")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
MiR-218 Inhibits CSE-Induced Apoptosis and Inflammation in BEAS-2B by Targeting BRD4
Authors Liu X, Wang J, Luo H, Xu C, Chen X, Zhang R
Received 24 August 2020
Accepted for publication 9 November 2020
Published 31 December 2020 Volume 2020:15 Pages 3407—3416
DOI https://doi.org/10.2147/COPD.S278553
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Xiaoli Liu,1 Junchen Wang,2,3 Huiling Luo,1 Chengxu Xu,1 Xingyu Chen,1 Rongxuan Zhang1
1Department of Respiratory, The Second People’s Hospital of Lanzhou City, Lanzhou City, Gansu Province, People’s Republic of China; 2Department of Interventional Medicine and Oncology, The Affiliated Hospital of Northwest Minzu University, Lanzhou City, Gansu Province, People’s Republic of China; 3Department of Interventional Medicine and Oncology, Gansu Second People ‘S Hospital, Lanzhou City, Gansu Province, People’s Republic of China
Correspondence: Rongxuan Zhang
Department of Respiratory, The Second People’s Hospital of Lanzhou City, No. 388 Jiangyuan Road, Chengguan District, Gansu Province 730030, People’s Republic of China
Tel +86-18194260886
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is an age-related disease, and its incidence rate is increasing every year. MicroRNAs (miRNAs) play critical roles in the COPD process and function as key biomarkers or potential therapeutic targets for patients with COPD. However, the potential roles and functional effects of miR-218 in COPD remain undefined.
Methods: The expression levels of miR-218 and bromodomain protein 4 (BRD4) were assessed by real-time quantitative polymerase chain reaction (RT-qPCR) or Western blot, respectively. In addition, a COPD cell model was established using cigarette smoke extract (CSE) in bronchial epithelial cell line (BEAS-2B). Enzyme-linked immunosorbent assay (ELISA) kit was applied to measure the concentrations of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-8 (IL-8) in cell supernatants of BEAS-2B cells. Moreover, cell apoptosis was examined by flow cytometry assay. The association relationship between miR-218 and BRD4 was confirmed by dual-luciferase reporter and RNA immunoprecipitation assay.
Results: MiR-218 was downregulated in COPD and CSE-induced BEAS-2B cells, and it was positively correlated with forced expiratory volume in 1 second (FEV1) % in COPD patients. Mechanically, overexpression of miR-218 or knockdown of BRD4 mitigated apoptosis and inflammation in BEAS-2B cells induced by CSE. Additionally, overexpression of BRD4 weakened the miR-218-mediated effects on CSE-induced BEAS-2B cells.
Conclusion: Overexpression of miR-218 inhibited CSE-induced apoptosis and inflammation in BEAS-2B cells by targeting BRD4 expression.
Keywords: MiR-218, BRD4, COPD, cigarette smoke extract, COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive destructive lung disease with persistent chronic inflammation, characterized by airflow limitation and severe respiratory failure.1,2 Generally speaking, long-term exposure to cigarette smoke is identified as a major risk factor for COPD, inducing an airway epithelium inflammatory response and apoptosis in COPD.3,4 As COPD is frequently exacerbated and often associated with severe complications, the clinical prognosis of COPD patients remains poor.5 Not only that, the incidence of COPD is rapidly increasing in China, and a large number of aging people and industrial pollutants are major contributors to the high incidence rate of COPD.6
MicroRNAs (miRNAs) could mediate gene expression at post-transcriptional level by either suppressing transcription of target genes or degrading them directly via matching with bases in the 3ʹ untranslated region (3ʹUTR).7 Recent findings showed that miRNAs played an essential role in the development and progression of COPD.8,9 For example, overexpression of miR-146-5p clearly reduced the release of interleukin-8 (IL-8), conversely, inhibition of miR-146-5p functions as a pro-inflammatory feature in COPD.10 Besides, miRNAs are considered to be therapeutic targets of pulmonary disease. For instance, a previous study reported that miR-155 acted as a novel therapeutic target for asthma.11 Moreover, Xu et al. reported that miR-218 acted as an anti-inflammatory via mediating activation of NF-κB, implying miR-218 was involved in inflammation in COPD.12 The above results confirmed that dysregulation of miRNAs was associated with the pathogenesis of COPD. Therefore, the functional effects of miR-218 have not been investigated thoroughly in COPD.
Bromodomain protein 4 (BRD4), a primary member of the BET family proteins, is an important epigenetic regulator by binding to acetylated histones.13 Additionally, BRD4 also regulated inflammatory gene NF-κB expression via combining acetylated RELA, which in turn increased transcriptional transactivation activity and stability of NF-κB in the nucleus.14 Similarly, Huang et al. also revealed that BRD4 could activateNF-κB via Specific Binding to Acetylated RelA.15 Therefore, we hypothesized that the expression of BRD4 was closely associated with inflammation in COPD.
COPD is largely attributable to cigarette smoke. In the present study, cigarette smoke extract (CSE) exposure is employed to induce inflammation and apoptosis in bronchial epithelial cells. The association between miR-218 and BRD4 was investigated in CSE-induced bronchial epithelial cells.
Materials and Methods
Clinical Samples
The lung tissue specimens of patients with COPD and control specimens of smokers/non-smokers were collected from The Second People’s Hospital of Lanzhou City. The removed samples were promptly snap-frozen in liquid nitrogen and maintained at −80°C until further analyses. Written informed consent was achieved from patients prior to participation. All recruited subjects were subjected to physical examination, spirometry (airflow limitation available; FEV1/FVC less than 0.70 indicated presence of persistent airflow limitation and COPD using a post-bronchodilator), and assessment of exacerbation risk and comorbidities. Patients were excluded if they (1) were diagnosed with other complicated disorders; (2) received preoperative treatments before admission; (3) required home mechanical ventilation. Inclusion criteria were (1) first time diagnosis; (2) received no therapies before admission; (3) willing to participate in follow-up. All the procedures had approval from the Ethics Committee of Second People’s Hospital of Lanzhou City. The use of clinical specimens was conducted in accordance with the Declaration of Helsinki. The clinicopathologic features of these patients are displayed in Table 1.
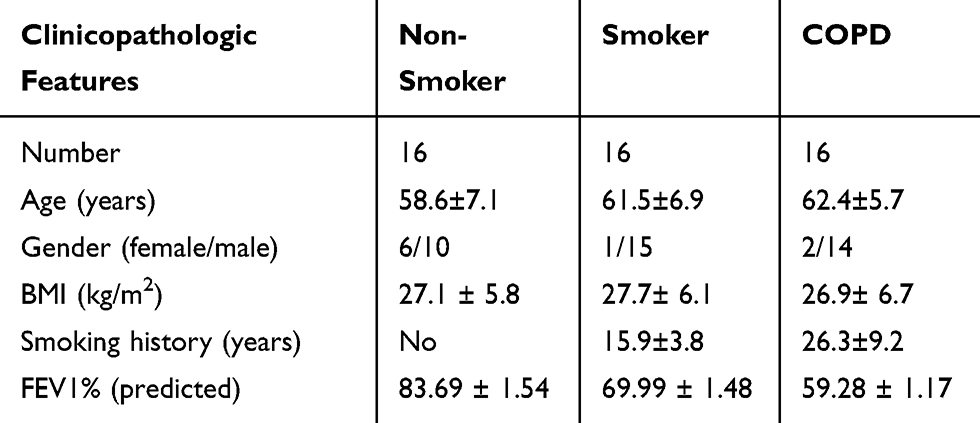 |
Table 1 Clinical Characteristics of the Recruited Subjects |
Cell Culture
Human bronchial epithelial cell line (BEAS-2B) was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). BEAS-2B cells were cultured in RPMI-1640 medium (GIBCO BRL, Grand Island, NY, USA) in a humidified atmosphere with 5% CO2 at 37°C. The 10% (v/v) fetal bovine serum (GIBCO BRL) was added to the medium. Additionally, CSE was prepared by commercial cigarettes (Liqun; Zhengzhou Tobacco Company, Henan, China; 11 mg of tar and 1 mg of nicotine). One cigarette was combusted with a vacuum pump, and then smoke was passed through the 5 mL medium to collect CSE as 100% CSE solution.
RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from BEAS-2B cells or tissues using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA). The OD260 nm/OD280 nm of samples value was measured to check RNA purity on Nanodrop 2000 (Applied Biosystems, Foster City, CA, USA). The special primers were used to analyze expression levels of miR-218 and BRD4 with SYBR Green Master Mix (Invitrogen, Carlsbad, CA, USA) under an ABI 7500 HT machine (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)/endogenous small nuclear RNA U6 was employed as the internal control. The primers were: miR-218 (Forward, 5ʹ-GCCGAGTTGTGCTTGATC-3ʹ; Reverse, 5ʹ-CTCAACTGGTGTCGTGGA-3ʹ);
BRD4 (Forward, 5ʹ-CCCCTCGTGGTGGTGAAG-3ʹ; Reverse, 5ʹ-GCTCGCTGCGGATGATG-3ʹ);
GAPDH (Forward, 5ʹ-TCCCATCACCATCTTCCAGG-3ʹ; Reverse, 5ʹ-GATGACCCTTTTGGCTCCC-3ʹ);
U6 (Forward, 5ʹ-CTCGCTTCGGCAGCACA-3ʹ; Reverse, 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ).
Transfection Assay
The mimics of miR-218 and negative control (miR-218 and Mir-NC), inhibitor of miR-218 and negative control (anti-miR-218 and anti-miR-NC), specific interference RNA against BRD4 (si-BRD4) and control (si-NC), and BRD4-overexpressing vector (BRD4) and control (pcDNA) were designed and obtained from Sangon (Shanghai, China). For transfection, BEAS-2B cells were seeded into 6-well plates with a density of 1×105 cells/well, followed by incubation overnight. 40 nM of aforementioned oligonucleotides or 1 μg of plasmids was transfected into BEAS-2B cells by Lipofectamine 2000 reagent (Invitrogen). Additionally, BEAS-2B cells were collected at 48 h post-transfection for subsequent experiments and transfection efficacy was determined by RT-qPCR assay.
Enzyme-Linked Immunosorbent Assay (ELISA)
The interleukin-6 (IL-6), interleukin-8 (IL-8), and tumor necrosis factor-α (TNF-α) in the supernatants of BEAS-2B cells were determined by ELISA kits (Invitrogen; #BMS213HS, #BMS204-3, and #BMS223HS). In brief, the BEAS-2B cell suspension was added to a 96-well plate at a density of 5 × l03 cells/well, and cultured overnight. Afterward, 50 μL standard or medium supernatant sample was added to another 96-well plate covered with goat anti-mouse IgM and then incubated at 37°C. After washing, each well was supplemented with blocking buffer and substrate. Finally, the absorbance was read at 492 nm under a multi-well scanning spectrophotometer (Bio-Rad, Hercules, CA, USA).
Analysis of Apoptosis
Apoptosis rate of BEAS-2B cells was assessed by Annexin V-FITC Apoptosis Detection Kit (Thermo Fisher Scientific). In brief, BEAS-2B cells were harvested and then incubated in staining buffer containing Annexin V labeled with fluorescein isothiocyanate (FITC) and propidium iodide (PI) for 30 min in dark conditions. Subsequently, flow cytometer (Applied Biosystems) was used to assess apoptotic cells.
Dual-Luciferase Reporter Assay
Online bioinformatics prediction software starBase v2.0 (http://starbase.sysu.edu.cn/starbase2/) was used to search the binding sites between BRD4 and miR-218. The 3ʹUTR segments of BRD4 were amplified by PCR and inserted into pGL3 vector (Ambion, Carlsbad, CA, USA), named as BRD4 WT. Mutations of miR-218-binding sites in BRD4 3ʹUTR sequence were generated using Mutagenesis Kit (Vazyme, Nanjing, China). BEAS-2B cells were co-transfected with 0.4 μg of luciferase reporter plasmid and 20 pmol of miR-218 or control by Lipofectamine 2000 (Invitrogen). Finally, fluorescence signals were tested with Dual-Luciferase Reporter kit (Sigma-Aldrich, Louis, Missouri, USA) in BEAS-2B cells post transfection 48 h, with Renilla luciferase activity as control.
RNA Immunoprecipitation (RIP) Assay
The RNA immunoprecipitation assay was conducted to confirm whether BRD4 could interact with miR-218. Transfected BEAS-2B cells were collected and then lysed in RIP buffer (Millipore, Billerica, MA, USA). The cell lysates were incubated with anti-Ago2 antibody (Ambion) and negative control normal IgG (Ambion) for 2 h at 4°C. After centrifugation, RT-qPCR assay was performed to test RNA enrichment in precipitate complexes.
Western Blot Assay
Radio-Immunoprecipitation assay (RIPA; Thermo Fisher Scientific) was utilized to extract protein from cells or tissues. The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membranes (Invitrogen). The membranes were blocked with 5% bovine serum albumin and then completely immersed in 1:1000 dilution primary antibody at 4°C, including BRD4 (#13440S; Cell Signaling Technology, Danvers, MA, USA) or β-actin (#4970S; 1:1000 dilution; Cell Signaling Technology). The secondary antibody with horseradish peroxidase conjugated (#7074S; Cell Signaling Technology) was added to the membrane and incubated for 2 h at 1:5000 dilution at room temperature. Finally, signal intensity of membranes was visualized with a chemiluminescence system.
Statistical Analysis
All statistical analyses were expressed as mean ± standard deviation with GraphPad Prism 7 (GraphPad Inc, La Jolla, CA, USA). A P value less than 0.05 was regarded as statistically significant. The difference between selected two groups was analyzed by the Student’s t-test. The multiple groups’ differences were estimated by one-way analysis of variance followed by Post-Hoc Test LSD. Pearson’s correlation analysis was used to determine the correlation relationship.
Results
MiR-218 Was Downregulated in COPD Patients
Expiratory volume in one second (FEV1) % is an essential index for assessment of living conditions of COPD patients. Our data suggested that COPD patients showed lower levels of FEV1% when compared with controls, including smokers and non-smokers (Figure 1A). The results of RT-qPCR assay suggested that miR-218 was apparently decreased in COPD patients compared with controls (Figure 1B). In addition, a notable positive correlation between FEV1% and miR-218 was observed in COPD patients (Figure 1C). Interestingly, we also found that miR-218 level was negatively correlated with serum inflammatory factor levels, including TNF-α, IL-6 and IL-8. All data implied that downregulation of miR-218 might be associated with COPD.
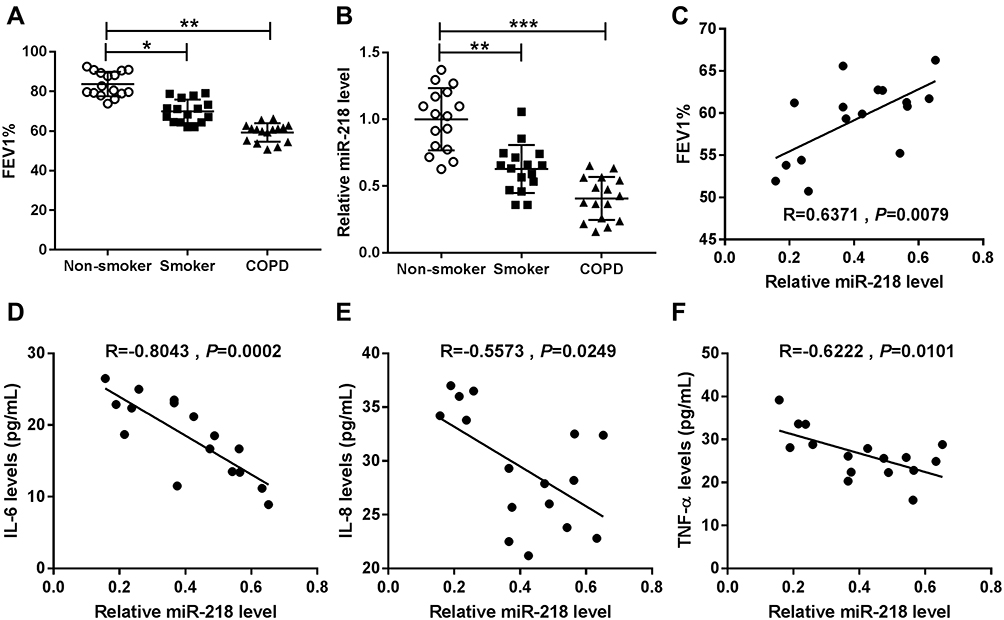 |
Figure 1 The expression level of miR-218 in chronic obstructive pulmonary disease. (A and B) Forced expiratory volume in one second (FEV1) % index and the relative level of miR-218 were displayed in non-smokers, smokers and COPD tissues. (C) The correlation relationship between FEV1% index and miR-218 level was analyzed in COPD patients by Pearson’s correlation analysis. (D–F) Pearson’s correlation analysis was used to analyze the correlation relationship between miR-218 and serum IL-6, IL-8, and TNF-α levels. Data are shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: FEV1, Forced expiratory volume in one second; COPD, chronic obstructive pulmonary disease; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6. |
Overexpression of miR-218 Suppressed Apoptosis and Inflammation in BEAS-2B Cells Caused by CSE
To assess the effect of miR-218 on COPD progression, BEAS-2B cells were treated with different concentrations of CSE. The results of RT-qPCR assay displayed that the expression level of miR-218 was greatly decreased in CSE-induced BEAS-2B cells with a dose/time dependent method (Figure 2A and B). Subsequently, we found that treatment with CSE led to the effective increase of TNF-α, IL-6 and IL-8 in cell supernatant, while their levels were significantly downregulated in the CSE+miR-218 group (Figure 2C–F). Furthermore, apoptosis was assessed by flow cytometry in transfected BEAS-2B cells. The data indicated that overexpression of miR-218 prominently suppressed CSE-induced apoptosis in BEAS-2B cells (Figure 2G). The results suggested that miR-218 reduced apoptosis and inflammation in BEAS-2B cells caused by CSE.
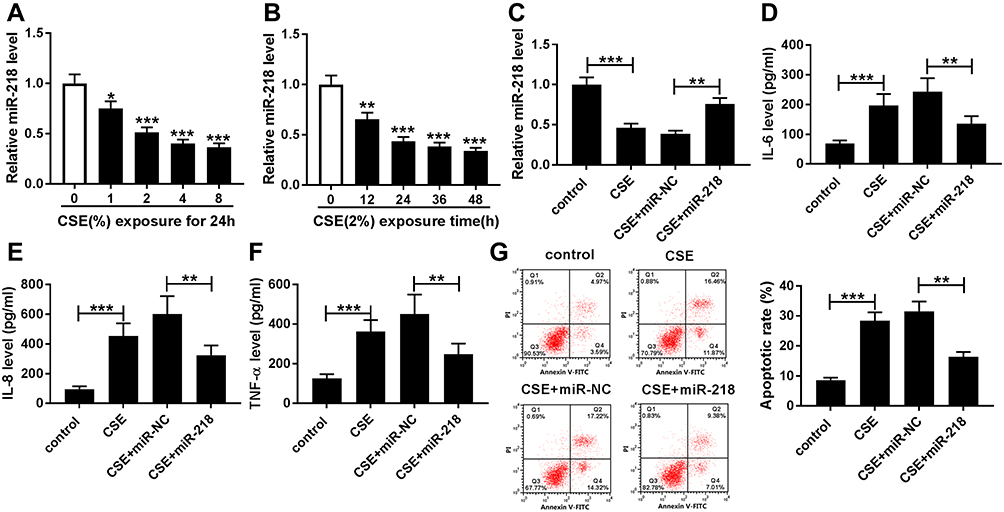 |
Figure 2 MiR-218 overexpression weakened apoptosis and inflammation in CSE-induced BEAS-2B cells. (A and B) The relative expression level of miR-218 was evaluated by RT-qPCR in BEAS-2B cells treated with different concentrations of CSE (0%, 1%, 2%, 4%, and 8%) for 24 h or treated with 2% CSE for different times (0 h, 12 h, 24 h, 36 h, and 48 h). (C) The expression of miR-218 was assessed by RT-qPCR assay in CSE-induced BEAS-2B cells transfected with miR-218 or miR-NC. (D–F) The levels of IL-6, IL-8, and TNF-α were measured by ELISA kit in the supernatants. (G) Apoptotic cells were monitored by flow cytometry assay. Data are shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: CSE, Cigarette smoke extract. |
BRD4 Was a Potential Target of miR-218
To investigate the target of miR-218 in BEAS-2B cells, a bioinformatics assay was performed. As shown in Figure 3A, miR-218 had complementary sequences in 3ʹUTR of BRD4. Dual-luciferase report assay exhibited that BEAS-2B cells co-transfected with luciferase reporter BRD4-WT and miR-218 mimic showed lower luciferase activity compared with control (Figure 3B). In addition, similar conclusions were confirmed with RIP assay, and the results revealed that overexpression of miR-218 led to an obvious enrichment of BRD4 in RIP-Ago2 group compared with control (Figure 3C). All data implied that BRD4 was a target of miR-218 in BEAS-2B cells.
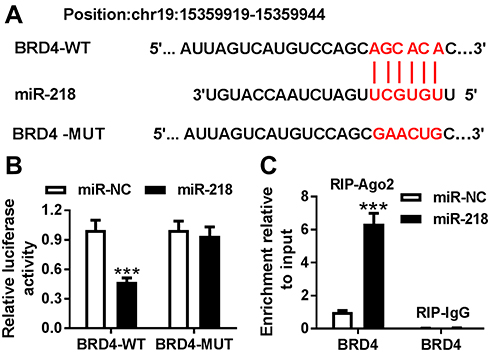 |
Figure 3 BRD4 was a target of miR-218. (A) The binding sites between miR-218 and 3ʹUTR of BRD4 are shown. (B) Luciferase activity was assessed by dual-luciferase report assay cells. (C) The RIP assay was conducted to test interaction between miR-218 and BRD4 BEAS-2B cells. Data are shown as mean ± SD from three independent experiments. ***P < 0.001. Abbreviation: BRD4, Bromodomain protein 4. |
BRD4 Was Upregulated in COPD Patients
The association between BRD4 and COPD was investigated. As shown in Figure 4A and B, the expression level of BRD4 was significantly increased in COPD tissues, and it was negatively correlated with miR-218 expression. Importantly, CSE induced the upregulation of BRD4 in BEAS-2B cells in a dose/time dependent way, no matter whether mRNA and protein (Figure 4C–F). Moreover, Western blot results revealed that miR-218 overexpression reduced protein level of BRD4, while protein level of BRD4 was effectively increased in BEAS-2B cells after transfection with anti-miR-218 (Figure 4G). Taken together, miR-218 negatively regulated BRD4 expression.
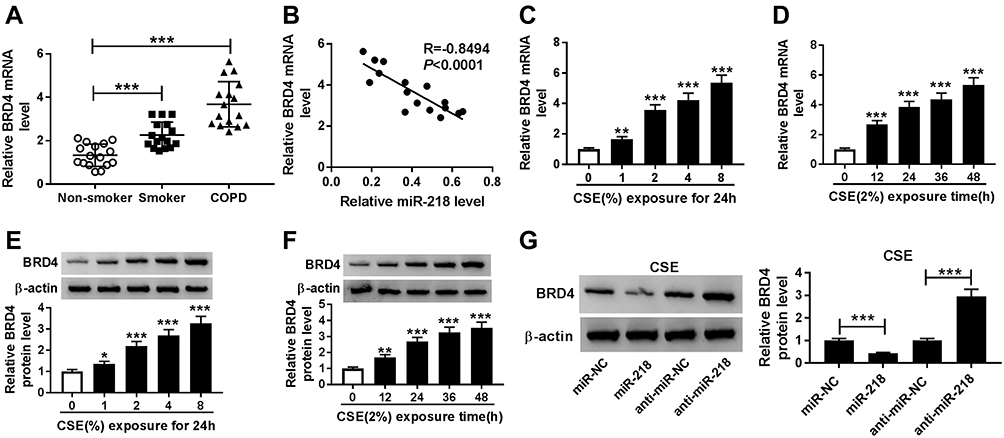 |
Figure 4 The expression level of BRD4 in COPD tissues and CSE-induced BEAS-2B cells. (A) RT-qPCR assay was performed to determine BRD4 level in COPD tissues and control groups. (B) Pearson’s correlation analysis was performed to analyze correlation relationship between miR-218 and BRD4 in COPD tissues. (C–F) RT-qPCR and Western blot assay were used to assess BRD4 levels in CSE-induced BEAS-2B cells. (G) The protein level of BRD4 was analyzed by Western blot assay in CSE-induced BEAS-2B cells transfected with miR-NC, miR-218, anti-miR-NC, anti-miR-218. Data are shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. |
CSE-Induced Apoptosis and Inflammation in BEAS-2B Cells Could Be Abolished by Silencing of BRD4
To further determine the functional effect of BRD4, BEAS-2B cells were transfected with si-BRD4 to knock down the expression of BRD4. As shown in Figure 5A, inhibition of BRD4 inhibited CSE-induced upregulation of BRD4 in BEAS-2B cells. Analogously, the levels of TNF-α, IL-6 and IL-8 were apparently enhanced in CSE-induced BEAS-2B cells, while BRD4 downregulation weakened this effect (Figure 5B–D). Besides, flow cytometry assay results confirmed that BRD4 knockdown protected BEAS-2B cells from CSE-induced apoptosis (Figure 5E). Overall, these results implied that inhibition of BRD4 abolished CSE-induced effects on BEAS-2B cells.
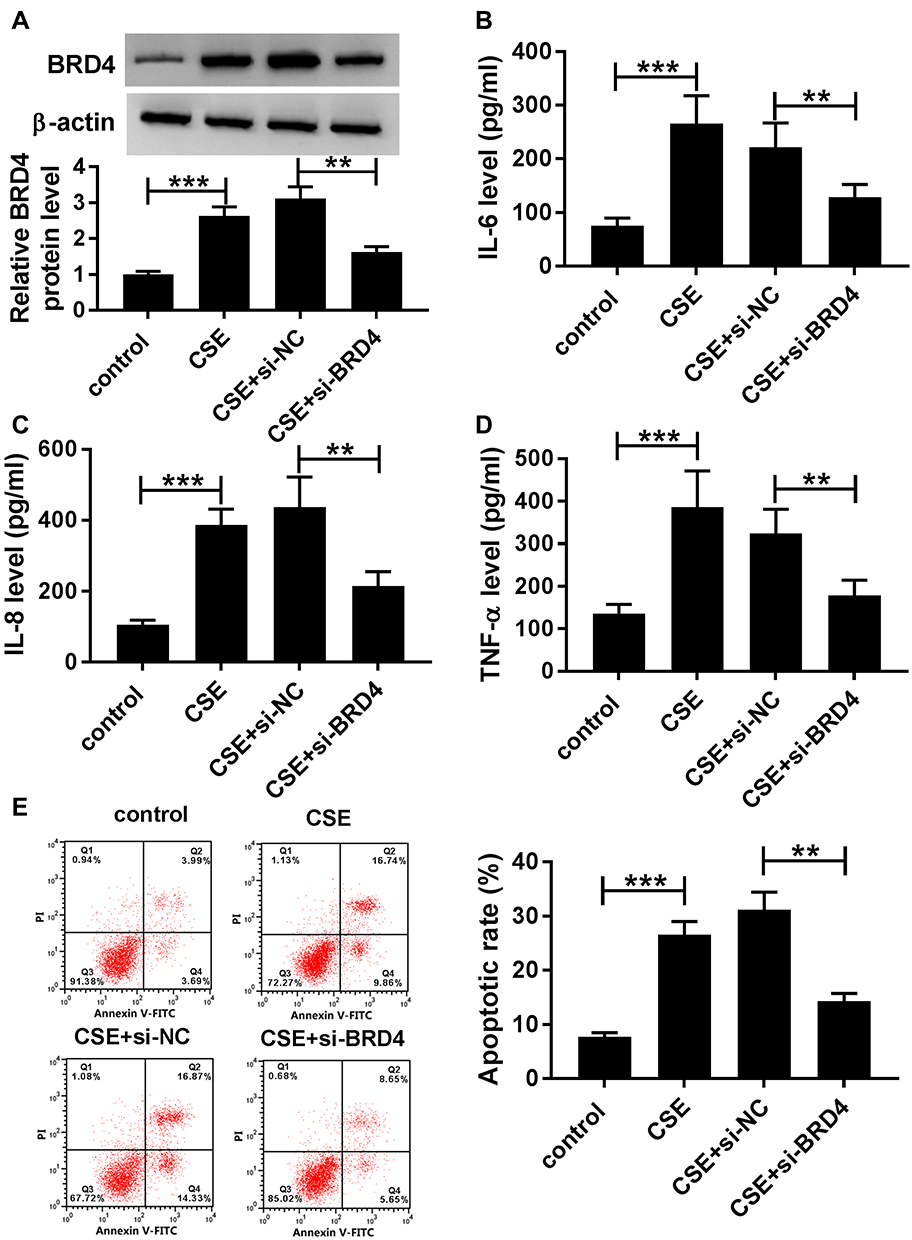 |
Figure 5 Knockdown of BRD4 inhibited CSE-induced apoptosis and inflammation in BEAS-2B cells. (A–E) BEAS-2B cells were divided into four groups: control, CSE, CSE+si-NC, and CSE+si-BRD4. (A) The protein level of BRD4 was measured by Western blot assay. (B–D) Inflammatory related factors (IL-6, IL-8, TNF-α) were measured by matched ELISA kits. (E) Flow cytometry assay was used to evaluate apoptosis of BEAS-2B cells. Data are shown as mean ± SD from three independent experiments. **P < 0.01, ***P < 0.001. |
Upregulation of BRD4 Abrogated the Effects of miR-218 Overexpression on CSE-Induced BEAS-2B Cells
As presented in Figure 6A, co-transfection with BRD4 and miR-218 apparently rescued the BRD4 expression compared with only transfection with miR-218 in CSE-induced BEAS-2B cells. Besides, gained functional experiences showed that overexpression of miR-218 reduced the levels of TNF-α, IL-6 and IL-8 in cell supernatants, which was abolished by overexpression of BRD4 (Figure 6B–D). Similarly, the upregulation of miR-218 resulted in a great inhibition of apoptosis in CSE-induced BEAS-2B cells, which could be restored by overexpression of BRD4, as demonstrated by flow cytometry analysis (Figure 6E). These results revealed that miR-218 inhibited CSE-induced apoptosis and inflammation in BEAS-2B by targeting BRD4.
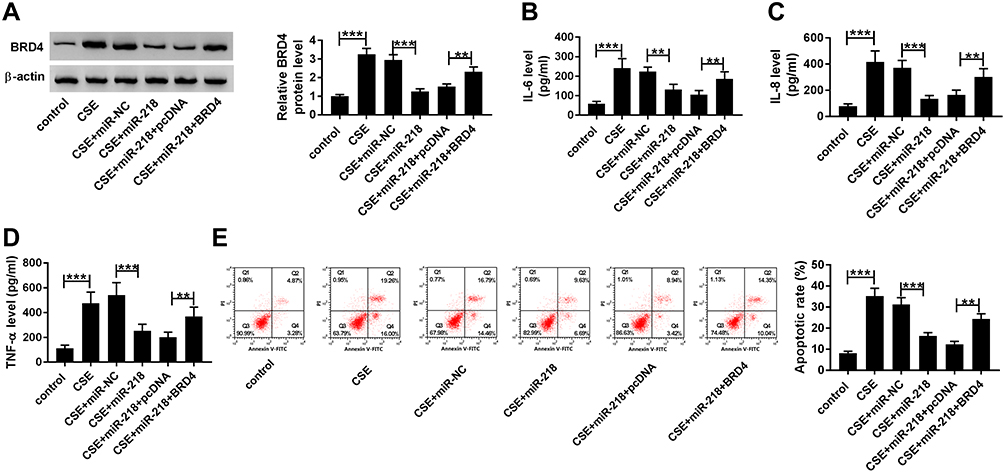 |
Figure 6 Overexpression of BRD4 attenuated effects of miR-218 upregulation on CSE-induced BEAS-2B cells. (A–E) BEAS-2B cells were divided into six groups: control, CSE, CSE+miR-NC, CSE+miR-218, CSE+miR-218+pcDNA, and CSE+miR-218+BRD4. (A) Western blot assay was carried out to assess level of BRD4. (B–D) The levels of IL-6, IL-8, TNF-α in cell supernatants were measured by ELISA kits. (E) The apoptosis of BEAS-2B cells was determined by flow cytometry assay. Data are shown as mean ± SD from three independent experiments. **P < 0.01, ***P < 0.001. |
Discussion
Conclusively, miR-218 was downregulated in COPD and in CSE-induced BEAS-2B cells; besides, the levels of miR-218 were closely correlated with secretion of inflammatory factor in COPD, revealing the anti-inflammatory properties of miR-218 in the development of COPD.
Cigarette smoke contributes to inflammation in the lung tissues via releasing of damage-associated molecular patterns from stressed or dying cells;16 besides, cigarette smoke-induced damage-associated molecular patterns contribute to the development of COPD.17 Additionally, Jin et al. reported that lung tissues persistently exposed to cigarette smoke could increase serum concentrations of TNF-α.18 Similarly, Gao et al. also disclosed that IL-6 and IL-8 were upregulated in lungs of COPD patients.19 Therefore, treatment with CSE is employed to induce inflammation and apoptosis in BEAS-2B cells as a cell model for COPD. Importantly, upregulation of miR-218 suppressed apoptosis and inflammation in BEAS-2B cells caused by CSE.
It has been confirmed that miRNAs regulated the expression of mRNA by targeting 3ʹUTR mRNAs;20 besides, miRNAs played crucial roles in the development and pathogenesis of lung diseases.21 The downregulation of miR-218-5p had been observed in human bronchial,22 small airway epithelium,23 and lung squamous cells.24 Wang et al. reported that miR-218 functioned as a tumor suppressor in lung cancer.25 Importantly, Schembri et al. also implied that miR-218-5p was associated with COPD processes.22 Similar to previous conclusions,12 the anti-inflammation function of miR-218 was confirmed in our results. The overexpression of miR-218 repressed apoptosis and inflammation in CSE-induced BEAS-2B cells.
BRD4 protein might have a promoter role in COPD because it is implicated in the inflammatory process by increasing pro-inflammatory cytokines.26,27 In addition, Song et al. found that BRD4 is implicated in inflammation in the development of COPD.28 Meanwhile, miRNA might participate in COPD processes by regulating inflammatory cytokine expression through targeting BRD4, such as miR-29b.29 Similarly, our data suggested that miR-218 inhibited CSE-induced apoptosis and inflammation in BEAS-2B by targeting BRD4.
Collectively, we found that BRD4 was a target of miR-218, and upregulation of BRD4 could attenuate effects of miR-218 overexpression on CSE-induced BEAS-2B cells, indicating that the miR-218/BRD4 axis might serve as a diagnostic target for COPD.
Conclusion
Collectively, our data showed that CSE could induce apoptosis and inflammation in BEAS-2B cells, which could be effectively weakened by enhancement of miR-218 or inhibition of BRD4. Mechanically, miR-218 regulated apoptosis and inflammation in CSE-induced BEAS-2B cells by targeting BRD4.
Patient Consent for Publication
Not applicable.
Data Sharing Statement
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The present study was approved by the ethical review committee of The Second People’s Hospital of Lanzhou City. Written informed consent was obtained from all enrolled patients.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Disclosure
The authors declare that they have no competing interests.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
2. Talay F, Tosun M, Yaşar ZA, et al. Evaluation of pregnancy-associated plasma protein-A levels in patients with chronic obstructive pulmonary disease and associations with disease severity. Inflammation. 2016;39(3):1130–1133.
3. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454.
4. Kim HJ, Baek S, Kim HJ, et al. The impact of smoking on airflow limitation in subjects with history of asthma and inactive tuberculosis. PLoS One. 2015;10(4):e0125020.
5. Criner RN, Han MK. COPD care in the 21st century: a public health priority. Respir Care. 2018;63(5):591.
6. Fang X, Wang X, Bai C. COPD in China: the burden and importance of proper management. Chest. 2011;139(4):920–929.
7. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233.
8. Sakao S, Tatsumi K. The importance of epigenetics in the development of chronic obstructive pulmonary disease. Respirology. 2011;16(7):1056–1063.
9. Osei ET, Florez-Sampedro L, Timens W, Postma DS, Heijink IH, Brandsma C-A. Unravelling the complexity of COPD by microRNAs: it’s a small world after all. Eur Respir J. 2015;46(3):807.
10. Osei ET, Florez-Sampedro L, Tasena H, et al. miR-146a-5p plays an essential role in the aberrant epithelial–fibroblast cross-talk in COPD. Eur Respir J. 2017;49(5):1602538. doi:10.1183/13993003.02538-2016
11. Zhou H, Li J, Gao P, Wang Q, Zhang J. miR-155: a novel target in allergic asthma. Int J Mol Sci. 2016;17(10):1773. doi:10.3390/ijms17101773
12. Xu H, Sun Q, Lu L, et al. MicroRNA-218 acts by repressing TNFR1-mediated activation of NF-κB, which is involved in MUC5AC hyper-production and inflammation in smoking-induced bronchiolitis of COPD. Toxicol Lett. 2017;280:171–180. doi:10.1016/j.toxlet.2017.08.079
13. Devaiah BN, Singer DS. Two faces of brd4: mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4(1):13–17. doi:10.4161/trns.22542
14. Hajmirza A, Emadali A, Gauthier A, Casasnovas O, Gressin R, Callanan BM. BET family protein BRD4: an emerging actor in NFκB signaling in inflammation and cancer. Biomedicines. 2018;6(1):16. doi:10.3390/biomedicines6010016
15. Huang B, Yang X-D, Zhou -M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29(5):1375. doi:10.1128/MCB.01365-08
16. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi:10.1016/S0140-6736(11)60988-4
17. Pouwels SD, Hesse L, Faiz A, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L881–L92.
18. Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. doi:10.1371/journal.pone.0111044
19. Gao W, Li L, Wang Y, et al. Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology. 2015;20(5):722–729. doi:10.1111/resp.12542
20. Tafrihi M, Hasheminasab E. MiRNAs: biology, biogenesis, their web-based tools, and databases. MicroRNA (Shariqah, United Arab Emirates). 2019;8(1):4–27.
21. Nana-Sinkam SP, Karsies T, Riscili B, Ezzie M, Piper M. Lung microRNA: from development to disease. Expert Rev Respir Med. 2009;3(4):373–385. doi:10.1586/ers.09.30
22. Schembri F, Sridhar S, Perdomo C, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci. 2009;106(7):2319. doi:10.1073/pnas.0806383106
23. Wang G, Wang R, Strulovici-Barel Y, et al. Persistence of smoking-induced dysregulation of MiRNA expression in the small airway epithelium despite smoking cessation. PLoS One. 2015;10(4):e0120824. doi:10.1371/journal.pone.0120824
24. Davidson MR, Larsen JE, Yang IA, et al. MicroRNA-218 is deleted and downregulated in lung squamous cell carcinoma. PLoS One. 2010;5(9):e12560.
25. Wang J, Li Z, Ge Q, et al. Characterization of microRNA transcriptome in tumor, adjacent, and normal tissues of lung squamous cell carcinoma. J Thorac Cardiovasc Surg. 2015;149(5):1404–14.e4.
26. Tian B, Yang J, Zhao Y, et al. BRD4 couples NF-κB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J Virol. 2017;91(6).
27. Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123.
28. Song J, Wang Q, Zong L. LncRNA MIR155HG contributes to smoke-related chronic obstructive pulmonary disease by targeting miR-128-5p/BRD4 axis. Biosci Rep. 2020;40(3).
29. Tang K, Zhao J, Xie J, Wang J. Decreased miR-29b expression is associated with airway inflammation in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2019;316(4):L621–l9.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.