")
Back to Journals » OncoTargets and Therapy » Volume 11
miR-185 suppresses progression of Ewing’s sarcoma via inhibiting the PI3K/AKT and Wnt/β-catenin pathways
Authors Zhang S, Li D, Jiao G, Wang H, Yan T
Received 9 March 2018
Accepted for publication 19 August 2018
Published 9 November 2018 Volume 2018:11 Pages 7967—7977
DOI https://doi.org/10.2147/OTT.S167771
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Jianmin Xu
Shuai Zhang,1 Dong Li,2 Guang-jun Jiao,1 Hong-liang Wang,1 Ting-bin Yan1
1Department of Orthopedics, Qilu Hospital of Shandong University, Jinan, Shandong Province, China; 2Department of Orthopedics, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong Province, China
Background: miRNAs are confirmed to play essential roles in tumorigenesis and progression of cancers, including Ewing’s sarcoma. miR-185 has been reported to be downregulated in some tumors, whereas the role of miR-185 in Ewing’s sarcoma remains unclear.
Purpose: The objective of this study was to investigate the role of miR-185 in the progression and metastasis of Ewing’s sarcoma and explore the associated mechanism.
Materials and methods: Ewing’s sarcoma cell line RD-ES was transfected with pCMV-MIR-miR185 vector to upregulate the expression of miR-185. Cell Counting Kit 8 and colony formation assays were used to assess the effect of miR-185 on cell proliferation. The effect of miR-185 on cell migration and invasion was detected by transwell assay. Flow cytometry assay was performed to detect apoptosis rate of RD-ES cells. The protein levels of apoptosis-related proteins was determined using Western blot assay or immunohistochemistry assay. Dual-luciferase reporter assay was used to validate the regulation between miR-185 and its target gene.
Results: Upregulation of miR-185 caused significant inhibition on cell growth capacity, migration and invasion of Ewing’s sarcoma cell RD-ES. Besides, upregulation of miR-185 was observed to accelerate cell apoptosis in a mitochondrial pathway through regulating Bcl-2/Bax, Caspase 3, and Caspase 9 in Ewing’s sarcoma in vitro. Moreover, upregulation of miR-185 was found to suppress the PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells. Furthermore, we identified that E2F6 was a target gene for miR-185, and the suppression on cell proliferation caused by overexpression of miR-185 was significantly rescued by the upregulation of E2F6 in RD-ES cells.
Conclusion: miR-185 is involved in cell growth, motility and survival of Ewing’s sarcoma as a tumor suppressor via suppressing PI3K/Akt/mTOR and Wnt/β-catenin pathways and targeting E2F6.
Keywords: miR-185, PI3K/Akt/mTOR pathway, Wnt/β-catenin pathway, E2F6, Ewing’s sarcoma
Introduction
Ewing’s sarcoma is a common malignant tumor, highly occurring in children and adolescents as the second common bone tumor after osteosarcoma.1,2 Ewing’s sarcoma is characterized by rapid progression, high postoperative recurrence, and high metastatic.3 It is reported that the 5-year survival rate of patients with localized disease is 60%–70%, whereas the 5-year survival rate of patients with metastatic Ewing’s sarcoma is only 20%–45% depending on the location of metastasis.4 And patients with a metastatic, recurrent form of Ewing’s sarcoma still have poor prognosis,5 leading to a particularly urgent to identify new biomarkers or therapeutic targets and to establish innovative treatment strategies.
MicroRNAs (miRNAs) are a class of evolutionarily conserved, small noncoding RNAs, which modulate target genes expression by binding to 3′UTR of mRNA to suppress mRNA translation or trigger mRNA degradation. It is demonstrated that the aberrant miRNAs expression profiles are involved in tumor initiation, progression and metastasis, including Ewing’s sarcoma.6–10 Recent studies have proved that various miRNAs have the potential to be a biomarker or target for tumor progression, prognosis, and therapy.11–14 These findings suggest that miRNAs provide a novel area for the research of progression, metastasis and therapy of Ewing’s sarcoma. Recent studies demonstrate that miR-185 is downregulated in some tumors and impairs tumor growth and metastasis.15–19 Li S et al reveals that miR-185 is downregulated in non-small-cell lung cancer, and overexpression of miR-185 could lead to significant inhibition on tumor growth, migration, and invasion.15 In nasopharyngeal carcinoma, upregulation of miR-185-3 p could reduce the cell metastasis, while inhibition of miR-185-3 p correspondingly accelerates cell metastasis.16 In addition, it is demonstrated that miR-185 is associated with the prognosis of patients with colorectal cancer, high expression of miR-185 is correlated with poor survival and metastasis.21 However, the accurate role of miR-185 in the progression and metastasis of Ewing’s sarcoma remains unclear, and the exact functional mechanism is also largely unknown.
In this study, Ewing’s sarcoma RD-ES cells were transfected with the pCMV-MIR-miR185 vector to establish stable endogenous overexpression of miR-185 in vitro. We confirmed that upregulation of miR-185 modulated growth, motility, and survival of RD-ES cells by suppressing PI3K/Akt/mTOR and Wnt/β-catenin pathways. E2F6 was identified as a target gene of miR-185. Upregulation of E2F6 could rescue the suppression on cell proliferation caused by miR-185 in RD-ES cells. To our knowledge, this study is the first attempt to demonstrate the role of miR-185 in the progression of Ewing’s sarcoma in vitro.
Materials and methods
Cells culture and transfection
Ewing’s sarcoma cells RD-ES, A673, SK-ES-1, and SCCH were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and cultured in RPMI-1640 (Hyclone, Thermo Fisher Scientific, Waltham, MA, USA) medium containing 10% FBS (Gibco, Thermo Fisher Scientific) at 37°C with 5% CO2. RD-ES cells were transiently transfected with the pCMV-MIR-miR185 vector (Ribobio, Guangzhou, China) or negative control vector (pCMV-MIR) by using Lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s protocol. After 24 hours of transfection, the cell function experiments were carried out.
Real-time polymerase chain reaction (qRT-PCR)
After 48 hours of transfection, total RNA was isolated by Ultrapure RNA Kit (CWBIO, Beijing, China) and reverse transcribed to cDNA by using the miRNA cDNA Synthesis Kit (CWBIO). qRT-PCR was carried out by miRNA qPCR Assay Kit (CWBIO). The primer for mature miR-185 and U6 RNA was obtained from Ribobio, and the primer sequences were as follows: miR-185, 5′-TGGAGAGAAAGGCAGTTCCTGA-3′ (forward) and the reverse primer was obtained from the miRNA qPCR Assay Kit; U6, 5′-CTCGCTTCGGCAGCACA-3′ (forward) and AACGCTTCACGAATTTGCGT (reverse). The comparative Ct (ΔΔct) method was adopted to calculate the obtained qRT-PCR data.
Proliferation and viability assay
For CCK8 assay, transfected cells were seeded in each well of a 96-well plate at a density of 1×103 cells/well. Cell proliferation and vitality was detected by adding 10 μL of CCK8 reagent (Beijing Solarbio Science & Technology, Beijing, China) and incubated at 37°C for 90 minutes, OD value of excitation light was detected every 24 hours by using enzyme standard instrument with 450 nm.
For colony formation assay, transfected cells at logarithmic growth phase were seeded in 6 cm plates at a density of 500 cells/well to culture at 37°C with 5% CO2 until cells had formed sufficiently large colonies. Then, the cells were fixed with 1 mL of 4% paraformaldehyde for 30 minutes followed by staining with crystal violet for 30 minutes. Finally, the colonies were counted and photographed.
Cell migration and invasion assays
Transwell chamber coated with Matrigel was used for cell invasion assay, while Matrigel was not needed for migration assay. After transfection for 24 hours, cells were trypsinized and resuspended in serum-free culture medium, and 1×104 cells were transferred into the upper chamber. Complete medium with 10% FBS was added in the lower chamber. After incubation for 24 hours, 4% paraformaldehyde was utilized to fix cells that had invaded or migrated to the lower surface of the filter, then 0.1% crystal violet was performed to stain cells for 5 minutes. The invaded and migrated cells were imaged and counted under the microscope.
Apoptosis assay
Flow cytometry was performed to determine cell apoptosis using the Annexin V-FITC-PI apoptosis detection kit (4A Biotech, China), according to the manufacturer’s instructions. Briefly, cells transfected with the pCMV-MIR-miR185 vector or pCMV-MIR were cultured with serum-free medium for 24 hours. Next, transfected cells were resuspended in 1× binding buffer (10 mM HEPES/NaOH [pH 7.4], 140 mM NaCl, 2.5 mM CaCl2) at a density of 1–5×106 cells/mL. Then 100 μL of cell suspension was incubated with 5 μL of Annexin V-fluorescein isothiocyanate (FITC) in the dark for 5 minutes. After staining with 10 μL of propidium iodide (PI) at room temperature, cells were analyzed using a FACS caliber instrument.
Western blot assay
After 48 hours of transfection, cells were harvested and lysed with ice-cold RIPA Lysis Buffer (CWBIO) at 4°C to extract proteins, and the protein concentration was measured using a BCA Protein Assay Kit (Beyotime, Beijing, China). Thereafter, 20 μg of protein of each sample was loaded to each lane on 10% SDS-PAGE gel, and transferred onto polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA) incubated with primary antibodies in blocking solution at 4°C overnight. Then, the membrane was incubated with the secondary antibody for 1 hour. An enhanced chemiluminescence kit (CWBIO) was used for signal development according to the manufacturer’s protocol. The following primary antibodies were used: anti-Bcl-2 (1:1,000), anti-Bax (1:1,000), anti-Caspase 9 (1:1,000), anti-p-Akt (1:1,000), anti-Akt (1:1,000), anti-mTOR (1:1,000), anti-p-mTOR (1:1,000), anti-P70 (1:1,000), anti-Cyclin D1 (1:1,000), anti-wnt3 (1:1,000), anti-β-catenin (1:1,000), anti-E-cad (1:1,000), and anti-GAPDH (1:1,000), which were obtained from Abcam (Cambridge, UK). Anti-Active Caspase 3 p17-specific (1:1,000) and secondary antibodies (1:5,000) were obtained from PTG Company (Proteintech Group Inc., Rosemont, IL, USA). The bands were analyzed using the ImageJ software. The relative expression of proteins in miR-185 overexpressed cells was normalized to that in the negative control (NC) group.
Immunohistochemistry assay
After 48 hours of transfection, cells were grown on a cover slip and fixed with 4% paraformaldehyde for 15 minutes. After incubation with primary antibodies for 1 hour at room temperature, cells on the cover slip were stained using an Enhanced DAB Colorimetric Kit (Maixin, Fouzhou, China). Then the cells were counterstained with hematoxylin for 2 minutes, rinsed with water, and returned to blue with tris buffered saline (TBS), and observed under the microscope.
Dual-luciferase reporter assay
The wide-type (wt) or mutated (mut) E2F6 3′UTR was constructed into pmirGLO vector. And the pmirGLO-E2F6 3′UTR (wt) or pmirGLO-E2F6 3′UTR (mut) vectors were co-transfected with pCMV-MIR-miR185 or NC (pCMV-MIR) into RD-ES cells using Lipofectamine 2000. After transfection for 48 hours, the luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Statistical analysis
The SPSS 18.0 software was used for statistical analysis. The data were represented from triplicate experiments and expressed as mean ± standard deviation. The comparisons between two groups were analyzed using Student’s t-test. Differences were considered statistically significant for values of P<0.05.
Results
Overexpression of miR-185 suppresses the growth capacity of RD-ES cells
We observed that the miR-185 had the lowest expression level in RD-ES cells (Figure 1A). To investigate the role of miR-185 in Ewing’s sarcoma, pCMV-MIR-miR-185 vector was used to transfect RD-ES cells as miR-185 group, and pCMV-MIR empty vector was transfected as the NC. As shown in Figure 1B, the expression of miR-185 was clearly upregulated in miR-185 group compared to the NC (P<0.01). The effect of overexpression of miR-185 on growth capacity of RD-ES cells was determined by CCK8 and colony formation assays. CCK8 data showed that after cultured for 48 hours, overexpression of miR-185 significantly attenuated the proliferation ability of RD-ES cell compared with the NC group (P<0.01, Figure 1C). And the suppression effect was still significant after 72 hours of incubation (P<0.01). Furthermore, overexpression of miR-185 also reduced the colony formation ability of RD-ES cells compared with the NC group (P<0.01, Figure 1D).
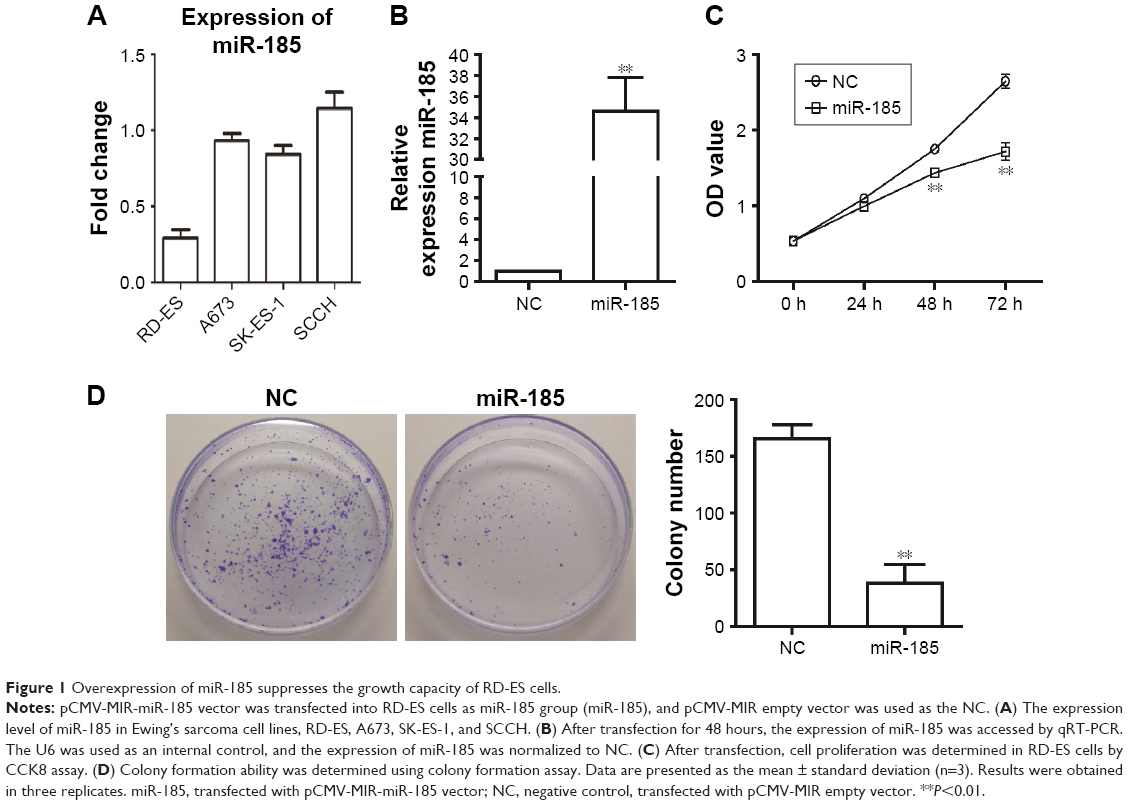 | Figure 1 Overexpression of miR-185 suppresses the growth capacity of RD-ES cells. |
Overexpression of miR-185 inhibits migration and invasion of RD-ES cells
To determine the effect of miR-185 on metastasis of Ewing’s sarcoma, Transwell chamber was performed. As shown in Figure 2A, the migratory ability was significantly decreased in miR-185 upregulation cells compared with the NC group (P<0.01). In addition, the increased expression of miR-185 also resulted in a significant decrease in invasion capability of RD-ES cells (P<0.01, Figure 2B).
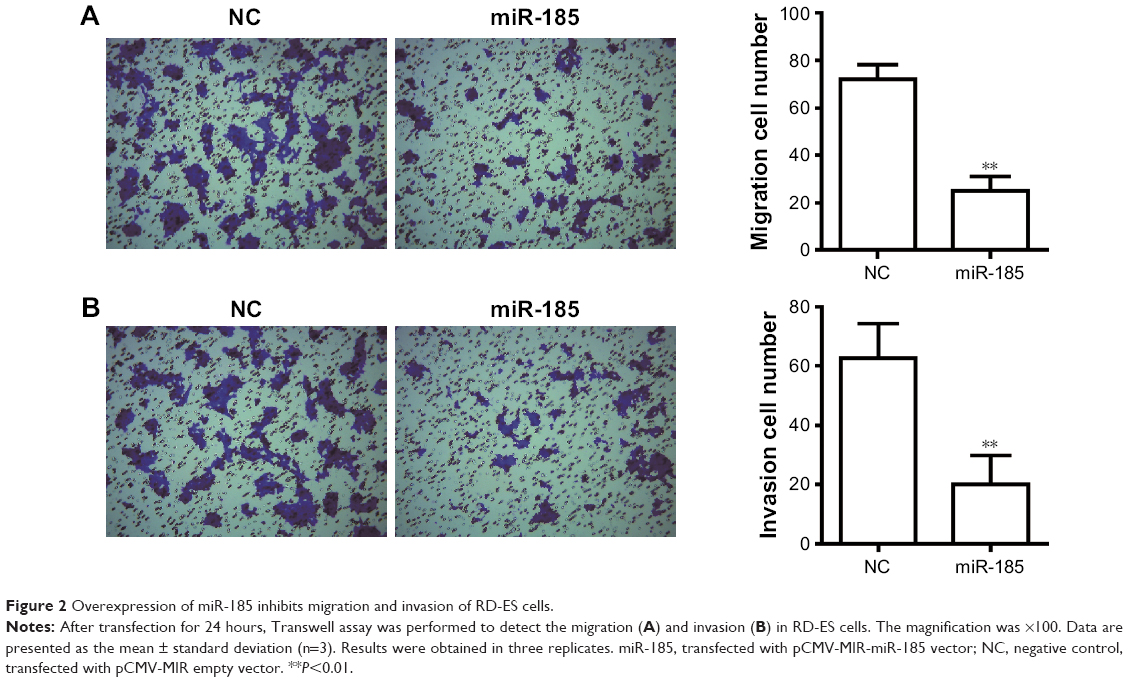 | Figure 2 Overexpression of miR-185 inhibits migration and invasion of RD-ES cells. |
Increased expression of miR-185 accelerates the apoptosis of RD-ES cells
Dysregulated apoptosis is the main hallmark of tumor cells, we determined whether miR-185 could impair apoptosis of RD-ES cells by flow cytometry assay. An obvious increase in the rate of cell apoptosis was observed in miR-185 upregulated cells compared with those in the NC group (Figure 3A, P<0.05). To further elucidate the relative mechanism underlying the elevated apoptosis, apoptosis-related proteins, antiapoptotic protein Bcl-2, and proapoptotic protein Bax, Caspase 9, and active Caspase 3 were detected by Western blot. We found that increased expression of miR-185 downregulated the expression of Bcl-2 and upregulated the expression of Bax, leading to a significant decrease of Bcl-2/Bax in the miR-185 upregulated group (P<0.05, Figure 3B). In addition, the expressions of Caspase 9 and active Caspase 3 were markedly increased by overexpression of miR-185 in RD-ES cells (P<0.05, Figure 3B). Therefore, increased expression of miR-185 could induce apoptosis of RD-ES cells by regulating the expression of apoptosis-related proteins.
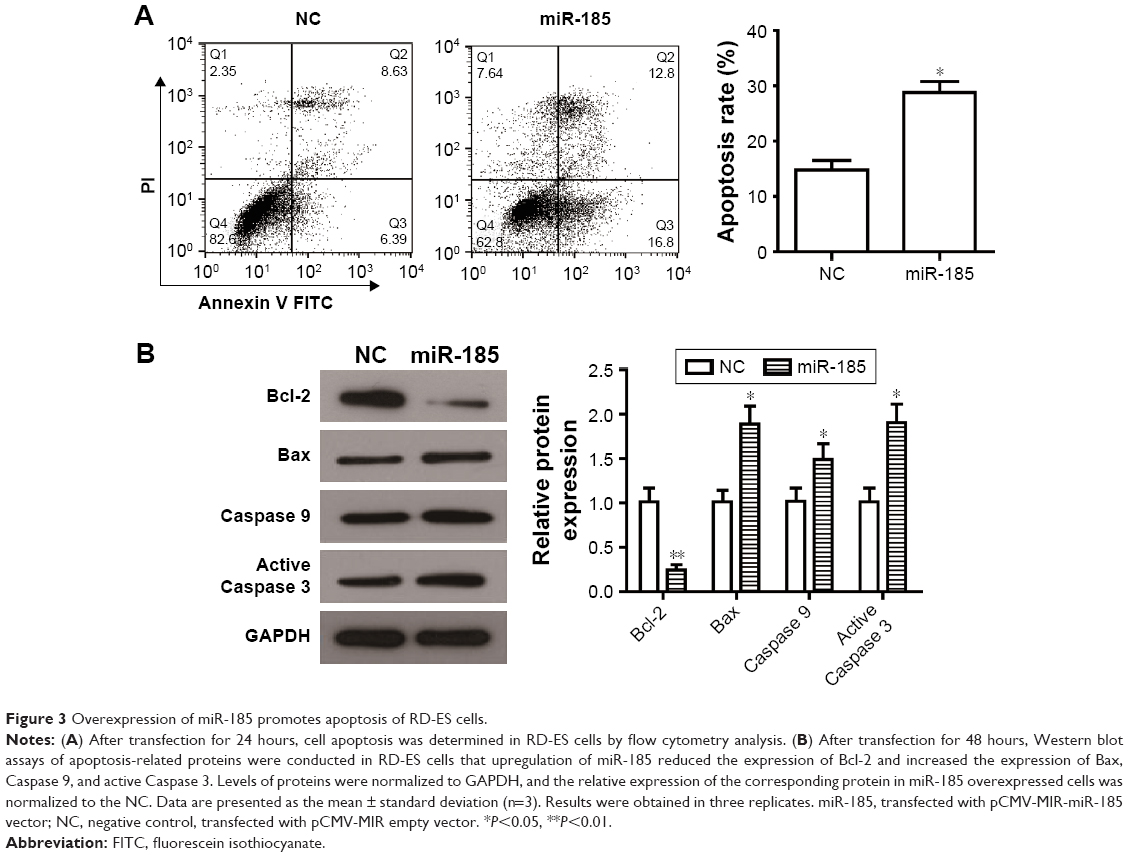 | Figure 3 Overexpression of miR-185 promotes apoptosis of RD-ES cells. |
Considering from the Western blot in Figure 3B, changes in Bcl-2 expression was most pronounced in miR-185 overexpressed cells. We checked whether overexpression of Bcl-2 can rescue the apoptosis or colony formation defect in miR-185 overexpressed cells. However, we found that miR-185 could not directly regulate the expression of Bcl-2 through the dual-luciferase assay (Figure S1A). Moreover, overexpression of Bcl-2 could abolish the proapoptotic effect of miR-185 on RD-ES cells (Figure S1B), suggesting that miR-185 regulated apoptosis of RD-ES cells mainly through the mitochondrial apoptotic pathway. Furthermore, colony formation assay showed that overexpression of Bcl-2 partial rescued the inhibition of miR-185 on the colony formation ability of RD-ES cells (Figure S1C), indicating that the inhibitory effect of miR-185 on proliferation of RD-ES cells was not only dependent on the Bcl-2 pathway. Taken together, these results indicate that besides the Bcl-2 pathway, there are other pathways involved in the antitumor function of miR-185.
Upregulation of miR-185 inhibits PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells
As well known, PI3K/Akt/mTOR pathway plays a crucial role in cell growth and survival by regulating cell cycle regulators and apoptosis-related proteins, and participates in tumor progression and metastasis. It has been reported that PI3K/Akt/mTOR pathway is constitutively activated in Ewing’s sarcoma and mediates survival signaling in Ewing’s sarcoma cells.22–24 In the present study, the expression levels of p-Akt and p-mTOR were decreased by overexpression of miR-185 in RD-ES cells (P<0.05, Figure 4A), however, the total of Akt or mTOR had no changes (P>0.05). Accordingly, the downstream proteins of the PI3K/Akt/mTOR pathway, Cyclin D1 and P70, were also decreased in the miR-185 upregulated group (P<0.05, Figure 4A).
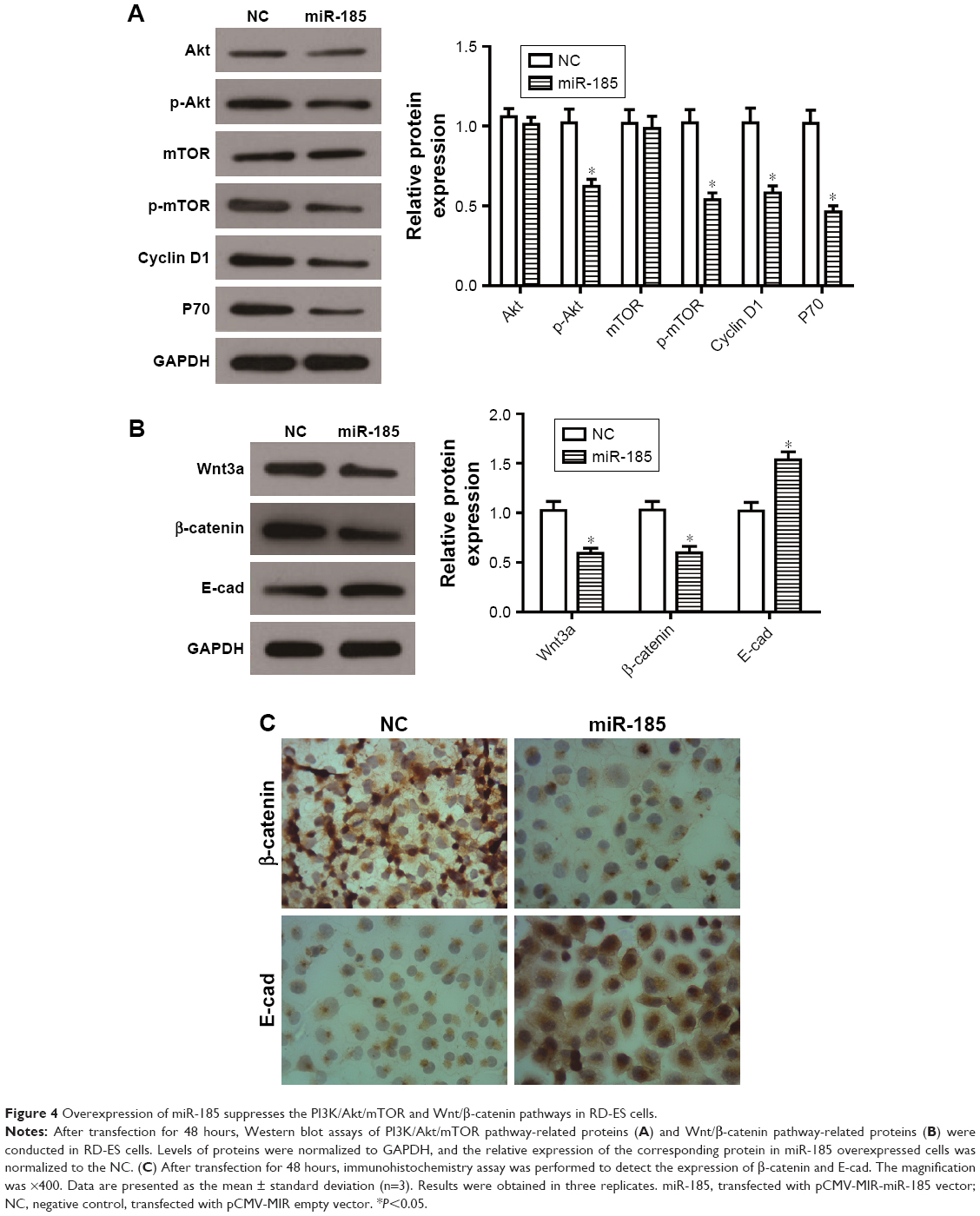 | Figure 4 Overexpression of miR-185 suppresses the PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells. |
Wnt/β-catenin pathway is also essential for several cellular processes, and its dysregulation will be involved in the progression of malignancy. It is reported that Wnt/β-catenin pathway might be activated in a subpopulation of Ewing’s sarcoma cells when some exogenous ligands present.25 Our data showed that the expressions of Wnt3a and β-catenin were both decreased by overexpression of miR-185 in RD-ES cells (P<0.05, Figure 4B), and its downstream protein, E-cad, was accordingly elevated (P<0.05). Furthermore, the immunohistochemistry assay showed that the expression level of nuclear β-catenin in RD-ES cells transfected with miR-185 was significantly decreased compared with the control group, while the expression of E-cad was obviously increased in RD-ES cells transfected with miR-185 (Figure 4C). Taken together, upregulation of miR-185 could suppress the PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells, indicating that miR-185 might act as an upstream mediator in the PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells.
miR-185 directly targets E2F6 in RD-ES cells
Bioinformatics analysis from Targetcan revealed that E2F6 3′UTR has a putative binding site for miR-185 (Figure 5A). To verify it, pmirGLO-E2F6 3′UTR and pmirGLO-E2F6 3′UTR (mut) vectors were constructed to transfect into miR-185 upregulated cells or NC cells. The dual-luciferase assay showed that miR-185 significantly reduced the luciferase activity of pmirGLO-E2F6–3′UTR in RD-ES cells (P<0.05, Figure 5B), while the luciferase activity of pmirGLO-E2F6 3′UTR (mut) did not impact by miR-185, indicating that E2F6 was the target gene of miR-185 in Ewing’s sarcoma cells. This result was further confirmed by the decreased expression of E2F6 in miR-185 overexpressed RD-ES cells (P<0.05, Figure 5C). To investigate whether miR-185 suppressed cell proliferation via regulating E2F6, E2F6 overexpressing vector (lacking its 3′UTR) was constructed and co-transfected into RD-ES cells with pCMV-MIR-miR185. As shown in Figure 5D, the suppression on cell proliferation via overexpression of miR-185 was significantly restored by the upregulation of E2F6 in RD-ES cells (P<0.05). These results suggest that the tumor suppressor role of miR-185 in Ewing’s sarcoma might be mediated by targeting E2F6. In addition, we observed that E2F6 knockdown (E2F6-KD) significantly reduced the expression of p-Akt and β-catenin (Figure 5E), resulting in an inhibition on Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells, while E2F6 overexpression (E2F6-OE) did no significant effect on expression of p-Akt and β-catenin.
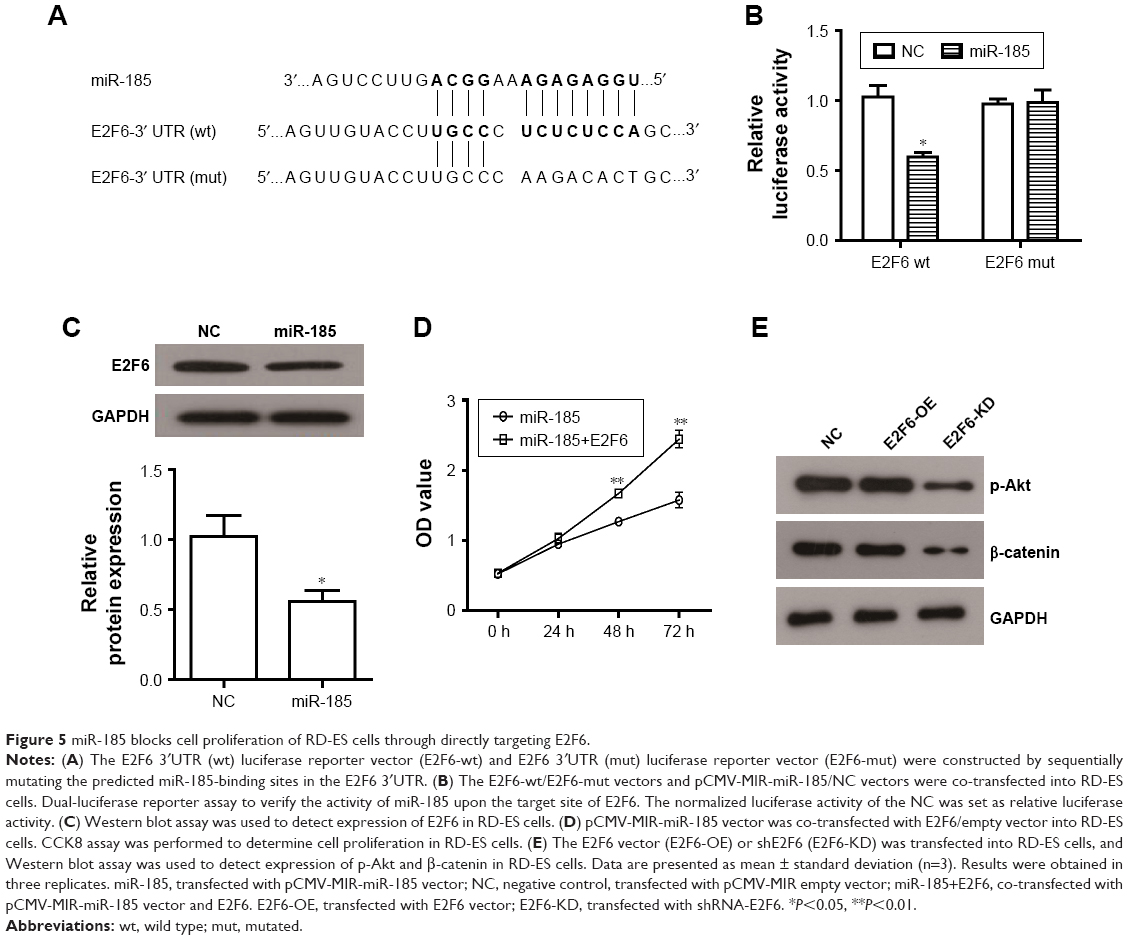 | Figure 5 miR-185 blocks cell proliferation of RD-ES cells through directly targeting E2F6. |
Discussion
It has been confirmed that a large number of miRNAs involved in tumorigenesis and tumor progression by regulating gene expression posttranscriptionally, therefore miRNA is thought to be a potential effective molecular tool that can be used in future therapeutic technologies for cancers. miR-185 is proved to be downregulated in a variety of tumors, and could play crucial role as a tumor suppressor in growth and metastasis of tumors.15–18,26 In the current study, our data validated that overexpression of miR-185 significantly suppressed cell growth capacity, migration, and invasion of Ewing’s sarcoma cell RD-ES in vitro, suggesting that miR-185 plays the role of tumor suppressor in the progression and motility of Ewing’s sarcoma.
Apoptosis is an essential regulatory mechanism to control cell growth, and deregulated apoptosis is observed in tumor cell. We found that increased expression of miR-185 significantly elevated the rate of apoptosis in RD-ES cells. To elucidate the relative mechanism underlying the elevated apoptosis, we detected apoptosis-related proteins in RD-ES cells. It is widely held that intrinsic mitochondrial pathway is one of the major apoptosis mechanisms, which is frequently evaded via regulation of anti-apoptotic or proapoptotic molecules in cancer cells.27,28 The caspase and Bcl-2 protein families play a key role in the process of apoptosis, among these families, Bcl-2, Bax, Caspase 3, and Caspase 9 are critical for initiating mitochondrial-dependent apoptosis pathway.29–31 Caspase 3 is the pivotal enzyme in the mitochondria-dependent apoptosis pathway to trigger the occurrence of apoptosis.29 The Bcl-2 family, containing Bcl-2 and Bax, plays an essential role in apoptosis induction by triggering the mitochondrial pathway.29–31 The ratio of Bcl-2/Bax determines the survival of the cells, and the decrease in the ratio results in loss of mitochondrial membrane potential which trigger initiation of apoptosis.32,33 The main mechanism of Bcl-2 regulation of apoptosis is mainly through Caspase 3 and Caspase 9 pathway.31,34 In the present study, overexpression of miR-185 downregulated the expression of Bcl-2, while upregulated Bax, active Caspase 3 and Caspase 9 in RD-ES cells, indicating that miR-185 triggered cell apoptosis in a mitochondrial pathway through regulating Bcl-2/Bax, Caspase 3, and Caspase 9 in Ewing’s sarcoma in vitro. In addition, the overexpression of Bcl-2 partially rescued the inhibitory effect of miR-185 on the growth of RD-ES cells, suggesting that besides the Bcl-2 pathway, there are other pathways involved in the antitumor function of miR-185. As described above, our data highlight the suppression effect of miR-185 on Ewing’s sarcoma progression and survival of Ewing’s sarcoma in vitro.
It is widely held that PI3K/Akt/mTOR pathway plays pivotal role in cellular progression, including proliferation, apoptosis, and survival. Activated Akt promotes cell growth and survival by inhibiting the expression of Bcl-2 proteins family. It is demonstrated that suppression of PI3K/Akt/mTOR pathway could promote cell apoptosis in Ewing’s sarcoma,24 and the activation is associated with cell resistance to actinomycin D in Ewing’s sarcoma.35 Moreover, PI3K/Akt/mTOR pathway is constitutively activated in Ewing’s sarcoma and mediates survival signaling in Ewing’s sarcoma cells.22–24 Li et al reveal that miR-125b inhibits cell proliferation, metastasis, and survival of Ewing’s sarcoma via suppressing PI3K/Akt/mTOR pathway.36 Another essential pathway involved in cellular processes, Wnt/β-catenin pathway, is also proved to be activated in a subpopulation of Ewing’s sarcoma cells when some exogenous ligands present.25 Hence, we examined the effect of miR-185 on PI3K/Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells. We found that overexpression of miR-185 markedly suppressed the PI3K/Akt/mTOR and Wnt/β-catenin pathways by inhibiting the expression level of key components, including p-Akt, p-mTOR, Wnt3a, and β-catenin. And the downstream proteins were correspondingly modulated by miR-185. P70 and Cyclin D1 are important proteins at downstream of PI3K/Akt/mTOR pathway. Overexpression of P70 could increase tumor cell proliferation and angiogenesis, and inhibit apoptosis.37 Cyclin D1 is closely associated with cell cycle, and also is one of the crucial regulators of the Wnt/β-catenin pathway.38 In addition to being a regulator of cellular signaling pathways, β-catenin is a multifunctional protein involved in cell adhesion and gene transcriptional regulation depending on its intracellular localization.39 Under normal circumstances, the Wnt pathway is in a dormant state, the β-catenin protein locates in the cell membrane, and the concentration in the cytoplasm is minuscule; when the cell undergoes pathologically changes such as carcinogenesis, the Wnt pathway is activated and the β-catenin protein in the cytoplasm accumulates continuously and transfers into the nucleus, resulting in the downstream gene expression. In the current study, from the immunohistochemistry assay, the expression level of nuclear β-catenin was significantly decreased in RD-ES cells followed by upregulation of miR-185. β-catenin is also reported to be a negative regulator of E-cad, a transmembrane protein involved in cell–cell adhesion, and the forming β-catenin/E-cad complex plays crucial role in maintaining the morphology and structural integrity of epithelial cells.40 Moreover, low expression of E-cad is closely related to the invasion and metastasis of tumor.41,42 Our data showed that upregulation of miR-185 could reduce the expression level of Cyclin D1 and P70, while enhance the expression of E-cad. As described above, miR-185 functions as a tumor suppressor to inhibit growth, motility and survival of Ewing’s sarcoma through regulation of PI3K/Akt/mTOR and Wnt/β-catenin pathways.
miRNAs regulate the behavior of malignant tumors mainly by binding the mRNA of target genes. It is reported that miR-185 inhibits growth and metastasis of non-small-cell lung cancer by targeting AKT1.15 Tang H et al shows that miR-185 suppresses invasion of glioma cell by targeting CDC42 and RhoA.43 In ovarian cancer, miR-185 is revealed to be associated with cisplatin resistance by targeting DNMT1 directly.44 In triple-negative breast cancer, miR-185 plays a role of tumor suppressor by targeting DNMT1 and E2F6.45 E2F6 is a member of E2F transcription factors family which play a crucial role in the regulation of cell proliferation, cell cycle, apoptosis, and differentiation through regulating the transcription of target genes.46 Recent studies demonstrate that E2F6 is upregulated in prostate cancer and breast cancer, and the decrease of E2F6 expression can inhibit the proliferation of triple-negative breast cancer cells.45,47 In the current study, we found that E2F6 was a possible target of miR-185 by bioinformatics predictions and dual-luciferase assay. Moreover, with the increase of miR-185 expression, E2F6 expression decreased significantly in RD-ES cells, further indicating that miR-185 directly targets E2F6. In addition, the suppression on cell proliferation of RD-ES cells via overexpression of miR-185 was significantly restored by the upregulation of E2F6, indicating that miR-185 blocked cell proliferation of RD-ES cells through directly targeting E2F6. We further revealed that loss of E2F6 could suppress the Akt/mTOR and Wnt/β-catenin pathways in RD-ES cells, while upregulation of E2F6 had no significant effect on these signaling pathway, indicating that the regulation of E2F6 on Akt/mTOR and Wnt/β-catenin signaling pathways requires synergy of other factors.
Conclusion
The present study demonstrates that miR-185 is involved in cell growth, motility, and survival of Ewing’s sarcoma as a tumor suppressor via suppressing PI3K/Akt/mTOR and Wnt/β-catenin pathways and targeting E2F6. To our knowledge, this study provides the first insights to validate that the miR-185/E2F6 axis plays a role in the progression and survival of Ewing’s sarcoma. All the evidence suggests that miR-185 might be a novel potential target for Ewing’s sarcoma therapy.
Acknowledgments
This study was jointly supported by grants from the Natural Science Foundation of Shandong Province for Young Scholars (No. 2015ZRE27529), China and the National Natural Science Foundation of China (No. 81602361).
Disclosure
The authors report no conflicts of interest in this work.
References
Riggi N, Stamenkovic I, Ross KA, Smyth NA, Murawski CD, Kennedy JG. The Biology of Ewing sarcoma. Cancer Lett. 2007;254(1):1. | ||
Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69. | ||
Taylor R, Knowles HJ, Athanasou NA. Ewing sarcoma cells express RANKL and support osteoclastogenesis. J Pathol. 2011;225(2):195–202. | ||
Gaspar N, Hawkins DS, Dirksen U, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol. 2015;33(27):3036–3046. | ||
Ladenstein R, Pötschger U, Le Deley MC, Dm L, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28(20):3284–3291. | ||
Medina-Villaamil V, Martínez-Breijo S, Portela-Pereira P, et al. Circulating MicroRNAs in blood of patients with prostate cancer. Actas Urol Esp. 2014;38(10):633–639. | ||
Li Y, Shao G, Zhang M. miR-124 represses the mesenchymal features and suppresses metastasis in Ewing sarcoma. Oncotarget. 2016;8(6):10274–10286. | ||
Schwentner R, Herrero-Martin D, Kauer MO, et al. The role of miR-17-92 in the miRegulatory landscape of Ewing sarcoma. Oncotarget. 2017;8(7):10980–10993. | ||
Kawano M, Tanaka K, Itonaga I, Iwasaki T, Tsumura H. MicroRNA-301a promotes cell proliferation via PTEN targeting in Ewing’s sarcoma cells. Int J Oncol. 2016;48(4):1531–1540. | ||
Kawano M, Tanaka K, Itonaga I, Iwasaki T, Tsumura H. MicroRNA-20b promotes cell proliferation via targeting of TGF-β receptor II and upregulates MYC expression in Ewing’s sarcoma cells. Int J Oncol. 2017;51(6):1842–1850. | ||
Mattie MD, Benz CC, Bowers J, et al. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer. 2006;5(1):24. | ||
Hwang JH, Voortman J, Giovannetti E, et al. Identification of microRNA-21 as a biomarker for chemoresistance and clinical outcome following adjuvant therapy in resectable pancreatic cancer. PLoS One. 2010;5(5):e10630. | ||
Zhou Q, Huang SX, Zhang F, et al. MicroRNAs: A novel potential biomarker for diagnosis and therapy in patients with non-small cell lung cancer. Cell Prolif. 2017;50(6):e12394. | ||
Huang LL, Huang LW, Wang L, Tong BD, Wei Q, Ding XS. Potential role of miR-139-5p in cancer diagnosis, prognosis and therapy. Oncol Lett. 2017;14(2):1215–1222. | ||
Li S, Ma Y, Hou X, et al. MiR-185 acts as a tumor suppressor by targeting AKT1 in non-small cell lung cancer cells. Int J Clin Exp Pathol. 2015;8(9):11854–11862. | ||
Liu C, Li G, Ren S, et al. miR-185-3p regulates the invasion and metastasis of nasopharyngeal carcinoma by targeting WNT2B in vitro. Oncol Lett. 2017;13(4):2631–2636. | ||
Li Q, Wang JX, He YQ, et al. MicroRNA-185 regulates chemotherapeutic sensitivity in gastric cancer by targeting apoptosis repressor with caspase recruitment domain. Cell Death Dis. 2014;5(4):e1197. | ||
Wang R, Tian S, Wang HB, et al. MiR-185 is involved in human breast carcinogenesis by targeting Vegfa. FEBS Lett. 2014;588(23):4438–4447. | ||
Ahmadi AA, Shadifar M, Ataee R, et al. The serotonin 5-HT2A receptor antagonist ritanserin induces apoptosis in human colorectal cancer and acts in synergy with curcumin. Int Biol Biomed. 2015;1(2):56–65. | ||
Malik S, Villanova L, Tanaka S, et al. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci Rep. 2015;5:9841. | ||
Liu M, Lang N, Chen X, et al. miR-185 targets RhoA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 2011;301(2):151–160. | ||
Ordóñez JL, Osuna D, Herrero D, de Alava E, Madoz-Gúrpide J. Advances in Ewing’s sarcoma research: where are we now and what lies ahead? Cancer Res. 2009;69(18):7140–7150. | ||
Machado I, López-Guerrero JA, Navarro S, et al. Epithelial cell adhesion molecules and epithelial mesenchymal transition (EMT) markers in Ewing’s sarcoma family of tumors (ESFTs). Do they offer any prognostic significance? Virchows Arch. 2012;461(3):333–337. | ||
Toretsky JA, Thakar M, Eskenazi AE, Frantz CN. Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing’s sarcoma family of tumors. Cancer Res. 1999;59(22):5745–5750. | ||
Pedersen EA, Scannell CA, Lawlor ER. Abstract 5031: R-spondin potentiates Wnt/β-catenin signaling in Ewing sarcoma cells. Cancer Res. 2013;73(8 Supplement):5031. | ||
Ma X, Shen D, Li H, et al. MicroRNA-185 inhibits cell proliferation and induces cell apoptosis by targeting VEGFA directly in von Hippel-Lindau-inactivated clear cell renal cell carcinoma. Urol Oncol. 2015;33(4):169.e1–11. | ||
Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5(6):a008672–249. | ||
Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2(4):277–288. | ||
Gui D, Guo Y, Wang F, et al. Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PLoS One. 2012;7(6):e39824. | ||
Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92–101. | ||
Wu R, Tang S, Wang M, Xu X, Yao C, Wang S. MicroRNA-497 induces apoptosis and suppresses proliferation via the Bcl-2/Bax-Caspase 9–Caspase 3 pathway and cyclin D2 protein in HUVECs. PLoS One. 2016;11(12):e0167052. | ||
Liu G, Wang T, Wang T, Song J, Zhou Z. Effects of apoptosis-related proteins caspase-3, Bax and Bcl-2 on cerebral ischemia rats. Biomed Rep. 2013;1(6):861–867. | ||
Meeran SM, Katiyar SK. Grape seed proanthocyanidins promote apoptosis in human epidermoid carcinoma A431 cells through alterations in Cdki-Cdk-cyclin cascade, and caspase-3 activation via loss of mitochondrial membrane potential. Exp Dermatol. 2007;16(5):405–415. | ||
Rahman M, Chan AP, Tang M, Tai IT. A peptide of SPARC interferes with the interaction between caspase8 and Bcl2 to resensitize chemoresistant tumors and enhance their regression in vivo. PLoS One. 2011;6(11):e26390. | ||
Yamamoto T, Ohno T, Wakahara K, et al. Simultaneous inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. J Cancer Res Clin Oncol. 2009;135(8):1125–1136. | ||
Li J, You T, Jing J. MiR-125b inhibits cell biological progression of Ewing’s sarcoma by suppressing the PI3K/Akt signalling pathway. Cell Prolif. 2014;47(2):152–160. | ||
Bian CX, Shi Z, Meng Q, Jiang Y, Liu LZ, Jiang BH. P70S6K 1 regulation of angiogenesis through VEGF and HIF-1alpha expression. Biochem Biophys Res Commun. 2010;398(3):395–399. | ||
Joo M, Lee HK, Kang YK. Expression of beta-catenin in hepatocellular carcinoma in relation to tumor cell proliferation and cyclin D1 expression. J Korean Med Sci. 2003;18(2):211. | ||
Terada N, Karim MR, Izawa T, Kuwamura M, Yamate J. Immunolocalization of β-catenin, E-cadherin and N-cadherin in neonate and adult rat kidney. J Vet Med Sci. 2017;79(11):1785–1790. | ||
Chen HN, Yuan K, Xie N, et al. PDLIM1 stabilizes the E-cadherin/β-catenin complex to prevent epithelial-mesenchymal transition and metastatic potential of colorectal cancer cells. Cancer Res. 2016;76(5):1122–1134. | ||
Yang YL, Chen MW, Xian L. Prognostic and clinicopathological significance of downregulated E-cadherin expression in patients with non-small cell lung cancer (NSCLC): a meta-analysis. PLoS One. 2014;9(6):e99763. | ||
Han T, Jiao F, Hu H, et al. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget. 2016;7(10):11194–11207. | ||
Tang H, Wang Z, Liu X, et al. LRRC4 inhibits glioma cell growth and invasion through a miR-185-dependent pathway. Curr Cancer Drug Targets. 2012;12(8):1032–1042. | ||
Xiang Y, Ma N, Wang D, et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: a novel epigenetic therapy independent of decitabine. Oncogene. 2014;33(3):378–386. | ||
Tang H, Liu P, Yang L, et al. miR-185 suppresses tumor proliferation by directly targeting E2F6 and DNMT1 and indirectly upregulating BRCA1 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(12):3185–3197. | ||
Li Q, Qiu XM, Li QH, et al. MicroRNA-424 may function as a tumor suppressor in endometrial carcinoma cells by targeting E2F7. Oncol Rep. 2015;33(5):2354–2360. | ||
Bhatnagar N, Li X, Padi SK, Zhang Q, Tang MS, Guo B. Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010;1(12):e105. |
Supplementary materials
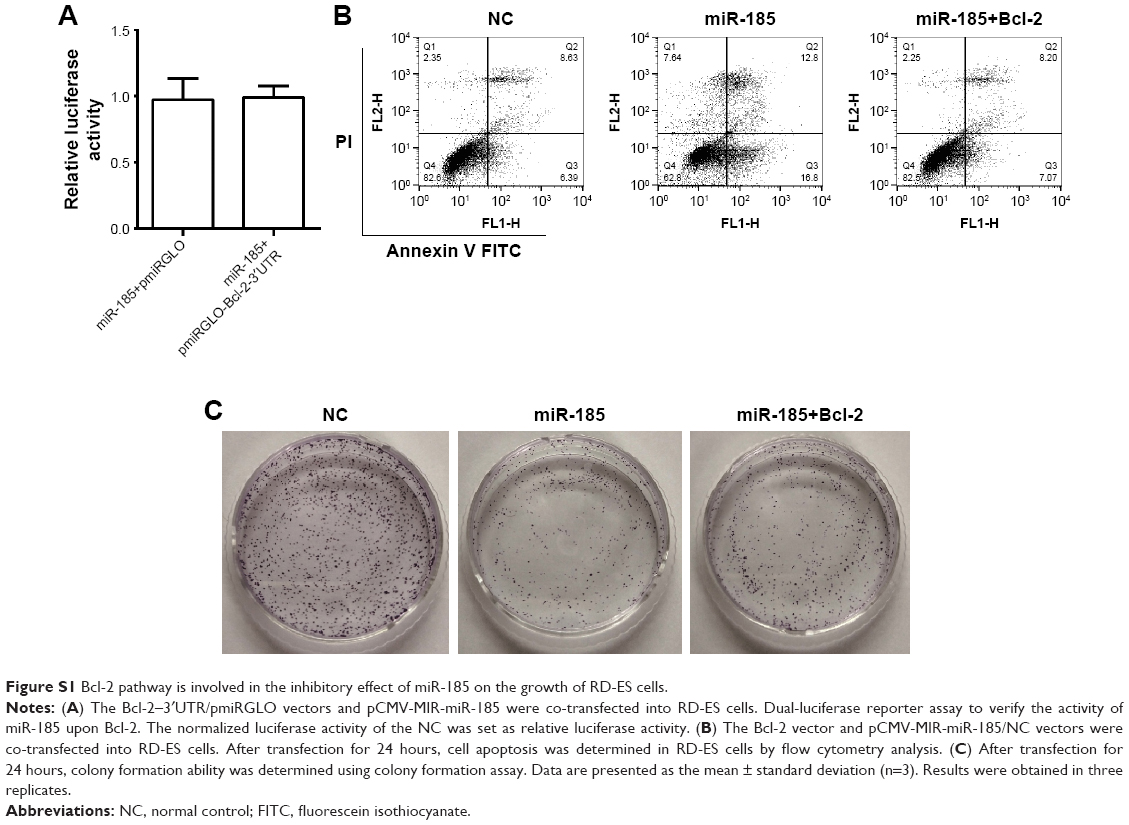 | Figure S1 Bcl-2 pathway is involved in the inhibitory effect of miR-185 on the growth of RD-ES cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.