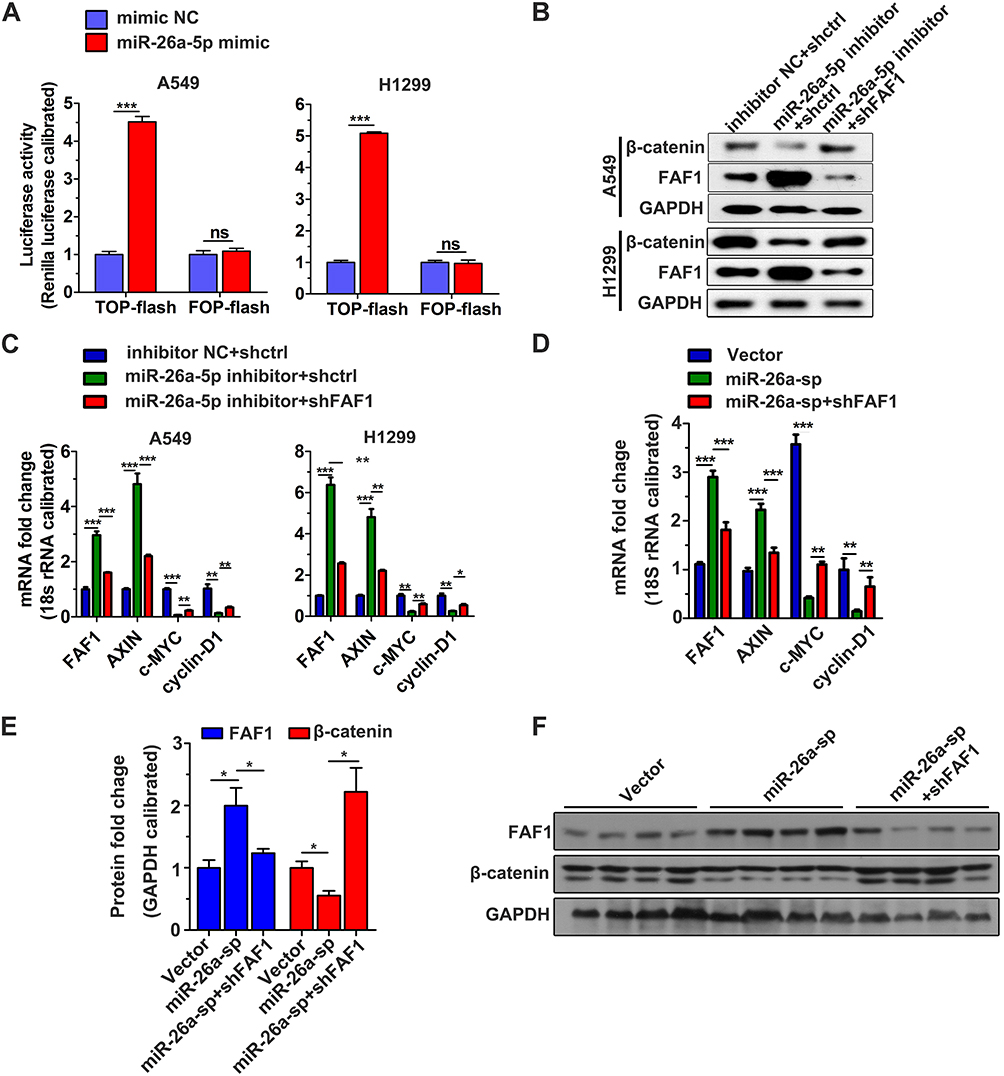 |
Figure 5 miR-26a-5p plays a crucial role in the Wnt/β-catenin signaling pathway. (A) TOP/FOP flash assay. The data represent mean ± s.d. values from triplicate wells. ***p < 0.001. (B) Blocking miR-26a-5p reduced protein levels of β-catenin. Representative data set from two independent experiments is shown. (C) Analysis of mRNA level of components belonging to the canonical Wnt signaling pathway in A549 and H1299 cells. qRT-PCR was performed 24 h after transfection. The data represent mean ± s.d. values from triplicate wells. *p < 0.05, **p < 0.01, ***p < 0.01 (D) qRT-PCR analysis of mRNA level of the signaling components in harvested tumors. **p < 0.01, ***p < 0.001. (E) Semiquantitative analysis of the protein levels of components in the harvested tumors. GAPDH was used as the loading control. *p < 0.05. (F) Representative image from Western blot analysis. Abbreviation: ns, not significant. |
Discussion
Lung cancer is one of the most common cancers worldwide, accounting for 13% of all new cancer cases.20 NSCLC comprises of 80–85% of all cases of lung cancer.5,6,12 Although diverse therapeutic strategies targeting NSCLC have been developed, lung cancer still has the highest mortality all over the world.1 In recent years, microRNA-targeted therapeutics has emerged as an important anti-cancer strategy. FAF1, a member of the death-inducing signaling complex, functions as a tumor suppressor in several types of cancers. Therefore, we hypothesized that mRNAs targeting FAF1 and playing important roles in cancer progression could be effective as potential therapeutic targets for the treatment of lung cancer. Indeed, our study revealed that miR-26a-5p directly targets FAF1 and downregulates its expression at both mRNA and protein levels. Moreover, blocking miR-26a-5p was found to inhibit the growth of NSCLC cells both in vitro and in vivo. In addition, miR-26a-5p inhibition also suppressed cell migration and invasion, but promoted cell apoptosis. These findings suggest that miR-26a-5p is an oncogenic microRNA and could be utilized as a target for oncotherapy.
Although we have identified miR-26a-5p as an oncogenic microRNA, the molecules downstream of miR-26a-5p/FAF1 remain unexplored. The Wnt/β-catenin signaling pathway is essential during embryonic development,21 and aberrations in this pathway have been implicated in various cancers.22 The result of TOP/FOP flash assay revealed that miR-26a-5p promotes activation of the Wnt/β-catenin signaling pathway (Figure 5A). In a canonical Wnt pathway, the nuclear translocation of β-catenin is initiated by the binding of the Wnt ligand to Frizzled receptors. Without Wnt ligands, cytoplasmic β-catenin is degraded by a degradation complex, composed of APC, AXIN, CK1α, and GSK-3β.23 In the present study, we found that blocking miR-26a-5p increased the mRNA expression level of AXIN and reduced the protein expression level of β-catenin (Figure 5A, B and D–F), indicating that miR-26a-5p inhibition promotes β-catenin degradation. In other words, miR-26a-5p activates the canonical Wnt signaling pathway through preventing β-catenin degradation. Moreover, FAF1 knockdown was found to reverse the above-mentioned changes (Figure 5A, B and D–F), which suggests that miR-26a-5p influences the Wnt signaling pathway by FAF1 regulation. After entry into the nucleus, β-catenin forms a transcriptional complex with LEF1/TCF and boosts the transcription of several Wnt-responsive genes, including c-MYC and cyclin-D1.24–26 This transcription would be inhibited on enhanced degradation of β-catenin. Indeed, our study showed that blocking miR-26a-5p downregulated the mRNA levels of both c-MYC and cyclin-D1 (Figure 5A and D). In conclusion, the miR-26a-5p/FAF1 axis plays an important role in the canonical Wnt signaling pathway by regulation of β-catenin degradation.
A miRNA may target multiple genes and regulate their expressions. A few target genes of miR-26-5p, such as TNRC6A, ITGβ8, and PTEN, have been reported. miR-26a-5p facilitates cell proliferation by targeting TNRC6A. Further, miR-26a-5p targets ITGβ8 and downregulates its expression, thereby potentiating metastasis of lung cancer cells. In addition, downregulation of PTEN by miR-26a-5p have been reported to promote the progression of bladder cancer.27–29 Therefore, we propose that miR-26a-5p exhibits its effects not only through the regulation of FAF1 expression, but also by other means.
In conclusion, our results support the existence of the miR-26a-5p/FAF1/β-catenin pathway. We have identified miR-26a-5p as an oncogenic miRNA that influences NSCLC progression by downregulating FAF1. Our findings would be useful in the development of novel drug targets for NSCLC treatment.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
All animal experiments were conducted in accordance with the guidelines for the humane treatment of animals (Fujian Medical University Laboratory Animal Center) and were approved by the Ethics Committee of Fujian Medical University.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi:10.3322/caac.21166
2. Vestergaard HH, Christensen MR, Lassen UN. A systematic review of targeted agents for non-small cell lung cancer. Acta Oncol. 2018;57(2):176–186. doi:10.1080/0284186X.2017.1404634
3. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637.
4. Alghamdi HI, Alshehri AF, Farhat GN. An overview of mortality & predictors of small-cell and non-small cell lung cancer among Saudi patients. J Epidemiol Glob Health. 2018;7:S1–S6.
5. Meza R, Meernik C, Jeon J, Cote ML. Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS One. 2015;10(3):e0121323. doi:10.1371/journal.pone.0121323
6. Dempke WC. Targeted Therapy for NSCLC–A Double-edged Sword? Anticancer Res. 2015;35(5):2503–2512.
7. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J Thorac Oncol. 2015;10(9):1240–1242.
8. Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin Cancer Biol. 2018;52(Pt 1):103–109. doi:10.1016/j.semcancer.2017.11.019
9. Jeremic B. Standard treatment option in stage III non-small-cell lung cancer: case against trimodal therapy and consolidation drug therapy. Clin Lung Cancer. 2015;16(2):80–85.
10. Lemjabbar-Alaoui H, Hassan OU, Yang YW, Buchanan P. Lung cancer: biology and treatment options. Biochim Biophys Acta. 2015;1856(2):189–210.
11. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
12. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
13. Ryu SW, Chae S, Lee KJ, Kim E. Identification and characterization of human Fas associated factor 1, hFAF1. Biochem Biophys Res Commun. 1999;262(2):388–394. doi:10.1006/bbrc.1999.1217
14. Ryu SW, Lee S, Park M-Y. Fas-associated factor 1, FAF1, is a member of Fas death-inducing signaling complex. J Biol Chem. 2003;278(26):24003–24010. doi:10.1074/jbc.M302200200
15. Jensen HH, Hjerrild M, Guerra B, Larsen MR, Hojrup P, Boldyreff B. Phosphorylation of the Fas associated factor FAF1 by protein kinase CK2 and identification of serines 289 and 291 as the in vitro phosphorylation sites. Int J Biochem Cell Biol. 2001;33(6):577–589. doi:10.1016/S1357-2725(01)00039-5
16. Ryu SW, Kim E. Apoptosis induced by human Fas-associated factor 1, hFAF1, requires its ubiquitin homologous domain, but not the Fas-binding domain. Biochem Biophys Res Commun. 2001;286(5):1027–1032. doi:10.1006/bbrc.2001.5505
17. Bjorling-Poulsen M, Seitz G, Guerra B, Issinger OG. The pro-apoptotic FAS-associated factor 1 is specifically reduced in human gastric carcinomas. Int J Oncol. 2003. doi:10.3892/ijo.23.4.1015
18. Nohata N, Hanazawa T, Kikkawa N, et al. Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol. 2011;38(1):209–217.
19. Xue J, Yang J, Luo M, Cho WC, Liu X. MicroRNA-targeted therapeutics for lung cancer treatment. Expert Opin Drug Discov. 2017;12(2):141–157. doi:10.1080/17460441.2017.1263298
20. Merabishvili V, Arseniev A, Barchuk A, Shcherbakov A, Demin E, Merabishvili E. Lung cancer morbidity and mortality. Siberian J Oncol. 2019;17:15–26. doi:10.21294/1814-4861-2018-17-6-15-26
21. Munoz-Descalzo S, Hadjantonakis AK, Arias AM. Wnt/ss-catenin signalling and the dynamics of fate decisions in early mouse embryos and embryonic stem (ES) cells. Semin Cell Dev Biol. 2015;47:101–109.
22. Duchartre Y, Kim Y-M, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2016;99:141–149. doi:10.1016/j.critrevonc.2015.12.005
23. Nakamura T, Hamada F, Ishidate T, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3(6):395–403. doi:10.1046/j.1365-2443.1998.00198.x
24. Zhang S, Li Y, Wu Y, et al. Wnt/beta-catenin signaling pathway upregulates c-Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat Rec (Hoboken). 2012;295(12):2104–2113. doi:10.1002/ar.22592
25. Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi:10.1073/pnas.96.10.5522
26. Li VS, Ng SS, Boersema PJ, et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–1256. doi:10.1016/j.cell.2012.05.002
27. Song Q, Liu B, Li X, et al. MiR-26a-5p potentiates metastasis of human lung cancer cells by regulating ITGbeta8- JAK2/STAT3 axis. Biochem Biophys Res Commun. 2018;501(2):494–500. doi:10.1016/j.bbrc.2018.05.020
28. Kang L, Yang C, Wu H, et al. miR-26a-5p regulates TNRC6A expression and facilitates theca cell proliferation in chicken ovarian follicles. DNA Cell Biol. 2017;36(11):922–929. doi:10.1089/dna.2017.3863
29. Wang H, Hu Z, Chen L. Enhanced plasma miR-26a-5p promotes the progression of bladder cancer via targeting PTEN. Oncol Lett. 2018;16(4):4223–4228.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.