")
Back to Journals » Drug Design, Development and Therapy » Volume 10
miR-125b acts as a tumor suppressor in chondrosarcoma cells by the sensitization to doxorubicin through direct targeting the ErbB2-regulated glucose metabolism
Authors Tang X, Zheng W, Ding M, Guo K, Yuan F, Feng H, Deng B, Sun W, Hou Y, Gao L
Received 15 June 2015
Accepted for publication 2 September 2015
Published 24 February 2016 Volume 2016:10 Pages 571—583
DOI https://doi.org/10.2147/DDDT.S90530
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Xian-ye Tang,1,2,* Wei Zheng,1,2,* Min Ding,3 Kai-jin Guo,1,2 Feng Yuan,1 Hu Feng,1 Bin Deng,1 Wei Sun,1 Yang Hou,4 Lu Gao5,6
1Department of Orthopedic, The First Clinical Medical College, Nanjing Medical University, Nanjing, 2Affiliated Hospital of Xuzhou Medical College, Xuzhou, Jiangsu, 3Department of Emergency Trauma Surgery, East Hospital Affiliated to Tongji University, 4Department of Orthopedic, Changzheng Hospital, Shanghai, 5College of Life Sciences, Northeast Agricultural University, Harbin, Heilongjiang, People’s Republic of China; 6Department of Pathology, University of Maryland School of Medicine, Baltimore, MD, USA
*These authors contributed equally to this work
Abstract: Chondrosarcoma is the second most common type of primary bone malignancy in the United States after osteosarcoma. Surgical resections of these tumors are the only effective treatment to chondrosarcoma patients due to their resistance to conventional chemo- and radiotherapy. In this study, miR-125b was found to perform its tumor-suppressor function to inhibit glucose metabolism via the direct targeting of oncogene, ErbB2. We report miR-125b was downregulated in both chondrosarcoma patient samples and cell lines. The total 20 Asian chondrosarcoma patients showed significantly downregulated miR-125b expression compared with normal tissues. Meanwhile, miR-125 was downregulated in chondrosarcoma cells and doxorubicin resistant cells. Overexpression of miR-125 enhanced the sensitivity of both parental and doxorubicin resistant cells to doxorubicin through direct targeting on the ErbB2-mediated upregulation of glycolysis in chondrosarcoma cells. Moreover, restoration of the expression of ErbB2 and glucose metabolic enzymes in miR-125 pretransfected cells recovered the susceptibility to doxorubicin. Our study will provide a novel aspect on the overcoming chemoresistance in human chondrosarcoma cells and may help in the development of therapeutic strategies for the treatments of patients.
Keywords: miR-125b, chondrosarcoma, doxorubicin, glucose metabolism, sensitization
Introduction
Chondrosarcoma is a malignant tumor of the bone and it typically affects adults between the age of 20 and 60 years old.1–3 To date, surgical resection of these tumors remains the only curative treatment offered to patients since chondrosarcoma are notorious for their resistance to conventional chemo- and radiotherapy.2–5 Therefore, understanding and exploring the mechanisms of chemotherapy and radiation resistance in chondrosarcoma could lead us to develop novel therapeutic strategy for the treatments of chondrosarcoma patients. The mechanisms accounting for chemoresistance have been discussed before, the abnormal expression of P-glycoprotein in chondrosarcoma has been reported, and it has been proposed that the P-glycoprotein is an important mechanism in the development of chemoresistance.6,7 Moreover, the specific pharmacologic inhibitor of telomerase, BIBR1532, has been studied as an alteration to resensitize chondrosarcoma cells to traditional chemotherapy.8
microRNA (miRNA) is the noncoding, single-stranded RNA of approximately 22 nucleotides. miRNA has been well studied to regulate gene expression and constitutes a novel class of gene regulators.9 Mature miRNAs are partially complementary to multiple messenger RNA (mRNA) targets and induce the degradation of mRNAs of their target genes by direct binding to the 3′-UTR regions.10 So far, hundreds of miRNAs have been shown to play essential roles in a variety of biological processes including proliferation, differentiation, migration, cell cycle, and apoptosis.10,11 It has been reported that miRNAs are involved in drug resistance12 to act as potential oncogenes or tumor suppressors.13–15 miR-125b that belongs to miR-125 family has been reported to be implicated in a variety of carcinomas as either repressors or oncomiRs including ovarian cancer,16 bladder cancer,17 breast cancer,18–20 hepatocellular carcinoma,21,22 melanoma,23 cutaneous squamous cell carcinoma,24 and osteosarcoma.25 A recent miRNA array analysis described that miR-125b was downregulated in chondrosarcoma cells, indicating miR-125b might act as a tumor suppressor in human chondrosarcoma.26
In 1956, Warburg observed that the rate of glycolysis was abnormally high in cancer cells, yet a smaller fraction of this glucose is broken down by oxidative phosphorylation.27 The “Warburg effect” indicates that the metabolic properties of cancer cells are more dependent on aerobic glycolysis, fatty acid synthesis, and glutaminolysis for proliferation, which is quite different from those of normal cells.28 On the basis of this theory, targeting metabolic dependence of tumors could be a selective approach to treat clinical patients. In this study, we reported a novel function of miR-125b, which promotes chemotherapy in chondrosarcoma cells. miR-125 was downregulated in chondrosarcoma cells and doxorubicin resistant cells. Overexpression of miR-125 enhanced the sensitivity of both parental and doxorubicin resistant cells to doxorubicin through direct targeting on ErbB2-mediated glucose metabolism. Restoration of ErbB2 and glucose metabolic enzymes in miR-125 pretransfected cells recovered the susceptibility to doxorubicin.
Materials and methods
Cell lines and cell culture
CSPG, OUMS-27, CH-2879, JJ012, CS-1, and SW1353 are human chondrosarcoma cells. SNM83 cells are normal human chondrocyte cell line. All cells were cultured in Dulbecco’s Modified Eagle’s Medium/F12 (Gibco BRL, Karlsruhe, Germany) with 10% fetal bovine serum (Gibco BRL) in humidified atmosphere 5% CO2 in air at 37°C. Doxorubicin resistant clone 1, 2 (Doxo R1, R2) and Doxo RP (pooled clone) were developed from JJ012 cells by treating with gradually increasing concentrations of doxorubicin in cell culture medium. The resistant cells were reselected every month by the treatment of doxorubicin.
Chondrosarcoma patient tissues
All primary human conventional chondrosarcoma tissues and normal articular chondrocytes specimens were obtained from patients undergoing surgery for chondrosarcoma during 2012–2013 at the Department of Oncology, Changzheng Hospital, Shanghai, People’s Republic of China, and stored in liquid nitrogen until analysis. Tumors were obtained under institutional review board approved protocol by the Ethics Committee of the Department of Oncology, Changzheng Hospital, Shanghai, People’s Republic of China. All patients provided written informed consent.
Antibodies and reagents
Antibodies used from this project were purchased from: poly(ADP-ribose) polymerase (PARP) (Cell Signaling Technology, Danvers, MA, USA: #9532); Hexokinase II (Cell Signaling Technology: #2867); β-actin (Cell Signaling Technology: #4967); PDK1 (Cell Signaling Technology: #3820); ErbB2 (Cell Signaling Technology: #2242); EGFR (Cell Signaling Technology: #2963); and LDHA (Cell signaling Technology: #2012); Doxorubicin was purchased from Sigma-Aldrich (Hong-Kong, People’s Republic of China).
Pre-miRNA or anti-miRNA transfection
miRNA precursors (pre-miRNAs) and miRNAs antisense RNAs (anti-miRNAs) were purchased from Applied Biosystems (Waltham, MA, USA). Pre-miR negative and anti-miR-negative were used as negative controls. Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) was used for the transfection of pre-miRNAs or anti-miRNAs. Forty-eight hours after transfection, the expression of miR-125b was detected by real-time PCR, and the expression of ErbB2, a target of miR-125b, was tested by real-time PCR and/or Western blotting.
Plasmid DNA and siRNA transfections
Transfection was performed using the Lipofectamine 2000 Transfection reagent (Invitrogen) according to the manufacturer’s protocol. Overexpression vectors containing wild-type ErbB2 (RC212583) and LDHA (RC228293) were purchased from www.origene.com. The siRNA oligonucleotides for ErbB2 was purchased from Sigma-Aldrich, with a scrambled siRNA (Sigma-Aldrich) used as a control. Forty-eight hours after transfection, cells were collected or whole-cell lysates were prepared for further analysis.
Glucose uptake assay
Cells were seeded in 12-well plates at 1×105–3×105 cells per well. Culture media was collected at 48 hours and stored at −20° until assayed. Glucose uptake was measured using an Amplex Red Glucose/Glucose Oxidase assay kit (Molecular Probes, Eugene, OR, USA). Absorbance was measured at 563 nm using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA) and the results were normalized to the amount of total protein compared with the control cells.
Lactate production assay
Lactate production in the medium was detected by using a Lactate assay kit (BioVision Inc., Milpitas, CA, USA). Results were normalized to the amount of total protein compared with the control cells.
Generation of JJ012 doxorubicin resistant cell line
JJ012 cells were treated with gradually increasing concentrations of doxorubicin in regular cell culture conditions for selection of resistant cells. After successive treatments for up to 2 months, several resistant cell clones were developed from the parental cell line. Doxorubicin resistant clone 1, 2 and pooled clones were used for subsequent experiments in this study. The resistant cells were selected by doxorubicin treatments each month.
Quantitative real-time PCR (qRT-PCR)
RNA was isolated from cultured cells using the RNeasy mini-kit (Qiagen, Hilden, Germany) (with an on-column DNAse digestion step according to the manufacturer’s instructions). Briefly, lysates of cells were passed through a Qiashredder (Qiagen) and the eluted lysates mixed 1:1 with 70% ethanol. The lysates were applied to a mini-column and after washing and DNAse I digestion, the RNAs were eluted in 30–50 μL of RNAse-free water. The quantity and quality of total RNA samples were checked by agarose–gel electrophoresis and using the Bioanalyzer RNA 6000 Nano assay (Agilent, Waldbronn, Germany). For miRNA expression analysis, qRT-PCR was done by using the TaqMan miRNA reverse transcription kit (Applied Biosystems) and TaqMan miRNA assays kit (Applied Biosystems) following the manufacturer’s protocols. Precursor miR-125b RT primer: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCACG; RNU6B served as an internal control. All reactions were performed in triplicate. End-point PCR was performed to analyze the expression of miR-125b by using a mirVana qRT-PCR miRNA detection kit and mirVana qRT-PCR Primer Sets (Applied Biosystems) according to the manufacturer’s protocols. Human U6 served as an internal control. For the detection of mRNA expressions of HK II, PDK1, and LDHA, real-time PCR were performed. RNA was isolated from cultured cells using the RNeasy mini-kit (Qiagen) (with an on-column DNAse digestion step according to the manufacturer’s instructions). cDNA was mixed with 2× SYBR Green PCR Master Mix (Applied Biosystems) and various sets of gene-specific primers and then subjected to real-time PCR quantification by using the iQ5 real-time PCR system (Bio-Rad Laboratories Inc., Hercules, CA, USA). The sequences of the primers used were as follows:
HK II, 5′ primer (5′-CCGTGGTGGACAAGATAAGAGAGAACC-3′) and 3′ primer (5′-GGACACGTCACATTTCGGAGCCAG-3′); PDK1, 5′ primer (5′-ACAAGGAGAGCTTCGGGGTGGATC-3′) and 3′ primer (5′-CCACGTCGCAGTTTGGATTTATGC-3′); LDHA, 5′ primer (5′-ATCTTGACCTACGTGGCTTGGA) and 3′ primer (5′-CCATACAGGCACACTGGAATCTC-3′).
The PCR conditions were 25–30 cycles at 95°C for 30 seconds, 56°C for 30 seconds, and 72°C for 1 minute. The PCR products were separated on a 2% agarose gel. All reactions were performed in triplicate. The relative amounts of mRNA were calculated by using the comparative CT method.
Luciferase reporter assay
The pMIR-reporter luciferase vector containing the wild-type 3′-UTR and binding site mutant 3′-UTR of ErbB2 and the empty vector were constructed according to the methods previously described.29 Briefly, the 3′-UTR sequence (618 bp) of ErbB2 was obtained from UTRdb (http://utrdb.ba.itb.cnr.it/) and EST clone containing the 3-UTR sequences from ErbB2 cDNA was purchased from Invitrogen and used as templates for PCR amplification of the full-length or binding-sites mutation (deletion of the position 37–44 of ErbB2 3′-UTR: CUCAGGGA). For the luciferaseassay, cells at the density of 2×105 per well in 24-well plates were cotransfected with pMIR-REPORT luciferase reporters with 3′-UTR of wild-type ErbB2 or binding site mutant ErbB2, pre-miR-125b, or pre-miR-negative using Lipofectamine 2000 reagent. Forty-eight hours later, cells were harvested and lysed with passive lysis buffer (Promega Corporation, Fitchburg, WI, USA). Luciferase activity was measured by using a dual luciferase reporter assay (Promega). The pRL-TK vector (Promega) was used as an internal control. The results were expressed as relative luciferase activity (firefly Luc/Renilla Luc).
Cell viability assay
A total of 1×104 cells for each well were seeded in 48-well plates for overnight. The medium was replaced with fresh medium with or without doxorubicin at the indicated concentrations and incubated for 48 hours. Cell viability was measured using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Absorbance was measured spectrophotometrically at 570 nm by the Universal Microplate Reader EL800 (BIO-TEK instruments, Inc., Vermont, MA, USA).
Western blot analysis
Whole cells were lysed in 1× SDS sample buffer and resolved by electrophoresis using SDS-PAGE and transferred to nitrocellulose membranes. The membranes were probed with primary antibodies overnight, and then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 3 hours, followed by detection with a Super Signal Enhanced Chemiluminescence kit (Pierce, Rockford, IL, USA). For sequential blotting, the membranes were stripped with Stripping Buffer (Pierce) and reprobed with proper antibodies.
Statistical analysis
Statistical evaluation for data analysis was determined by unpaired Student’s t-test. All data were shown as the means. A statistical difference of P<0.05 was considered significant.
Results
miR-125b is downregulated in human chondrosarcoma cells and doxorubicin resistant cells
Since miR-125b has been reported to act as a tumor suppressor gene in multiple tumor types, we started to check the expression of miR-125b in human chondrosarcoma cells and normal chondrocyte cell. As we expected, miR-125b was significantly downregulated in multiple chondrosarcoma cell lines compared with normal chondrocyte cell (Figure 1A).Consistently, the expressions of miR-125 in 20 Asian human conventional chondrosarcoma tissue were lower than normal articular chondrocytes (Figure 1B and Table 1), suggesting miR-125b might be a tumor suppressor in human chondrosarcoma. It has been known that inactivation of tumor suppressor genes contributes to chemoresistance. We generated doxorubicin resistant chondrosarcoma cell line using JJ012 cell by gradually treated JJ012 cells at elevated drug concentrations for 3 month. The JJ012 doxorubicin resistant colons were picked and pooled as the JJ012 Doxo R. Figure 1C showed resistant cells were insensitive to regular doxorubicin treatments compared with the parental cells, which displayed a proximate IC50 at 50 μM, while the IC50 of RP was much higher than parental cells. We next measured the expression of miR-125b in doxorubicin resistant clone 1, clone 2, and clone pool. Our results showed miR-125b were downregulated in all doxorubicin resistant chondrosarcoma cells, which further supported that miR-125 possessed the function of a tumor suppressor gene (Figure 1D). The protein expression of the cleaved PARP (c-PARP) is an important marker of caspase-mediated apoptosis. The resistance to doxorubicin was confirmed by the detection of c-PARP after the cells were treated with doxorubicin at 50 μM for 48 hours. We found much higher levels of uncleaved PARP and correspondingly much lower levels of c-PARP in doxorubicin resistant cells compared to parental cells (Figure 1E).
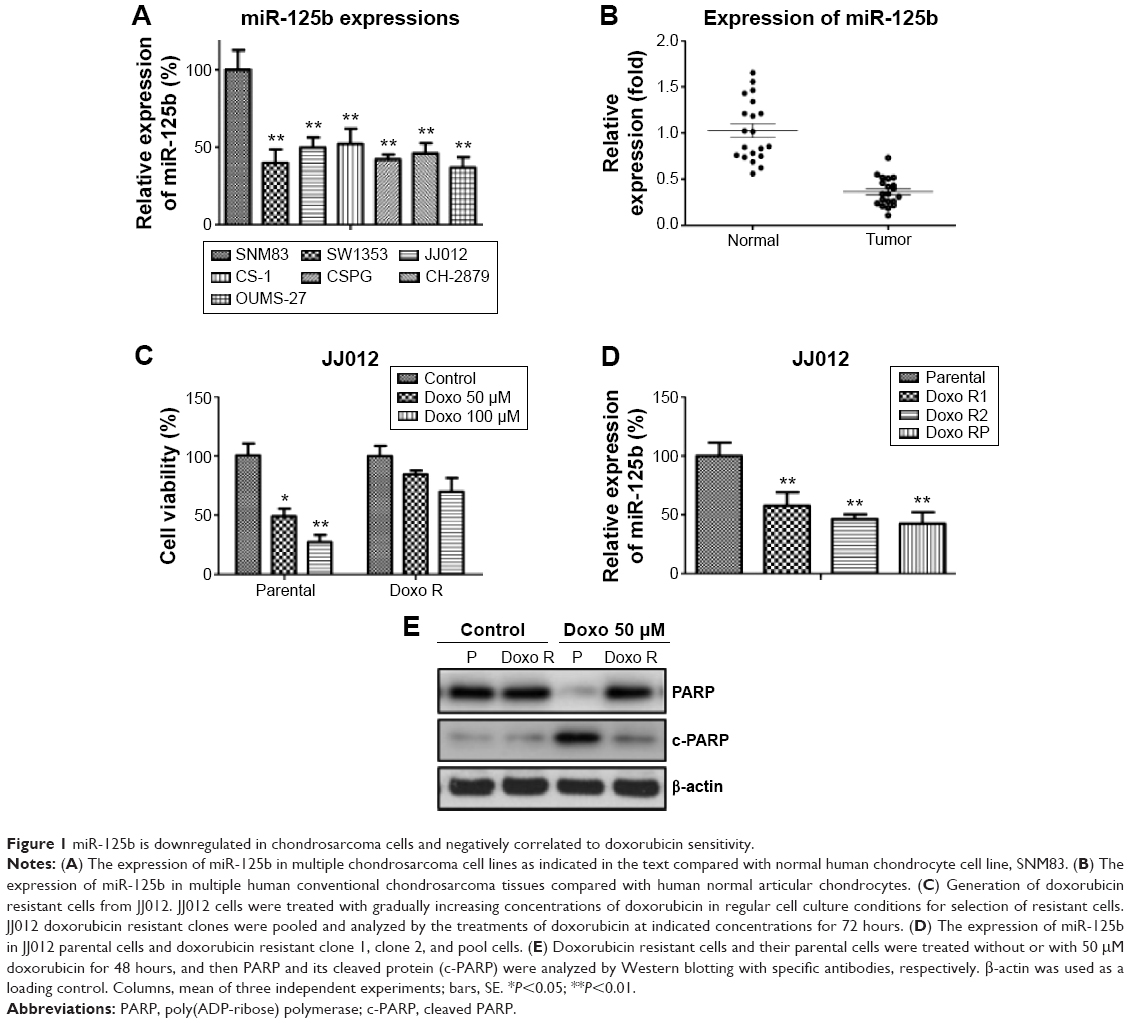 | Figure 1 miR-125b is downregulated in chondrosarcoma cells and negatively correlated to doxorubicin sensitivity. |
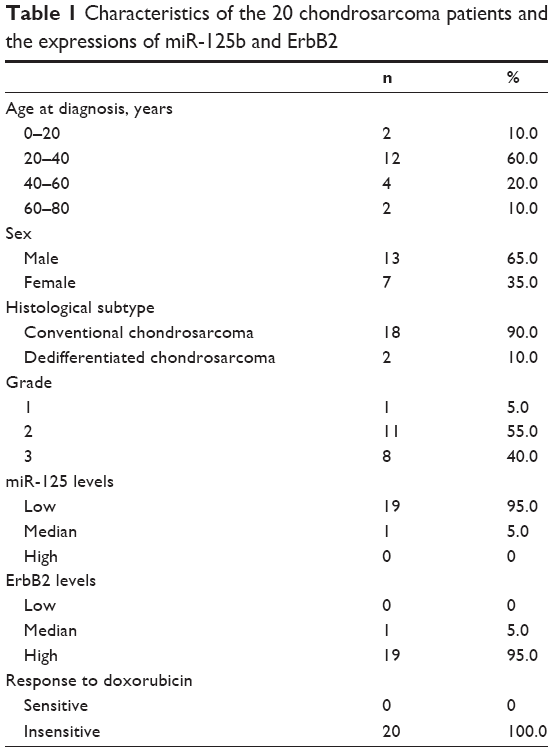 | Table 1 Characteristics of the 20 chondrosarcoma patients and the expressions of miR-125b and ErbB2 |
Overexpression of miR-125b inhibits chondrosarcoma cell proliferation and sensitizes chondrosarcoma cells to doxorubicin
The observation that miR-125b was downregulated in chondrosarcoma cells triggered us to explore the detailed functions of miR-125b by transient transfection of miR-125b into cancer cells (Figure 2A, upper). As we expected, the cells growth rates were significantly decreased in miR-125b overexpression chondrosarcoma cells (Figure 2A, lower), suggesting miR-125b acts as a tumor suppressor in chondrosarcoma. Consequently, we measured the sensitivity of cells with overexpression of miR-125b in response to doxorubicin treatments. In all three cell lines, JJ012, CH-2879, and SW1353 displayed sensitive to doxorubicin treatments when they were transfected with miR-125b compared with control miRNAs (Figure 2B). Taken together, our results demonstrated strong inhibitory effects on tumor behaviors by miR-125b in chondrosarcoma cells.
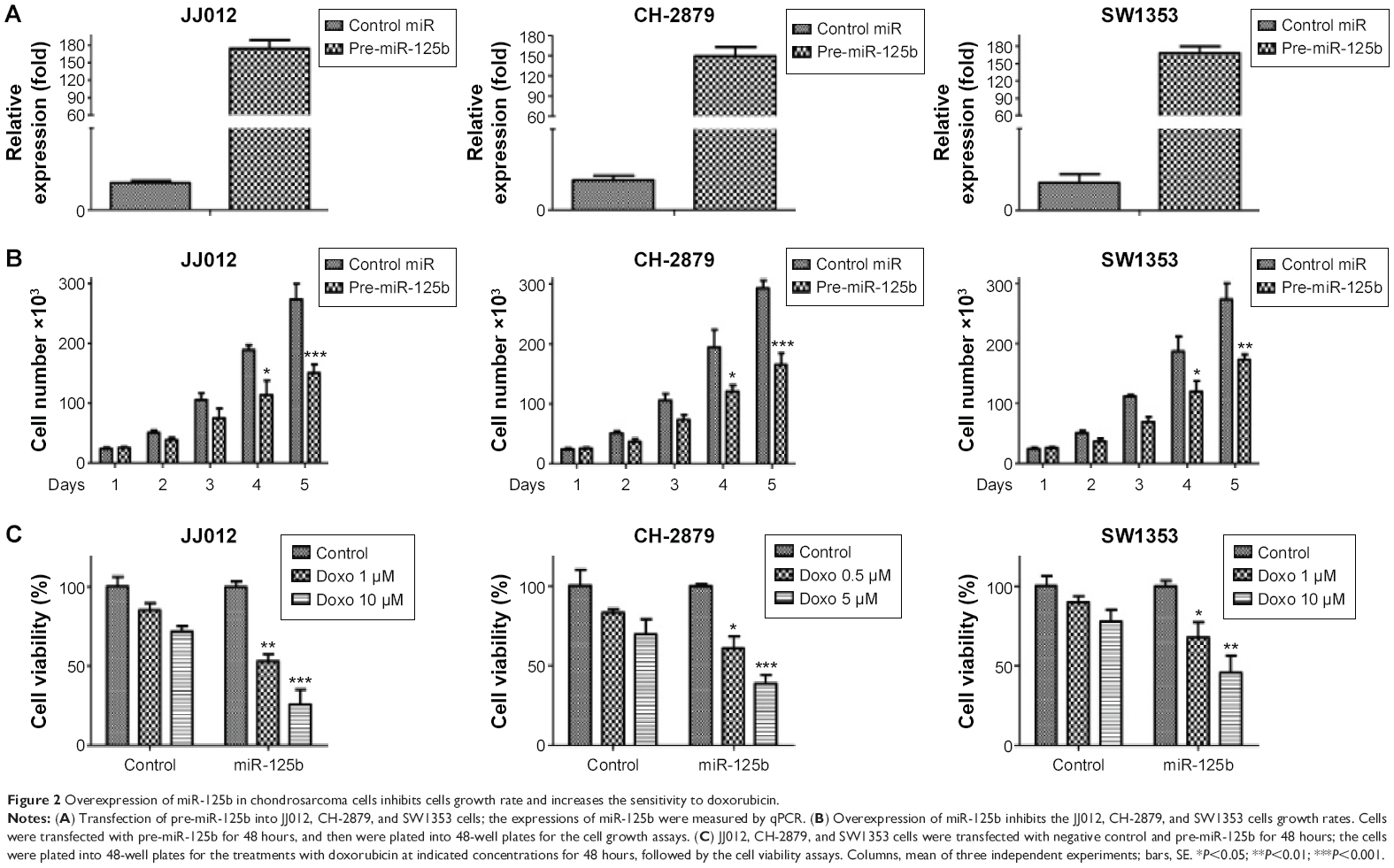 | Figure 2 Overexpression of miR-125b in chondrosarcoma cells inhibits cells growth rate and increases the sensitivity to doxorubicin. |
Doxorubicin resistant cells exhibit elevated glucose metabolism
To explore the mechanisms accounting for the miR-125b-mediated sensitization to chemotherapy in chondrosarcoma cells, we screened multiple pathways that might involve in the chemosensitivity. Interestingly, the glucose metabolism that contributes to the gain of resistance of multiple anticancer drugs was significantly changed in doxorubicin resistant cells. Currently, increasing evidence supports the idea that dysregulated cellular metabolism is linked to drug resistance in cancer therapy. In our assays, both of glucose uptake (Figure 3A, left) and lactate product (Figure 3B) were increased in doxorubicin resistant cells, indicating the dysregulated metabolism might contribute to chemoresistance. Consistently, the key enzymes in the glucose metabolism were upregulated in doxorubicin resistant cells at both protein (Figure 3C) and mRNA (Figure 3D) levels. These results further supported the observation that doxorubicin resistant chondrosarcoma cells exhibited elevated glucose metabolism.
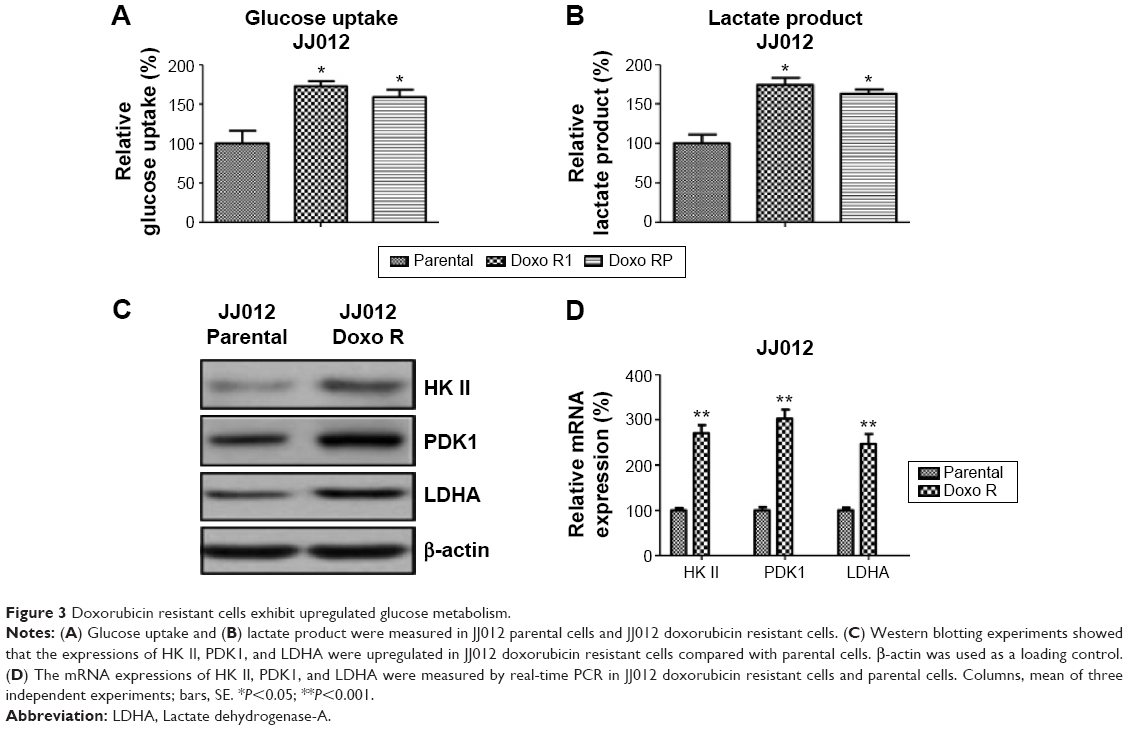 | Figure 3 Doxorubicin resistant cells exhibit upregulated glucose metabolism. |
Overexpression of miR-125b in chondrosarcoma downregulates glucose metabolism
To figure out whether miR-125b regulates glucose metabolism in chondrosarcoma cells, we measured the glucose uptake and lactate product in JJ012 and CH-2879 miR-125b overexpressing cells compared with the transfection of control miRNA. Our data demonstrated significant inhibitions of glucose metabolism in the miR-125b overexpressing cells (Figure 4A and B), indicating miR-125b might sensitize the chondrosarcoma cells to doxorubicin through the downregulation of glucose metabolism. Consistent results in Figure 4C showed the expressions of HK II, PDK1, and LDHA were decreased by overexpression of miR-125b in JJ012 and CH-2879 cells. In general, our results from Figures 3 and 4 revealed a correlation between miR-125 mediated doxorubicin sensitization and the altered glucose metabolism.
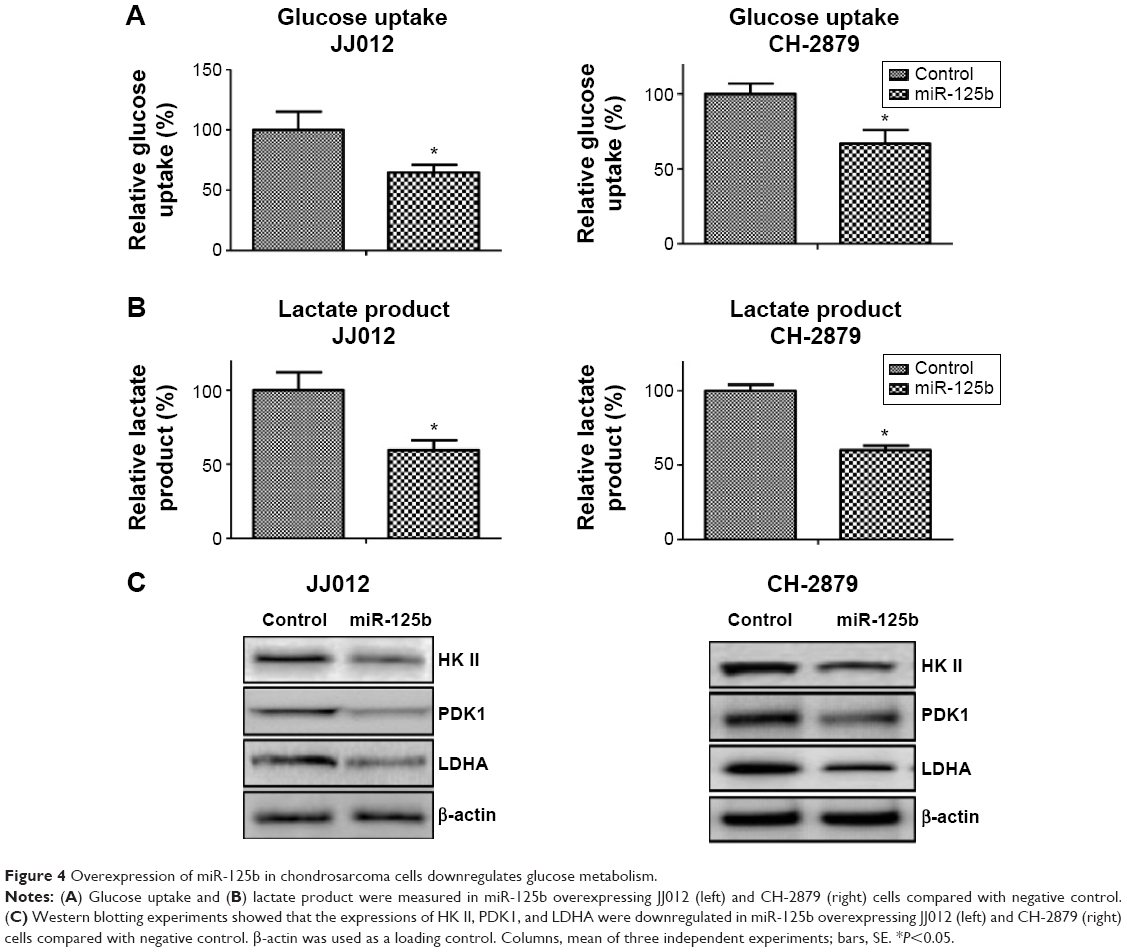 | Figure 4 Overexpression of miR-125b in chondrosarcoma cells downregulates glucose metabolism. |
ErbB2 is a direct target of miR-125b in chondrosarcoma cells
Because miR-125b is capable of suppressing glucose metabolism and increasing the sensitivity of chondrosarcoma cells to doxorubicin, we searched miRNA databases for potential miR-125b targets that may possibly contribute to doxorubicin resistance. The three public miRNA databases (TargetScan, Pictar, and MicroRNA) all predicted that ErbB2 might be a target for miR-125b, and the 3′-UTR of ErbB2 contains a highly conserved binding site for miR-125b. So far, no publication reported that ErbB2 is a miR-125b target in chondrosarcoma cells. To determine whether miR-125b could target ErbB2 in chondrosarcoma cells, we transfected the pre-miR-125b into JJ012 and CH-2879 cells. The expression of miR-125b significantly downregulated ErbB2 proteins in both cells (Figure 5A). Moreover, knockdown of miR-125b upregulated the expression of ErbB2 in both cells. The expression of EGFR, which is another family member of ErbB2, was not changed by transfection of miR-125b, indicating ErbB2 is a specific target of miR-125b. We next investigated whether miR-125b directly targets the 3′-UTR of ErbB2 mRNA, and we performed luciferase reporter analysis by cotransfecting a vector containing plasmid microRNA (pMIR) reporter-luciferase fused with original sequence or predicted binding site mutant sequence of the 3′-UTR of ErbB2 mRNA and pre-miR-125b or control miRNA. Overexpression of miR-125b decreased the luciferase activity of the reporter with wild-type 3′-UTR of ErbB2 by approximately 60% in both JJ012 and CH-2879 cells (Figure 5B). However, no inhibitory effects of miR-125b on the activity of the reporter with binding site mutant of 3′-UTR of ErbB2 were detected (Figure 5B). Taken together, our results demonstrated that ErbB2 is a direct target of miR-125b in chondrosarcoma cells.
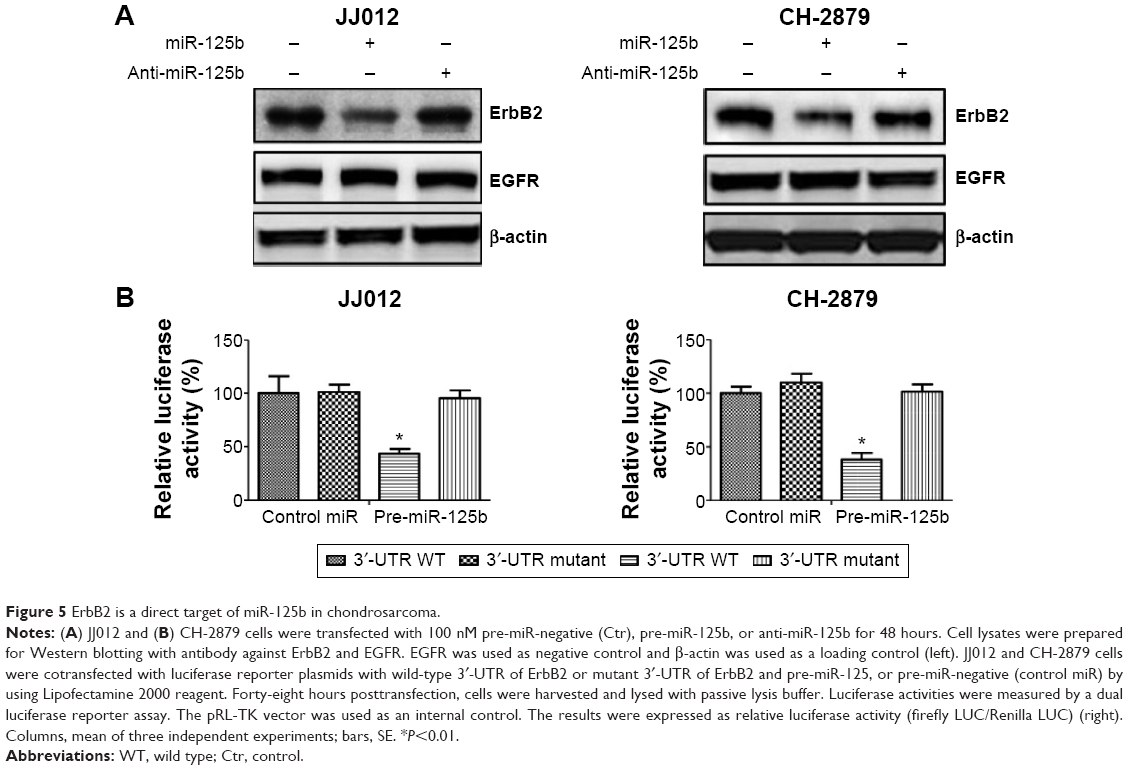 | Figure 5 ErbB2 is a direct target of miR-125b in chondrosarcoma. |
Restoration of ErbB2 in miR-125b overexpressing chondrosarcoma cells recovers glucose metabolism and doxorubicin sensitivity
Since it has been reported ErbB2 upregulates glucose metabolism in breast cancer cells and confers to chemoresistance,30,31 our aforementioned results revealed a correlation between the downregulation of ErbB2 and the miR-125-mediated downregulation of glucose metabolism and sensitivity of doxorubicin. To examine whether the downregulation of glucose metabolism by miR-125b was through the inhibition of ErbB2, we transfected overexpression vector containing wild-type ErbB2 into miR-125 pretransfected chondrosarcoma cells. Exogenous overexpression of ErbB2 restored the ErbB2 to the original level resulted in the glucose metabolism enzymes recovered to the nontransfection levels (Figure 6A). Similar results obtained from glucose uptake and lactate product assays showed both of them were recovered by overexpression of ErbB2 in miR-125b pretransfected cells (Figure 6B), indicating the metabolism enzymes were regulated by ErbB2, not directly by miR-125b. To figure out whether miR-125b-mediated sensitization to doxorubicin was through the targeting on ErbB2, we measured the chemosensitivity. Overexpression of ErbB2 in miR-125b pretransfected cells led to a significant resistance compared with control vector transfected into miR-125b pretransfected cells (Figure 6C). To further support our points that ErbB2 promotes glycolysis of chondrosarcoma cells, JJ012 cells were transfected with siErbB2 to knock down the expression of ErbB2 (Figure 6D, middle). Figure 6D shows the glucose uptake and lactate product were significantly decreased by the knocking down of ErbB2. Taken together, our results demonstrate both of the glucose metabolism and chemosensitivity changed by overexpression of miR-125b in chondrosarcoma cells were due to the direct targeting on ErbB2.
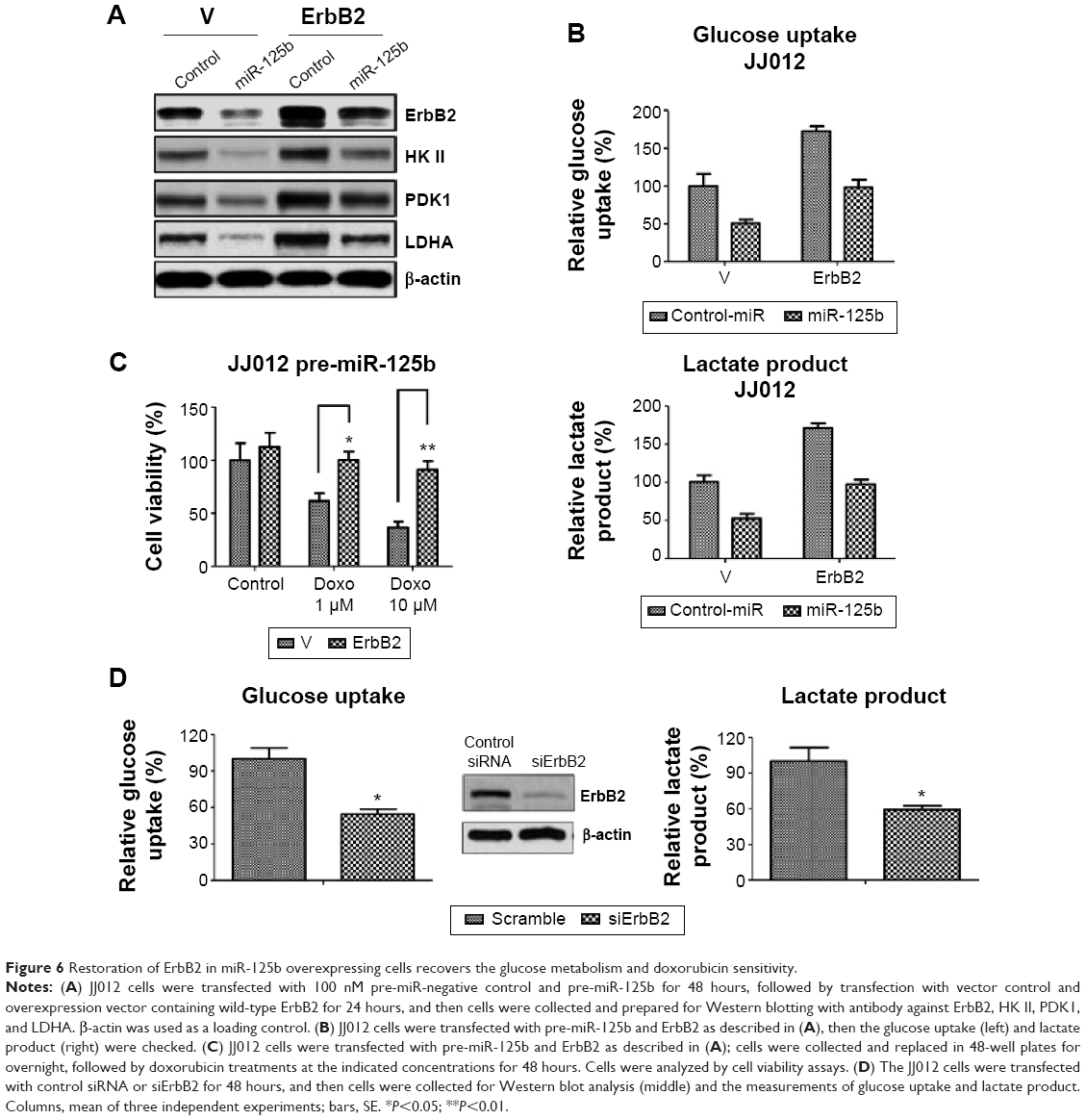 | Figure 6 Restoration of ErbB2 in miR-125b overexpressing cells recovers the glucose metabolism and doxorubicin sensitivity. |
Overexpression of miR-125b sensitizes doxorubicin resistant cells through the inhibition of glucose metabolism
The aforementioned results revealed that overexpression of miR-125b contributed to the sensitivity of doxorubicin. Here we showed overexpression of miR-125b in doxorubicin resistant chondrosarcoma cells resensitized the resistant cells to doxorubicin (Figure 7A). We transfected miR-125b and control miRNA into JJ012 doxorubicin resistant cells, and then treated the cells with indicated concentrations of doxorubicin for 48 hours. Transfection with the miR-125b significantly inhibited the cell viability at 50 and 100 μM (Figure 7A). Meanwhile the protein levels of ErbB2 and glucose metabolic enzymes were approximately decreased to the parental levels (Figure 7B). The similar results in Figure 7C showed the glucose uptake and lactate product were also decreased to the similar levels of parental cells, indicating that the miR-125b-mediated resensitization to doxorubicin was through the ErbB2-regulated glucose metabolism pathway. Finally, we overexpressed the key enzymes of glucose metabolism in miR-125b pretransfected JJ012 cells (Figure 7D left) and measured the sensitivity to doxorubicin (Figure 7D, right). The results demonstrated that exogenous upregulation of glucose metabolism rendered miR-125b overexpressing chondrosarcoma cells resistant to doxorubicin, indicating overexpression of miR-125b resulted in cells susceptive to doxorubicin through the inhibition of glucose metabolism.
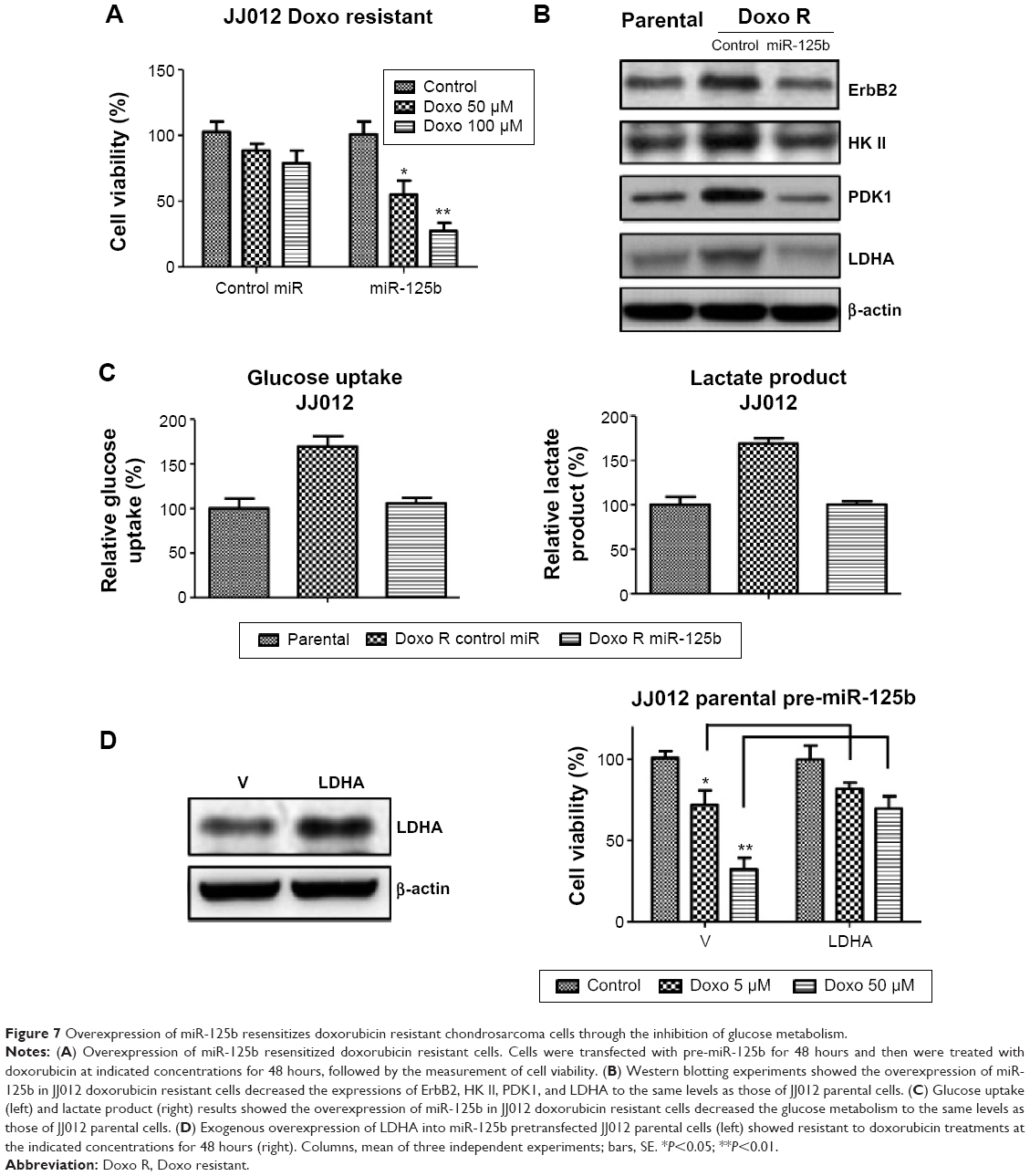 | Figure 7 Overexpression of miR-125b resensitizes doxorubicin resistant chondrosarcoma cells through the inhibition of glucose metabolism. |
Discussion
Chondrosarcoma is the second most common type of primary bone malignancy after osteosarcoma. Currently, very limited systemic treatment can be offered for high-grade and metastatic tumors since chondrosarcomas turned to be resistant to conventional chemo- and radiotherapy.4,5 So far the mechanisms for chemoresistance in chondrosarcoma have been revealed. The most prominent mechanism of chemoresistance is the abnormal expression of P-glycoprotein in chondrosarcoma. Moreover, hypoxia induces angiogenesis via increased levels of VEGF32 and the loss of tumor suppressor p16 in chondrosarcoma33 have been reported to leads to chemo- and radioresistance. A recent study reported that the growth of chondrosarcoma cells can be inhibited by mTOR inhibitor in an in vivo syngeneic rat model,5 suggesting a putative chemotherapeutic approach for clinical applications. However, very limited study focused on the glucose metabolism in chondrosarcoma. Therefore, the purpose of this study is to explore putative regulators in the alterations of glucose metabolism pathways in chondrosarcoma cells, which can be selected as targets for the development of therapeutics.
The “Warburg effects” described that cancer cells reprogram metabolic pathways for their bioenergetic and biosynthetic requirements. Increased aerobic glycolysis, fatty acid synthesis, and glutamine metabolism has been linked to therapeutic resistance in cancer. Many enzymes of glucose metabolism such as glucose transporters, hexokinase, pyruvate kinase M2, and LDHA involved in the reactions required for the glycolytic breakdown of glucose for a center energy supply.34 Currently, inhibition of glucose metabolism as an anticancer strategy has been widely studied. Multiple well-studied oncogenes such as AKT,35 ErbB2,30 Src,36 and Ras37 have been reported to promote glycolysis of cancer cells. On the other way, tumor suppressors such as P5338 and PTEN39 have been shown to decrease glycolysis rate. Therefore, we classified miR-125b into tumor suppressor in this project based on its ability to inhibit ErbB2 and negatively regulate anaerobic glycolysis in chondrosarcoma cells. We described a metabolic upregulation in doxorubicin resistant chondrosarcoma cells and inhibition of glucose metabolism by overexpression of miR-125b resensitized resistant cells to doxorubicin. The key enzymes in glucose metabolism were downregulated by the miR-125-induced inhibition of ErbB2, which might be served for clinical drug development. This will be the first time to link dysfunctioned glucose metabolism and chemoresistance in chondrosarcoma and might trigger us to explore detailed mechanisms to develop metabolism inhibitors for overcoming chemoresistance.
Many miRNAs have been identified as having an oncogenic or tumor suppressor like function shown to be involved in cell proliferation, differentiation, apoptosis, and drug resistance. miR-125 has been reported to act as a tumor-suppressor functions in several cancers including ovarian cancer,16 bladder cancer,17 hepatocellular carcinoma,22 cutaneous squamous cell carcinoma,24 and osteosarcoma.25 Moreover, miR-125b has been reported to promote cell proliferation in prostate cancer cells,40 enhances invasive potential in urothelial carcinomas,41 and suppresses p53-dependent apoptosis in human neuroblastoma cells.42 A recent miRNA array analysis described that miR-125b was downregulated in chondrosarcoma cells, indicating miR-125b might act as a tumor suppressor in human chondrosarcoma.26 However, little is known about their role in chemoresistance, such as to doxorubicin, and currently, the explicit functions of miR-125b in the regulation of metabolism is still under investigation. As an important miRNA, miR-125 possessed an inhibitory function in chondrosarcoma in this study since it negatively regulated glucose metabolism and promotes chemosensitivity to doxorubicin. miR-125b-induced downregulation of ErbB2 and ErbB3 has been reported to reduce cell motility and invasiveness of breast cancer.29 In our study, we aimed at examining the role of miR-125b in doxorubicin resistance in chondrosarcoma cells through the comparison between doxorubicin resistant and sensitive cells. We found that miR-125b was downregulated in chondrosarcoma cells compared with normal human chondrocytes. More importantly, miR-125b was downregulated in doxorubicin resistant cancer cells, with its overexpression enhancing doxorubicin-induced cytotoxicity and apoptosis, subsequently increasing the sensitivity of chondrosarcoma cells to doxorubicin. Moreover, we demonstrated that ErbB2 was a direct target of miR-125b in chondrosarcoma cells. The inhibition of ErbB2 by overexpression of miR-125b led to suppression of glucose metabolism, which rendered chondrosarcoma cells susceptible to doxorubicin. Restoring the expression of ErbB2 and glucose metabolic enzymes recovered doxorubicin resistance in counteracting miR-125b-mediated sensitivity. Taken together, miR-125b plays a critical role in doxorubicin resistance through suppression of ErbB2-induced glucose metabolism, and it may serve as a potential target for overcoming chemoresistance in human chondrosarcoma. We are the first to demonstrate that ErbB2 is a direct target of miR-125b in chondrosarcoma cells and showed that miR-125b increased doxorubicin sensitivity, acting primarily as a tumor suppressor in chondrosarcoma cells. However, further investigations with clinical patient samples need to be performed in the further project to identify the role of miR-125b in human chondrosarcoma development.
Acknowledgment
The work was supported by the project (LW201004) by the Public Health Bureau of Jiangsu Province (GUO).
Disclosure
The authors report no conflicts of interest in this work.
References
Bertoni F, Bacchini P, Hogendoorn PCW. Chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organisation Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002:247–251. | ||
Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13(3):320–329. | ||
Fiorenza F, Abudu A, Grimer RJ, et al. Risk factors for survival and local control in chondrosarcoma of bone. J Bone Joint Surg Br. 2002;84(1):93–99. | ||
van Oosterwijk JG, Anninga JK, Gelderblom H, et al. Update on targets and novel treatment options for high-grade osteosarcoma and chondrosarcoma. Hematol Oncol Clin North Am. 2013;27(5):1021–1048. | ||
Onishi AC, Hincker AM, Lee FY. Surmounting chemotherapy and radioresistance in chondrosarcoma: molecular mechanisms and therapeutic targets. Sarcoma. 2011;2011:381564. | ||
Kim DW, Kim KO, Shin MJ, et al. Sirna-based targeting of antiapoptotic genes can reverse chemoresistance in p-glycoprotein expressing chondrosarcoma cells. Mol Cancer. 2009;8:28. | ||
Kim SG, Jeon CH, Suh HS, et al. P-glycoprotein expression in extracellular matrix formation of chondrogenic differentiation of human adult stem cells. Cell Biol Int. 2007;31(9):1042–1048. | ||
Parsch D, Brassat U, Brummendorf TH, et al. Consequences of telomerase inhibition by bibr1532 on proliferation and chemosensitivity of chondrosarcoma cell lines. Cancer Invest. 2008;26(6):590–596. | ||
Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14(8):475–488. | ||
Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. | ||
Bushati N, Cohen SM. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. | ||
Hong L, Yang Z, Ma J, et al. Function of mirna in controlling drug resistance of human cancers. Curr Drug Targets. 2013;14(10):1118–1127. | ||
Shenouda SK, Alahari SK. MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009;28(3–4):369–378. | ||
Zhang B, Pan X, Cobb GP, et al. MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1–12. | ||
Wang D, Qiu C, Zhang H, et al. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One. 2010;5(9):e13067. | ||
Guan Y, Yao H, Zheng Z, et al. Mir-125b targets bcl3 and suppresses ovarian cancer proliferation. Int J Cancer. 2011;128(10):2274–2283. | ||
Huang L, Luo J, Cai Q, et al. MicroRNA-125b suppresses the development of bladder cancer by targeting e2f3. Int J Cancer. 2011;128(8):1758–1769. | ||
Akhavantabasi S, Sapmaz A, Tuna S, et al. Mir-125b targets arid3b in breast cancer cells. Cell Struct Funct. 2012;37(1):27–38. | ||
Wang HJ, Guo YQ, Tan G, et al. Mir-125b regulates side population in breast cancer and confers a chemoresistant phenotype. J Cell Biochem. 2013;114(10):2248–2257. | ||
Wang H, Tan G, Dong L, et al. Circulating mir-125b as a marker predicting chemoresistance in breast cancer. PLoS One. 2012;7(4):e34210. | ||
Alpini G, Glaser SS, Zhang JP, et al. Regulation of placenta growth factor by microRNA-125b in hepatocellular cancer. J Hepatol. 2011;55(6):1339–1345. | ||
Kim JK, Noh JH, Jung KH, et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors mir-125a-5p and mir-125b. Hepatology. 2013;57(3):1055–1067. | ||
Kappelmann M, Kuphal S, Meister G, et al. MicroRNA mir-125b controls melanoma progression by direct regulation of c-jun protein expression. Oncogene. 2013;32(24):2984–2991. | ||
Xu N, Zhang L, Meisgen F, et al. MicroRNA-125b down-regulates matrix metallopeptidase 13 and inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion. J Biol Chem. 2012;287(35):29899–29908. | ||
Liu LH, Li H, Li JP, et al. Mir-125b suppresses the proliferation and migration of osteosarcoma cells through down-regulation of stat3. Biochem Biophys Res Commun. 2011;416(1–2):31–38. | ||
Yoshitaka T, Kawai A, Miyaki S, et al. Analysis of microRNAs expressions in chondrosarcoma. J Orthop Res. 2013;31(12):1992–1998. | ||
Vander HM, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | ||
Najafov A, Alessi DR. Uncoupling the warburg effect from cancer. Proc Natl Acad Sci U S A. 2010;107(45):19135–19136. | ||
Scott GK, Goga A, Bhaumik D, et al. Coordinate suppression of erbb2 and erbb3 by enforced expression of micro-rna mir-125a or mir-125b. J Biol Chem. 2007;282(2):1479–1486. | ||
Zhao YH, Zhou M, Liu H, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28(42):3689–3701. | ||
Zhao Y, Liu H, Liu Z, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71(13):4585–4597. | ||
Sun X, Charbonneau C, Wei L, et al. Cxcr4-targeted therapy inhibits VEGF expression and chondrosarcoma angiogenesis and metastasis. Mol Cancer Ther. 2013;12(7):1163–1170. | ||
Asp J, Sangiorgi L, Inerot SE, et al. Changes of the p16 gene but not the p53 gene in human chondrosarcoma tissues. Int J Cancer. 2000;85(6):782–786. | ||
Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. | ||
Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. | ||
Valle-Casuso JC, Gonzalez-Sanchez A, Medina JM, et al. Hif-1 and c-src mediate increased glucose uptake induced by endothelin-1 and connexin43 in astrocytes. PLoS One. 2012;7(2):e32448. | ||
Gaglio D, Metallo CM, Gameiro PA, et al. Oncogenic k-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. | ||
Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol. 2010;22(2):186–191. | ||
Blouin MJ, Zhao Y, Zakikhani M, et al. Loss of function of pten alters the relationship between glucose concentration and cell proliferation, increases glycolysis, and sensitizes cells to 2-deoxyglucose. Cancer Lett. 2010;289(2):246–253. | ||
Shi XB, Xue L, Ma AH, et al. Mir-125b promotes growth of prostate cancer xenograft tumor through targeting pro-apoptotic genes. Prostate. 2011;71(5):538–549. | ||
Ratert N, Meyer HA, Jung M, et al. Reference mirnas for mirnaome analysis of urothelial carcinomas. PLoS One. 2012;7(6):e39309. | ||
Le MT, Teh C, Shyh-Chang N, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23(7):862–876. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.