")
Back to Journals » OncoTargets and Therapy » Volume 12
miR-193b Increases the Chemosensitivity of Osteosarcoma Cells by Promoting FEN1-Mediated Autophagy
Authors Dong S, Xiao Y, Ma X, He W, Kang J, Peng Z, Wang L, Li Z
Received 19 June 2019
Accepted for publication 7 November 2019
Published 22 November 2019 Volume 2019:12 Pages 10089—10098
DOI https://doi.org/10.2147/OTT.S219977
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Suwei Dong,1 Yanbin Xiao,1 Xiang Ma,1 Wei He,2 Jianping Kang,1 Zhuohui Peng,1 Lei Wang,1 Zhen Li3
1Department of Orthopaedics, The Third Affiliated Hospital of Kunming Medical University (Tumor Hospital of Yunnan Province), Kunming, Yunnan, People’s Republic of China; 2Medical Services Section, The First People’s Hospital of Yunnan Province, Kunming, Yunnan, People’s Republic of China; 3Department of Cancer Biotherapy Center, The Third Affiliated Hospital of Kunming Medical University (Tumor Hospital of Yunnan Province), Kunming, Yunnan, People’s Republic of China
Correspondence: Yanbin Xiao; Zhen Li
The Third Affiliated Hospital of Kunming Medical University (Tumor Hospital of Yunnan Province), No. 519 Kunzhou Road, Kunming, Yunnan, People’s Republic of China
Email [email protected]; [email protected]
Background: Osteosarcoma (OS) is one of the most common malignant bone tumors and specific microRNAs (miRNAs) are closely associated with malignant OS progression. In this study, we examined the role of microRNA-193b-3p (miR-193b) and the involvement of autophagy and apoptosis in the chemosensitivity of OS cells.
Methods: We employed qRT-PCR, Western blot, and immunohistochemistry to examine the expression levels of miR-193b, flap endonuclease 1 (FEN1), and autophagy-related proteins. Apoptosis was determined by flow cytometry using an Annexin V-FITC/PI apoptosis detection kit. Luciferase reporter assays confirmed the relationship between miR-193b and FEN1.
Results: miR-193b was downregulated in OS compared to adjacent normal tissues (p < 0.05). miR-193b overexpression in the OS cell lines induced autophagy and apoptosis, as shown by Western blotting and flow cytometry. Knockdown of FEN1, a structure-specific nuclease overexpressed in OS tissues (p < 0.001), induced apoptosis through activation of autophagy. Luciferase reporter assays confirmed that FEN1 is a direct target of miR-193b, FEN1 knockdown reinforced miR-193b induced apoptosis. Moreover, miR-193b expression enhanced epirubicin-induced autophagy and apoptosis.
Conclusion: Collectively, the results showed that miR-193b/FEN1 may serve as a novel therapeutic target for OS aimed mainly at the induction of autophagy and apoptosis. The miR-193b/FEN1 axis increased the chemosensitivity of OS cells, while activation of autophagy enhanced the anticancer effects of epirubicin.
Keywords: miR-193b, FEN1, apoptosis, autophagy, osteosarcoma
Introduction
Osteosarcoma (OS) is one of the most common malignant bone tumors, with an incidence of 4–5 per million among children and teenagers.1,2 Despite great advances, the efficacy of OS treatment remains unsatisfactory, especially for patients diagnosed at advanced stages of the disease.3 Therefore, understanding the molecular mechanisms that contribute to the carcinogenesis and development of OS is essential for developing novel therapeutic approaches.
The expression and function of specific microRNAs (miRNAs) are reported to be closely associated with malignant OS progression,4,5 and dysregulation of miR-193b has been reported in various human malignancies. However, little is known about the roles of miR-193b in OS and drug sensitivity.
Flap structure-specific endonuclease 1 (FEN1) participates in numerous DNA processing pathways, including Okazaki fragment maturation, stalled replication fork rescue, telomere maintenance, long-patch base excision repair, and apoptotic DNA fragmentation,6,7 and disruption of FEN1 function leads to the accumulation of DNA double-strand breaks.8 Moreover, FEN1 is considered a marker for metastasis and poor prognosis in multiple cancers;9–11 additionally, recent studies have revealed that FEN1 has a role in regulating sensitivity to chemotherapy.12,13
Autophagy is a stress-relieving/homeostatic cellular recycling process involved in the maintenance of cellular homeostasis through the lysosomal degradation of various proteins and damaged organelles.14 Accumulating evidence has shown that autophagy can promote survival in patients undergoing chemotherapy, radiotherapy, and targeted therapies.15–17
The aim of the present study was to elucidate the potential role of miR-193b in apoptosis and autophagy of OS cells. We show that miR-193b regulates apoptosis and autophagy in OS cells by regulating FEN1 expression, which may lead to novel treatment methods and management strategies for OS.
Materials and Methods
Patient Tissue Samples
The present study was approved by the Institutional Ethics Committee of The Third Affiliated Hospital of Kunming Medical University, China. Paired tumor and adjacent normal formalin-fixed paraffin-embedded (FFPE) biopsy samples were obtained from 53 patients, collected between 2016 and 2017. Informed consent was obtained from each patient for the use of their tissues for research purposes. Patients with a diagnosis of relapse and who had received preoperative radiation, chemotherapy, or biotherapy were excluded from the study to avoid changes in tumor marker determination resulting from treatment. The miRNeasy FFPE Kit (Qiagen, Germany) was used to extract RNA from paraffin-embedded specimens.
Cell Culture and Transfection
The human MG-63, U2OS and 143B cell lines were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, USA.) and 1% penicillin and streptomycin and maintained at 37°C in a humidified incubator with 5% CO2. The miR-193b mimic, miR-193b inhibitor, and negative control were designed and synthesized by Guangzhou RiboBio Co., Ltd (Guangzhou, China). The small interfering RNA (siRNA) targeting FEN1 (si-FEN1) and negative control (NC) siRNA were also synthesized by Guangzhou RiboBio Co., Ltd. Plasmids for FEN1 and NC overexpression were purchased from Shanghai GenePharma Co., Ltd. All plasmid constructs were verified by sequencing. Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used for miRNA or siRNA transfection into OS cells according to the manufacturer’s protocols. Cells were harvested for subsequent experiments 48 h after transfection.
Luciferase Reporter Assay
The online prediction algorithm TargetScan (http://www.targetscan.org/) was used to predict the direct targets of miR-193b. The wild-type (WT) and mutant (MUT) FEN1 3ʹ-UTRs were cloned into the pGL3-basic luciferase reporter vector (Promega Cooperation, Madison, WI, USA) and named FEN1 WT and FEN1 MUT, respectively. Briefly, OS cells were seeded into a 24-well plate at a density of 1 × 105 cells/well. The cells were co-transfected with FEN1 WT or FEN1 MUT and the miR-193b mimic or NC miRNA using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Luciferase activity was measured 48 h after co-transfection using the Dual-Luciferase Reporter Assay kit (Promega) with Renilla luciferase activity as the internal control, according to the manufacturer’s protocol. Each experiment was performed independently at least three times.
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and purified with the RNeasy Maxi kit (Qiagen GmbH). To determine miR-193b expression, the purified RNA was reverse transcribed using the miScript Reverse Transcription kit (Qiagen GmbH) in a Roche Lightcycler 480 Real-Time PCR system (Roche Diagnostics, Basel, Switzerland). The relative miR-193b expression levels in tissue specimens and cells were calculated using the 2-ΔΔCq method,18 with U6 as the internal control. The primer sequences were as follows: FEN1 forward, 5ʹ- GTGAAGGCTGGCAAAGTCTA-3ʹ and reverse, 5ʹ-GTGAAGGCTGGCAAAGTCTA-3ʹ; GAPDH forward, 5ʹ- ACTCCCATTCTTCCACCTTTG-3ʹ and reverse, 5ʹ- CCCTGTTGCTGTAGCCATATT-3ʹ.
Immunoblotting
Cells were harvested 72 h after transfection and lysed in RIPA buffer (89900, Pierce, USA). The lysates were centrifuged at 14,000 rpm for 20 min at 4°C and the supernatants collected. Protein concentrations were measured using the BCA assay (23227, Thermo, USA). For each sample, 50 μg of protein lysate was loaded per well. Samples were electrophoresed on 10% SDS-PAGE gels and transferred onto polyvinylidene fluoride (PVDF) membranes (ISEQ00010, Millipore, USA) by electroblotting. The membranes were pretreated with 5% nonfat dry milk in Tris-buffered saline + Tween 20 (TBS-T) for 2 h, followed by overnight incubation with primary antibodies for 16 h at 4°C. The following primary antibodies were used: anti-FEN1, ab17994; anti-LC3I/II, ab51520; anti-p62, ab91526; anti-Beclin 1, ab62557; and anti-Cleaved Caspase-3, ab49822, all from Abcam, USA. The membranes were then incubated with a horseradish peroxidase (HRP)-labeled secondary antibody (1:10,000, #7076, Cell Signaling Technology, USA) for 1 h before detection by electrochemiluminescence (ECL) (RPN2135, GE healthcare, UK). GAPDH was used as the internal loading control (1:1000, ab181602; Abcam).
Cell Apoptosis Analysis
Cell apoptosis was analyzed using an Annexin V-fluorescein isothiocyanate (FITC) Apoptosis kit (BD Biosciences, San Diego, CA, USA) following the manufacturer’s protocol. Transfected cells were seeded into 24-well plates (1 × 105 cells/well) and cultured in a humidified incubator containing 5% CO2 at 37°C for 24 h. The cells were subsequently resuspended in 500 µL of binding buffer containing 1% FITC-labeled Annexin V and propidium iodide. After incubation in the dark for 30 min, apoptosis levels were evaluated using the FACS Aria system (BD Immunocytometry Systems, San Jose, CA, USA) and analyzed by Cell Quest software (Becton Dickinson Ltd). All the samples were assayed three times.
Immunohistochemical (IHC) Analysis
Samples were processed for IHC analysis to determine FEN1 expression levels and distribution patterns. Paraffin-embedded tissue sections (4 μm) were mounted on charged glass slides and baked at 60°C for 2 h. The slides were then allowed to cool to room temperature, deparaffinized in xylene, and rehydrated in a graded alcohol series. Sections were microwave-treated for 10 min in citrate buffer (pH 6.0) for antigen retrieval, and endogenous peroxidase activity was blocked by incubation in 0.3% hydrogen peroxide for 10 min. Rabbit polyclonal antibodies (ab17994, Abcam) diluted 1:250 in phosphate-buffered saline (PBS) were used to detect the FEN1 protein. After two washes in PBS, the slides were incubated with ABC (Vector Laboratories, Burlingame, CA, USA), washed, overlaid with 3-30-diaminobenzidine (DAB; Dako Corporation, Carpinteria, CA, USA), and counterstained with hematoxylin. Human lung squamous carcinoma tissue was used as a positive control, while negative controls were obtained by replacing the primary antibody with non-immunized serum. Tissue was considered positive for FEN1 protein expression if more than 10% of the tumor cells showed nuclear staining. All the slides were independently evaluated for protein expression by three different observers and slides with an incongruent grading were re-evaluated and a consensus was reached.
Statistics
The correlation between immunocytochemical labeling of FEN1 and other clinical pathology parameters was analyzed by the χ2 test. Comparisons between groups were performed using independent t-tests. A p-value <0.05 was considered significant. All statistical analyses were performed in SPSS for Windows version 18.0 (SPSS Inc., Chicago, IL, USA). Results are expressed as the mean ± standard deviation (SD).
Results
miR-193b Induces Apoptosis and Autophagy in OS Cells
To determine whether miR-193b is dysregulated in OS specimens, we RT-qPCR to analyze the expression of miR-193b in 20 pairs of tumor and matched adjacent normal specimens from OS patients. miR-193b expression was lower in the tumor specimens than in adjacent normal tissues (Figure 1A). To examine the role of miR-193b in OS cell apoptosis, OS cell lines were transfected either with an miR-193b mimic or with an inhibitor. miRNA RT-PCR was utilized to assess miR-193b expression following transfection (Figure 1B), while flow cytometry was used to examine the effects of miR-193b on apoptosis. A higher rate of apoptosis was detected in OS cells treated with the miR-193b mimic than in control cells, whereas OS cells treated with the miR-193b inhibitor showed the opposite trend (Figure 1C). The cleaved caspase 3 level was also increased following transfection with an miR-193b mimic (Figure 1D). In addition, Western blotting showed that the miR-193b mimic increased Beclin 1 expression and the LC3-II/I ratio and decreased the expression of p62; however, these miR-193b mimic-induced effects were blocked by treatment with the autophagy inhibitor 3-MA. In contrast, OS cells treated with the miR-193b inhibitor exhibited the opposite trend (Figure 1E). These results suggest that miR-193b may induce apoptosis via an autophagy-dependent signaling pathway.
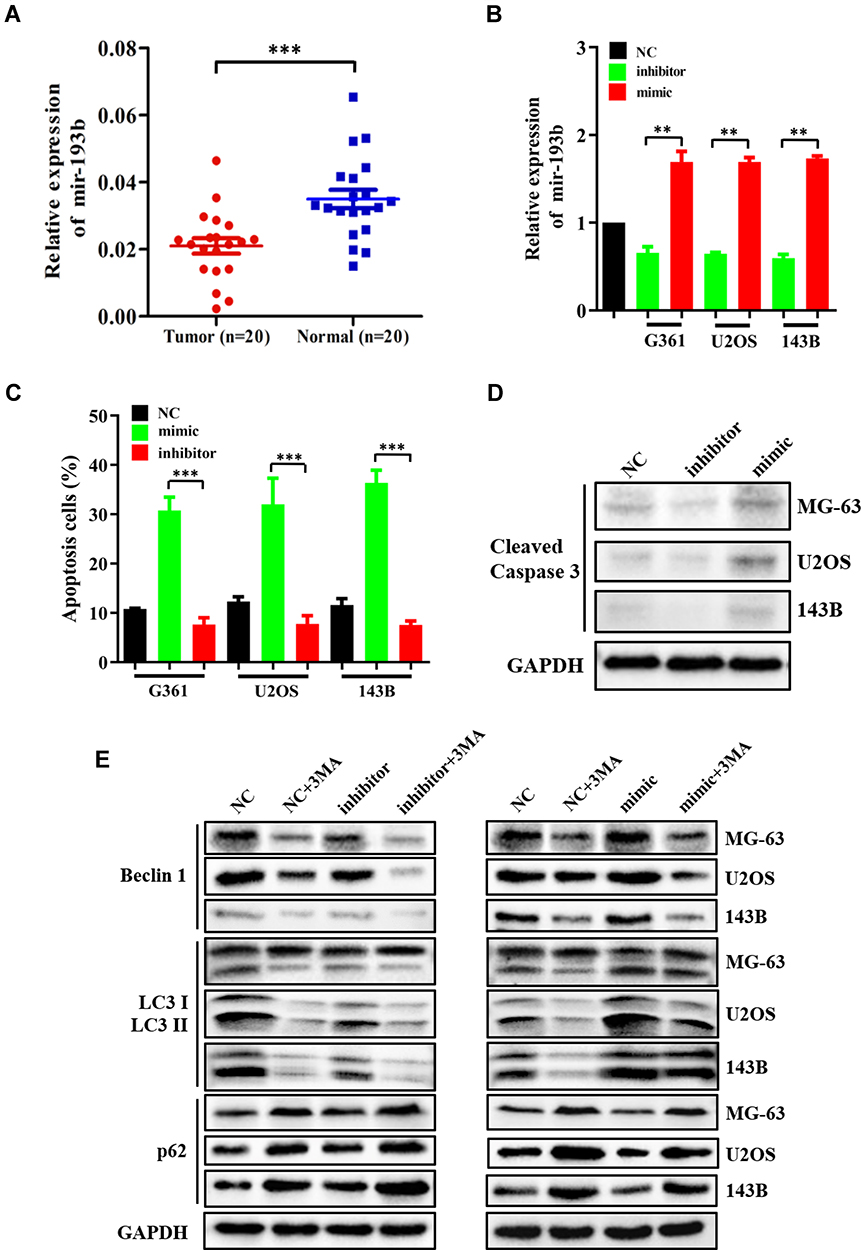 |
Figure 1 miR-193b is downregulated in OS tissues, which induces apoptosis and autophagy in OS cells. (A, B) miR-193b expression in 20 pairs of OS and paired adjacent normal specimens using miRNA RT-PCR. (C, D) The effects of altered miR-193b expression on apoptosis. (E) Western blot of the levels of Beclin 1, p62, and LC3-I/II protein expression following the transfection of OS cells with either a mimic or an inhibitor of miR-193b. 3-MA blocked miR-193b mimic-induced effects but reinforced the expression patterns of proteins associated with miR-193b inhibitor-induced autophagy. **p < 0.01, ***p < 0.001 vs control. |
FEN1 Is a Direct Target of miR-193b
TargetScan prediction indicated that FEN1 was a potential target for miR-193b. In the dual-luciferase reporter assays using pGL3 firefly luciferase reporter plasmids, both WT and MUT 3ʹ-UTR sequences of FEN1 were structured based on potential binding sites. Compared to that in the control, miR-193b overexpression greatly decreased luciferase activity when FEN1 WT was co-transfected into 293T cells. Nevertheless, no marked inhibitory effect of miR-193b on luciferase activity was observed following co-transfection with FEN1 MUT. Furthermore, miR-193b overexpression decreased FEN1 protein expression in OS cells, whereas the miR-193b inhibitor elicited the opposite effect (Figure 2A). These data suggest that FEN1 is a direct target of miR-193b in OS cells.
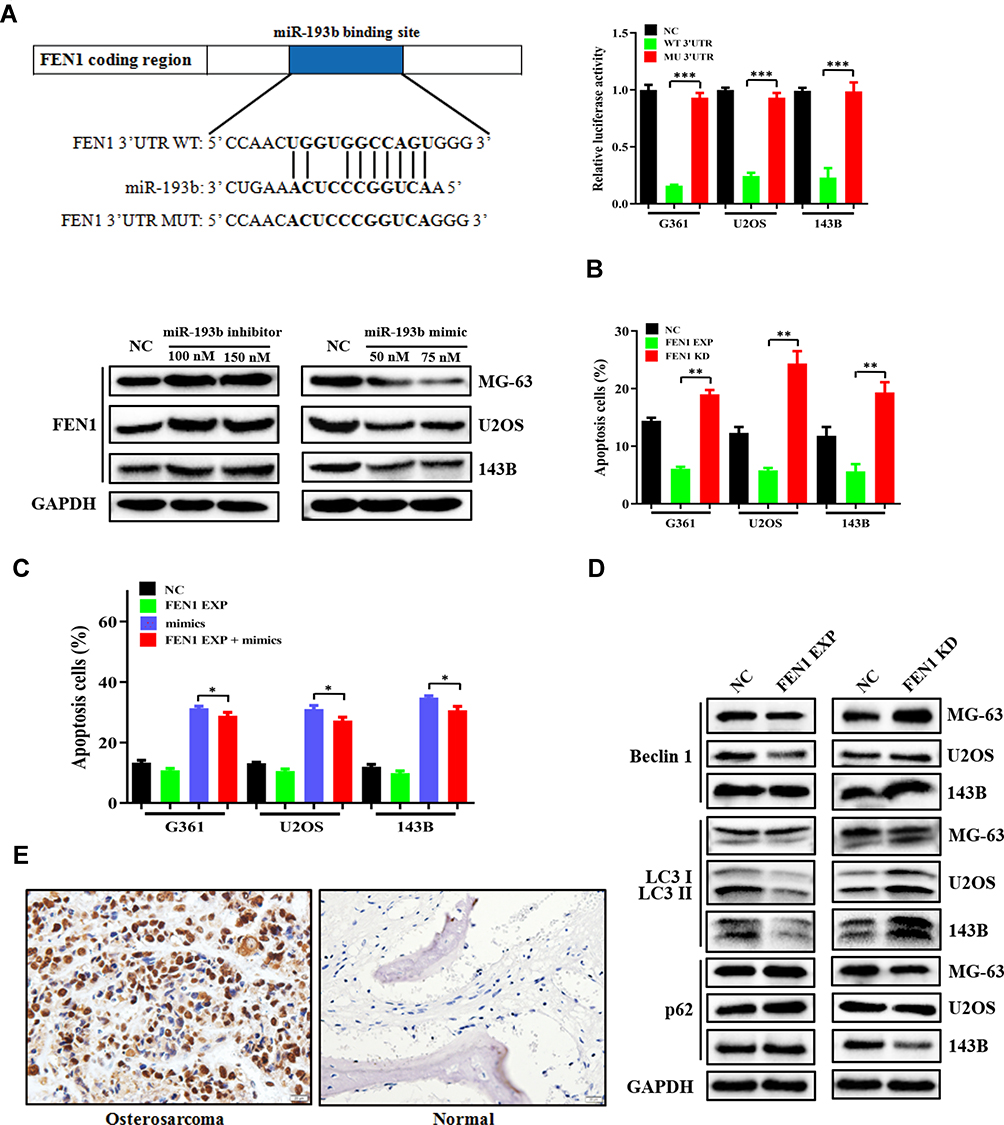 |
Figure 2 FEN1 is a direct target of miR-193b and exhibits upregulated expression in OS tissues. (A) The miR-193b binding site in the 3ʹ-UTR of FEN1 was identified by TargetScan and matched mutations. A luciferase reporter assay was conducted in OS cells to verify the interaction between miR-193b and the FEN1 binding site. The level of FEN1 protein expression in OS cells transfected with either a mimic or an inhibitor of miR-193b. (B) The roles of altered FEN1 expression on apoptosis. (C) FEN1 over expression decreased miR-193b induced apoptosis. (D) The expression levels of Beclin 1, p62, and LC3-I/II under altered FEN1 expression. (E) Immunohistochemical staining of FEN1 in OS and paired adjacent normal specimens. *p < 0.05, **p < 0.01, ***p < 0.001 vs control. |
To further determine the effect of FEN1 on apoptosis and autophagy, si-FEN1 and FEN1 expression vectors were transfected into OS cells. si-FEN1 induced a higher rate of apoptosis compared to control cells, whereas OS cells transfected with the FEN1 expression vectors showed the opposite trend (Figure 2B). In FEN1 expression vector transfected OS cells, FEN1 over-expression decreased miR-193b mimics induced apoptosis compared with control group, indicated that miR-193b induces OS cells apoptosis depend at least partly on FEN1 (Figure 2C). Furthermore, si-FEN1 transfection decreased autophagy levels relative to the control group (Figure 2D). Immunohistochemistry further revealed that FEN1 expression was considerably higher in the tumor specimens than in those of adjacent normal tissue (Figure 2E, Table 1). Together, these data indicate that miR-193b induces apoptosis and autophagy by downregulating FEN1 expression.
 |
Table 1 Expression of FEN1 in Human OS and Paired Adjacent Normal Specimens |
miR-193b Enhances the Chemosensitivity of OS Cells
A complicated interplay exists between autophagy and apoptosis that is highly dependent on the cellular environment. To further delineate the relationship between autophagy and apoptosis, we first examined the effects of epirubicin on miR-193b-induced autophagy. Western blotting analysis showed that 3-MA inhibited miR-193b mimic-induced autophagy but reinforced the opposite effect elicited by the miR-193b inhibitor. More importantly, miR-193b increased epirubicin-induced autophagy (Figure 3A). Next, a viability assay was performed with OS cells pretreated with 10 mM 3-MA, miR-193b mimic, or vehicle alone before the addition of epirubicin. Flow cytometric analysis showed that miR-193b overexpression significantly increased epirubicin-induced apoptosis, while 3-MA-mediated inhibition of autophagy moderately abrogated miR-193b- and epirubicin-induced apoptosis (Figure 3B). These findings suggest that miR-193b mediates autophagy and enhances epirubicin-induced apoptosis in OS cells.
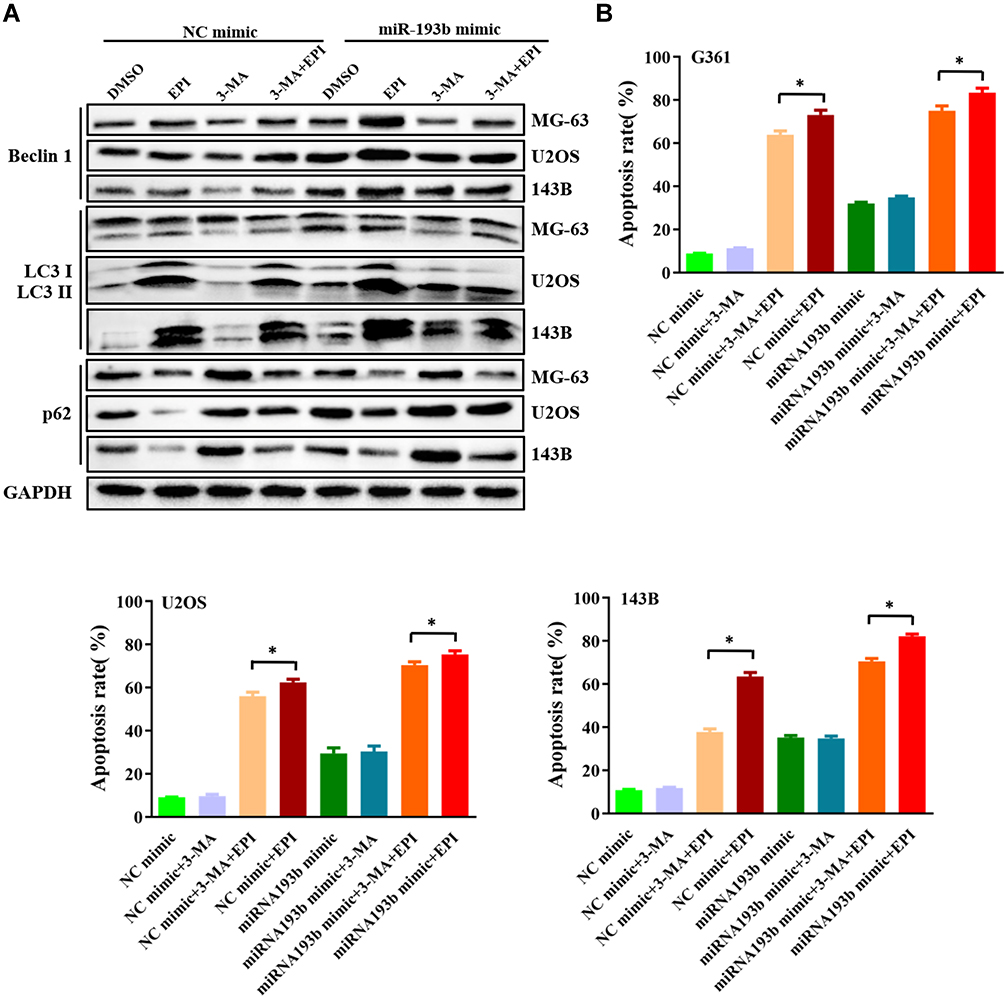 |
Figure 3 miR-193b enhances the chemosensitivity of OS cells in an autophagy-dependent manner. (A) An miR-193b mimic enhanced epirubicin-induced autophagy. (B) An miR-193b mimic promoted epirubicin-induced cell death in OS cells, which was largely abolished by addition of 3-MA. *p < 0.05 vs control. |
Discussion
Emerging evidence suggests that miRNAs can function as tumor suppressors or oncogenes depending on their target mRNAs, thereby markedly affecting the biology of cancer.19–21 Several miRNAs have been identified as being associated with different cancers, including OS,22,23 and there is evidence that miR-193b acts as a tumor suppressor in various types of cancer.24–30 In the present study, we demonstrated that miR-193b expression is lower in OS specimens than in adjacent normal tissue. To better characterize the role of miR-193b, we then conducted functional studies, and showed that miR-193b overexpression induces apoptosis in OS cells, while miR-193b knockdown yields the opposite effect in vitro. However, the mechanism by which miR-193b induces apoptosis in OS cells remains poorly known, although miR-193b upregulation has been reported to result in activation of autophagy and promotion of cell death.31
Autophagy is a self-degradative process that involves autophagosome formation and maintains cellular homeostasis.32,33 Autophagy may function either as a cell survival mechanism under specific conditions, or as a second type of programmed cell death.34,35 Several reports have highlighted the participation of autophagy regulation in different cancers, including OS.36,37 In the present study, we showed that miR-193b significantly increases the expression of autophagy-related proteins in OS cells and promotes cell sensitivity to epirubicin treatment, indicating that miR-193b-induced OS cell apoptosis may be autophagy-dependent.
In determining the mechanisms underlying the effect of miR-193b on the induction of apoptosis in OS cells, we revealed that miR-193b targets the 3ʹ-UTR of FEN1 directly and negatively regulates FEN1 expression in OS cells. As a multifunctional nuclease, FEN1 plays a critical role in DNA repair, and is frequently overexpressed in various human cancers; consequently, FEN1 overexpression is considered to be a potential therapeutic target and biomarker to monitor cancer progression.38,39 Consistent with the observation in the OS cells, we demonstrated that FEN1 expression is upregulated in OS tissues. FEN1 knockdown has been shown to significantly sensitize cancer cells to DNA damaging agents such as taxol, mitomycin C, and cisplatin.40,41 Epirubicin, commonly used as a treatment for OS, acts by intercalating into DNA strands, resulting in DNA cleavage by topoisomerase II.42 More importantly, FEN1 was shown to participate in the repairment of DNA damage agent induced Top2-SSB complexes then maintain DNA stability,43 and disruption of FEN1 function leads to the accumulation of DNA double-strand breaks.8 Autophagy is reportedly activated in response to DNA damage, which may mediate survival at low levels of DNA damage; however, excessive lesions that cannot be repaired may lead to persistent, unrestrained autophagy, that in turn induces a form of cell death known as “autophagic cell death” (ACD).44–46 Consequently, we suspect that FEN1 knockdown can sensitize OS cells to epirubicin through the disruption of DNA damage repair processes.
In summary, our results indicated that miR-193b/FEN1 is a potential novel therapeutic target for OS aimed mainly at the induction of apoptosis. Moreover, the results also showed that the miR-193b/FEN1 axis increases the chemosensitivity of OS cells. These findings reveal a novel pathway and suggest a potential strategy to reduce the chemoresistance of OS cells.
Ethical Statement
The study was approved by the Ethics Committee of the Third Affiliated Hospital of Kunming Medical University, and all patients gave written informed consent and authorization for use of biological specimens, in accordance with the Declaration of Helsinki.
Availability of Data and Materials
We declare that the materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.
Acknowledgments
This study was supported in part by grants from the National Natural Science Foundation of China (#81760495), Applied Basic Research of Yunnan Science & Technology Agency (Joint Funds of Yunnan Science & Technology Agency and Kunming Medical University) (#2017FE468[-160], 2017FE467[-082], 2018FE001[-149], 2018FE001[-057]), and Youth doctoral fund of Yunnan Tumor Hospital (#BSKY-201704).
Author Contributions
Suwei Dong wrote the paper, analyzed the data, and performed the luciferase reporter assay and analyzed data. Yanbin Xiao participated in research design and revised Discussion section for this paper. Xiang Ma performed the tissue test and analyzed and interpreted tissue data and revised Materials and Methods section for this paper. Wei He cultured the cells, prepared the protein and analyzed and interpreted PCR data and revised Materials and Methods section for this paper. Jianping Kang performed the cell apoptosis test and analyzed and interpreted apoptosis data and revised Abstract section for this paper. Zhuohui Peng collected and analyzed and interpreted patient data and revised Results section for this paper. Lei Wang performed Western blotting and analyzed and interpreted data and revised Materials and Methods section for this paper. Zhen Li participated in research design and performed PCR and revised Introduction section for this paper. All authors drafted and revised the article, read and approved the final manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125(1):229–234. doi:10.1002/ijc.v125:1
2. Messerschmitt PJ, Garcia RM, Abdul-Karim FW, Greenfield EM, Getty PJ. Osteosarcoma. J Am Acad Orthop Surg. 2009;17(8):515–527. doi:10.5435/00124635-200908000-00005
3. Briccoli A, Rocca M, Salone M, Guzzardella GA, Balladelli A, Bacci G. High grade osteosarcoma of the extremities metastatic to the lung: long-term results in 323 patients treated combining surgery and chemotherapy, 1985-2005. Surg Oncol. 2010;19(4):193–199. doi:10.1016/j.suronc.2009.05.002
4. Izadpanah S, Shabani P, Aghebati-Maleki A, et al. Insights into the roles of miRNAs; miR-193 as one of small molecular silencer in osteosarcoma therapy. Biomedi Pharmacother. 2019;111:873–881. doi:10.1016/j.biopha.2018.12.106
5. Shekhar R, Priyanka P, Kumar P, Ghosh T, Khan MM, Nagarajan P. The microRNAs miR-449a and miR-424 suppress osteosarcoma by targeting cyclin A2 expression. J Biol Chem. 2019;294:4381–4400. doi:10.1074/jbc.RA118.005778
6. Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi:10.1146/annurev.biochem.73.012803.092453
7. Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–138. doi:10.1146/annurev-biochem-072511-122603
8. Ward TA, McHugh PJ, Durant ST. Small molecule inhibitors uncover synthetic genetic interactions of human flap endonuclease 1 (FEN1) with DNA damage response genes. PLoS One. 2017;12(6):e0179278.
9. Isohookana J, Haapasaari KM, Soini Y, Leppanen J, Karihtala P. Proteins of the retinoblastoma pathway, FEN1 and MGMT are novel potential prognostic biomarkers in pancreatic adenocarcinoma. Pathol Res Pract. 2018;214(6):840–847. doi:10.1016/j.prp.2018.04.016
10. Zhang K, Keymeulen S, Nelson R, et al. Overexpression of flap endonuclease 1 correlates with enhanced proliferation and poor prognosis of non-small-cell lung cancer. Am J Pathol. 2018;188(1):242–251. doi:10.1016/j.ajpath.2017.09.011
11. He L, Luo L, Zhu H, et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol Oncol. 2017;11(9):1302–1303. doi:10.1002/mol2.2017.11.issue-9
12. Ma L, Cao X, Wang H, Lu K. Discovery of myricetin as a potent inhibitor of human flap endonuclease 1, which potentially can be used as sensitizing agent against ht-29 human colon cancer cells. J Agric Food Chem. 2019;67(6):1656–1665.
13. He L, Luo L, Zhu H, et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol Oncol. 2017;11(6):640–654. doi:10.1002/mol2.2017.11.issue-6
14. Limpert AS, Lambert LJ, Bakas NA, et al. Autophagy in cancer: regulation by small molecules. Trends Pharmacol Sci. 2018;39(12):1021–1032. doi:10.1016/j.tips.2018.10.004
15. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. doi:10.1124/mol.114.091850
16. Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1–11. doi:10.1038/onc.2015.99
17. Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clinl Cancer Res. 2011;17(4):654–666. doi:10.1158/1078-0432.CCR-10-2634
18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
19. Jasinski-Bergner S, Kielstein H. Adipokines regulate the expression of tumor-relevant MicroRNAs. Obes Facts. 2019;12(2):211–225. doi:10.1159/000496625
20. Pardini B, Calin GA. MicroRNAs and long non-coding RNAs and their hormone-like activities in cancer. Cancers. 2019;11(3):378. doi:10.3390/cancers11030378
21. Kwok GT, Zhao JT, Weiss J, et al. Translational applications of microRNAs in cancer, and therapeutic implications. Non-Coding RNA Res. 2017;2(3–4):143–150. doi:10.1016/j.ncrna.2017.12.002
22. Viera GM, Salomao KB, de Sousa GR, Baroni M, Delsin LEA, Pezuk JA. miRNA signatures in childhood sarcomas and their clinical implications. Clin Transl Oncol. 2019;21:1583–1623.
23. Sampson VB, Yoo S, Kumar A, Vetter NS, Kolb EA. MicroRNAs and potential targets in osteosarcoma: review. Front Pediatr. 2015;3:69. doi:10.3389/fped.2015.00069
24. Khordadmehr M, Shahbazi R. miR-193: a new weapon against cancer. J Cell Physiol. 2019;234:16861–16872. doi:10.1002/jcp.28368
25. Hashemi ZS, Forouzandeh Moghadam M, Khalili S, Ghavami M, Salimi F, Sadroddiny E. Additive effect of metastamiR-193b and breast cancer metastasis suppressor 1 as an anti-metastatic strategy. Breast Cancer. 2019;26(2):215–228. doi:10.1007/s12282-018-0915-z
26. Song B, Du J, Song DF, Ren JC, Feng Y. Dysregulation of NCAPG, KNL1, miR-148a-3p, miR-193b-3p, and miR-1179 may contribute to the progression of gastric cancer. Biol Res. 2018;51(1):44. doi:10.1186/s40659-018-0192-5
27. Mazzu YZ, Hu Y, Shen Y, Tuschl T, Singer S. miR-193b regulates tumorigenesis in liposarcoma cells via PDGFR, TGFbeta, and Wnt signaling. Sci Rep. 2019;9(1):3197. doi:10.1038/s41598-019-39560-0
28. Kang M, Li Y, Zhu S, Zhang S, Guo S, Li P. MicroRNA-193b acts as a tumor suppressor gene in human esophageal squamous cell carcinoma via target regulation of KRAS. Oncol Lett. 2019;17(4):3965–3973. doi:10.3892/ol.2019.10039
29. Karmakar S, Kaushik G, Nimmakayala R, Rachagani S, Ponnusamy MP, Batra SK. MicroRNA regulation of K-Ras in pancreatic cancer and opportunities for therapeutic intervention. Semin Cancer Biol. 2019;54:63–71. doi:10.1016/j.semcancer.2017.11.020
30. Zhang J, Qin J, Su Y. miR-193b-3p possesses anti-tumor activity in ovarian carcinoma cells by targeting p21-activated kinase 3. Biomedi Pharmacother. 2017;96:1275–1282. doi:10.1016/j.biopha.2017.11.086
31. Nyhan MJ, O’Donovan TR, Boersma AW, Wiemer EA, McKenna SL. MiR-193b promotes autophagy and non-apoptotic cell death in oesophageal cancer cells. BMC Cancer. 2016;16:101. doi:10.1186/s12885-016-2123-6
32. Rein T. Is autophagy involved in the diverse effects of antidepressants? Cells. 2019;8(1):44. doi:10.3390/cells8010044
33. Eskelinen EL. Autophagy: supporting cellular and organismal homeostasis by self-eating. Int J Biochem Cell Biol. 2019;111:1–10. doi:10.1016/j.biocel.2019.03.010
34. Buccarelli M, Marconi M, Pacioni S, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018;9(8):841.
35. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26(4):605–616. doi:10.1038/s41418-018-0252-y
36. Deng S, Shanmugam MK, Kumar AP, Yap CT, Sethi G. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer. 2019;125(8):1228–1246. doi:10.1002/cncr.31978
37. O’Farrill JS, Gordon N. Autophagy in osteosarcoma. Adv Exp Med Biol. 2014;804:147–160.
38. Singh P, Yang M, Dai H, et al. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol Cancer Res. 2008;6(11):1710–1717. doi:10.1158/1541-7786.MCR-08-0269
39. Abdel-Fatah TM, Russell R, Albarakati N, et al. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol Oncol. 2014;8(7):1326–1338. doi:10.1016/j.molonc.2014.04.009
40. Wang Y, Li S, Zhu L, et al. Letrozole improves the sensitivity of breast cancer cells overexpressing aromatase to cisplatin via down-regulation of FEN1. Clin Transl Oncol. 2019;21:1026–1033. doi:10.1007/s12094-018-02019-1
41. Wang J, Zhou L, Li Z, et al. YY1 suppresses FEN1 over-expression and drug resistance in breast cancer. BMC Cancer. 2015;15:50. doi:10.1186/s12885-015-1043-1
42. Tarpgaard LS, Qvortrup C, Nygård SB, et al. A phase II study of Epirubicin in oxaliplatin-resistant patients with metastatic colorectal cancer and TOP2A gene amplification. BMC Cancer. 2016;16:91. doi:10.1186/s12885-016-2124-5
43. Kametani Y, Takahata C, Narita T, Tanaka K, Iwai S, Kuraoka I. FEN1 participates in repair of the 5ʹ-phosphotyrosyl terminus of DNA single-strand breaks. Carcinogenesis. 2016;37(1):56–62. doi:10.1093/carcin/bgv159
44. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: a meaningful partnership. Front Genet. 2016;7:204. doi:10.3389/fgene.2016.00204
45. Gomes LR, Menck CFM, Leandro GS. Autophagy roles in the modulation of DNA repair pathways. Int J Mol Sci. 2017;18(11):2351. doi:10.3390/ijms18112351
46. Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. Autophagy in DNA damage response. Int J Mol Sci. 2015;16(2):2641–2662. doi:10.3390/ijms16022641
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.