Back to Journals » OncoTargets and Therapy » Volume 13
Minicircle DNA-Mediated CAR T Cells Targeting CD44 Suppressed Hepatocellular Carcinoma Both in vitro and in vivo
Authors Wang H ![]() , Ye X, Ju Y, Cai Z, Wang X
, Ye X, Ju Y, Cai Z, Wang X ![]() , Du P, Zhang M, Li Y, Cai J
, Du P, Zhang M, Li Y, Cai J
Received 31 January 2020
Accepted for publication 17 April 2020
Published 4 May 2020 Volume 2020:13 Pages 3703—3716
DOI https://doi.org/10.2147/OTT.S247836
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Hezhi Wang, 1 Xueshuai Ye, 1, 2 Yi Ju, 3 Ziqi Cai, 2 Xiaoxiao Wang, 4 Pingping Du, 2 Mengya Zhang, 2 Yang Li, 1 Jianhui Cai 1, 5
1Department of Surgery, Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China; 2Hebei Engineering Technology Research Center for Cell Therapy, Hebei HOFOY Biotech Corporation Ltd., Shijiazhuang, Hebei, People’s Republic of China; 3Department of Medicine, Medical College of Hebei University of Engineering, Handan, Hebei, People’s Republic of China; 4Department of Internal Medicine, Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China; 5Department of Surgery & Oncology, Hebei General Hospital, Shijiazhuang, Hebei, People’s Republic of China
Correspondence: Jianhui Cai
Department of Surgery & Oncology, Hebei General Hospital, 348 West Heping Road, Shijiazhuang, Hebei 050051, People’s Republic of China
Tel +86-13315124890
Fax +86-0311-85511699
Email [email protected]
Purpose: Based on the continuous exploration of solid tumor immunotherapy, we focused on hepatocellular carcinoma with a high level of morbidity and mortality. We confirm the stability of mcDNA-based CAR T cell generating platform, and investigate the antitumor activity of CD44-CAR T cells against hepatocellular carcinoma both in vitro and in vivo.
Materials and Methods: We fused anti-CD44 scFv structure with transmembrane domain and intracellular domain. Using a non-viral mcDNA vector to load CD44-CAR gene, then transfected the mcDNA-CD44-CAR into human T cells by electroporation. We exhibited the transfection efficacy of CAR T cells and the CD44 expression of tumor cell lines by flow cytometry. The antitumor efficacy of CD44-CAR T cells in vitro and in vivo was detected through CCK-8 and ELISA assays, and xenograft mouse models, respectively.
Results: We obtained mcDNA-CD44-CAR with a high level of density after repeated extraction and purification. The expression efficacy of CD44-CAR in T cells was more than 50% after seven days electroporation and the phenotype of CD44-CAR T cells was no difference compared with normal T cells. For CD44-positive hepatocellular carcinoma xenograft mice, CD44-CAR T cells had stronger tumor growth suppression compared to normal T and mock T cells. The same results occurred on the in vitro experiments including cytokine secretion and cytotoxicity assays. H&E staining graphs revealed that CD44-CAR T cells did not induce side effects in xenograft mice.
Conclusion: The strategy for generating CAR T cells targeting cancer stem cell antigens was efficient and concise. The mcDNA had superior transgene ability without virus-related adverse effects. CD44-CAR T cells had strong suppression capacity against hepatocellular carcinoma.
Keywords: cancer immunotherapy, chimeric antigen receptor, nonviral vector, cancer stem cell antigen, tumor suppression
Corrigendum for this paper has been published
Introduction
Hepatocellular carcinoma (HCC) is the majority type of primary liver cancer. Worldwide, primary liver cancer is the fourth leading cause of cancer- related mortality and the sixth most common cancer.1 Chronic liver disease and liver cirrhosis due to alcoholic liver disease, nonalcoholic fatty liver disease and chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection are the major causes of HCC.2 For patients with HCC, the 5-year survival is less than 16% and the overall survival (OV) is less than one year.3,4 Surgery, radiotherapy and chemotherapy as traditional methods cannot significantly improve life quality and prolong survival time.
With great success in the treatment of hematological malignancies, immunotherapy has become an attractive weapon against cancers.5,6 Many promising immuno-oncology strategies emerged, such as checkpoint inhibitors, dendritic cell (DC) vaccines, tumor infiltrating lymphocytes (TILs), cytokine induced killer (CIK) T cell receptor (TCR) T cells and chimeric antigen receptor (CAR) T cells.7 We focus on CAR T cells treatment because it has been proved as the most remarkable progress in cancer therapy by the American Society of Clinical Oncology.8 We have achieved well-pleasing outcomes on non-viral mcDNA-mediated CAR T cells treated solid tumors, like PSCA-CAR T cells against prostatic cancer and NKG2D–CART cells against colorectal cancer.9,10 CAR T cells generated by transferring the target gene (CAR cassette) into T cells consist of three main structures. An extracellular domain, single-chain variable fragment (scFv), specifically bind tumor-associated antigens (TAAs) through human leukocyte antigen (HLA)-independent pathway. A hinge and transmembrane domain allow the scFv scalable in all spatial directions. An intracellular domain, one or more costimulatory molecules, promotes T cell proliferation and releases antitumor cytokines.
However, virus-related risks and appropriate target antigens selection are still challenging for CAR T cells.11 Recently, the use of cancer stem cell (CSC) antigens as targets for CAR T cells has received increasing attention from researchers. Cancer stem cells possess the ability of self-renew, trigger tumor differentiation and metastasis, and play a key role in tumor initiation and immune regulation.12,13 We take CD44 as the target antigen to guide CAR T cells reach to HCC cells because it has been profoundly studied,14 and as a CSC antigen, CD44 is highly expressed in HCC.15 To eliminate virus-related side effects on transfection procedures, minicircle DNA (mcDNA) was used as a vector. The integrase ΦC31 removes the bacterial backbone by recognizing the recombination sites attB and attP at both ends to obtain non-viral mcDNA with efficient gene transfection.16,17 In this study, we generate CAR T cells targeting CSC antigens of HCC with non-viral mcDNA vectors. Five types of HCC cell lines, CD44-positive or CD44-negative expression, are used to demonstrate whether CD44-CAR T cells obviously suppress the growth of HCC both in vitro and in vivo.
Materials and Methods
Parental Plasmid Construction and mcDNA Production
We designed a third-generation of CD44-CAR structure which was consisted of CD44 scFv, human CD8α hinge and transmembrane domain (nucleotides 412–609, GenBank NM 001768.6), human CD28 molecule (nucleotides 538–660, GenBank NM 006139.3), human CD137 molecule (nucleotides 640–765, GenBank NM 001561.5) and human CD3ζ molecule (nucleotides 154–492, GenBank NM 198253.2). We humanized corresponding monoclonal antibody (mAb) to acquire CD44 scFv sequence. Then we synthesized (Detai Biologics, Nanjing, China) the CAR structure after incorporating EcoRI and BamHI sites at both ends and confirmed it by genetic sequencing (Sango Biotech, Shanghai, China). We cloned the CD44-CAR structure into the parental plasmid pMC.CMV-EasyTM (System Biosciences, CA, USA) and then transferred it into E. coli strain ZYCY10P3S2T (System Biosciences). The inducer L- (+)-arabinose (Sigma Chemical, MO, USA) was added into the bacterial growth medium to generate CD44-CAR mcDNA by recombining attB and attP. We extracted the CD44-CAR mcDNA with an Endo-Free Plasmid DNA Maxi Kit (Omega Bio-tek, GA, USA) and confirmed it via restriction analysis.
Generation of CD44-CAR T Cells
Peripheral blood mononuclear cells (PBMCs) were obtained from the Hebei Blood Center. All procedures were performed under the guidelines approved by Hebei Medical University. Primary human T cells were isolated from PBMCs and cultured in RPMI-1640 medium (Thermo Fisher Scientific, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Thermo Fisher Scientific) at 37°C in 5% CO2. The activating factor, including 500 U/mL IL-2, 1 μg/mL anti-CD3 mAb, 1 μg/mL anti-CD28 mAb, 1000 U/L IFN-γ and 10 U/mL IL-15 were added for T cell expanding. After T cells were isolated, we added IFN-γ immediately, added IL-2 and CD3/CD28 mAbs on day 1, added IL-15 on day 3 and day 7. Then 5×106 T cells were transfected on day 4 via electroporation with a 4D-NucleofectorTM system (Lonza, Cologne, Germany). According to the manufacturer’s instructions, we added 3 μg of CD44-CAR mcDNA and 100 μL of P3 Primary Cell Buffer (Lonza) into T cells suspension, selected EO-115 program for electroporating. The CD44-CAR T cells were generated, and the medium was changed to fresh every other day.
Cell Lines
The human HCC cell lines Hep3B2, HepG2, and PLC8024 were obtained from American Type Culture Collection (ATCC, VA, USA) and cultured in minimal essential medium (MEM, Thermo Fisher Scientific). The human HCC cell lines MHCC97H and SMMC-7721 were obtained from the Shanghai Institute of cell bank (Shanghai, China) and cultured in RPMI-1640 medium (Thermo Fisher Scientific, MA, USA). All medium was supplemented with 10% FBS (Thermo Fisher Scientific) and 1% penicillin-streptomycin (Thermo Fisher Scientific). All cells cultured at 37°C in 5% CO2.
Flow Cytometry
The CAR expression was detected with human CD44 PE-conjugated antibody (Abcam, MA, USA). The phenotype of T cells was detected with human CD3-PC5, CD4-FITC and CD8-PE, all these antibodies were obtained from BD Bioscience. CD44 protein expressed on HCC was detected with human CD44-PE (BioLegend, CA, USA) antibody; isotype control group was stained with IgG1-PE (Abcam). The fluorescence analysis was performed with a BD FACSCanto™ flow cytometry system (BD Bioscience, CA, USA), statistics were conducted in FlowJo software (FlowJo, OR, USA).
Western Blot
The total protein in T cells and tumor tissues was extracted by RIPA lysate containing protease inhibitor. The protein concentration was determined by BCA method (Thermo Fisher Scientific) following the manufacturer’s instructions. Proteins were separated on 12% SDS-PAGE gel and transferred to polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was blocked in 5% skim milk for 2 hours and then incubated with the primary antibody at 4°C overnights. After washing the PVDF membrane, it was incubated with the corresponding secondary antibody for 50 minutes at room temperature. Proteins were visualized on a gel imager using a SuperSignalTM West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific). The primary antibodies used are as follows: anti-CD44 (1:1000, Abcam), anti-β-actin (1:5000, Abcam) and anti-CD3ζ (1:5000, Abcam).
Cytotoxicity Assays
We respectively cocultured effector cells with five types of HCC cells at ratios of 1:1, 5:1, 10:1 and 20:1 in 96-well plates (Corning, NY, USA). Culture conditions were 100 μL of medium per well in 5% CO2 at 37°C for 18 hours. We used the Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan) to exhibit the absorbance at 450 nm by Microplate Reader (BIOBASE, Shandong, China). Cytotoxicity results were calculated by the following formula: specific lysis (%) = [1– (mixture cell experiment–medium control)/(target cell spontaneous–medium control)] ×100%.
Cytokine Release Assays
For the in vitro experiment, after coculturing effector cells with target cells at a ratio of 10:1 for 24 hours, we harvested supernatants. Then we measured the cytokine release levels of IL-2, IFN-γ and TNF-α using ELISA-kits (Thermo Fisher Scientific). For the in vivo experiment, we collected 100 μL peripheral blood of xenograft mice after effector cells injection as samples.
Xenograft Mouse Models
We purchased 5- to 6-week-old NOD/SCID female mice from Beijing Vital River Laboratory Animal Technology Co., Ltd. All animals were raised and treated in specific pathogen-free (SPF)-grade cages of the Clinical Research Center of Hebei General Hospital. All animal experiments were approved by the Hebei Medical University Animal Care and Use Committee. Animal care and related procedures were approved by the Animal Care and Management Committee of Hebei General Hospital (license number: SCXK2016-0006) and complied with the International Regulations for the Administration of Laboratory Animals. We respectively inoculated five types of HCC cells (Hep3B2, HepG2, MHCC97H, SMMC-7721 and PLC8024) into the subcutaneous back of 75 mice. Each type contains 15 mice, which we randomly divided into three groups (n=5). After tumor volume reached 100mm3, five mice in each group were respectively injected with 1×107 of normal T, mock T and CD44-CAR T cells on day 3 and day 10. The formula: V =1/2 (length ×width2) was used to calculate tumor volume. After mice were sacrificed, we measured tumor weight and took tumor graph.
Histopathological and Immunofluorescence Assays
After anesthesia, the mice were perfused with saline and paraformaldehyde at the apex of the heart. The hearts, livers, lungs, brains and stomachs were taken out quickly and stored in paraformaldehyde fixative at 4 °C. After 24 hours of fixation, all tissues were embedded in paraffin and cut into 0.5μm thick sections. For hematoxylin and eosin (H&E) staining, paraffin sections were first dewaxed and stained with hematoxylin. Sections were then dehydrated in an ethanol gradient and stained with eosin. Finally, the sections were sealed and observed and imaged under an optical microscope (Tokyo, Japan, Nikon). For immunofluorescence staining, dewaxed the paraffin sections and placed them in the antigen retrieval solution (pH 8.0). Added AutoFluo Quencher to the sections and incubated with BSA for 30 minutes for serum blocking. The primary antibody was added dropwise to the sections and incubated at 4 ° C overnight. The sections were washed with PBS (pH7.4) and then incubated with secondary antibody for 50 minutes. Sections were incubated with 4ʹ, 6-diphenyl-2-phenylindole (DAPI) for 10 minutes to restain cell nuclei. Observed sections and acquired images on the fluorescence microscope (Tokyo, Japan, Nikon). The primary antibodies used are as follows: anti-CD44 (1:1000, Abcam) and anti-CD3 (1:200, Abcam).
Statistical Analysis
All data were mean ± SDs for statistical description. Statistical analysis was performed by GraphPad Prism 8.0 (GraphPad Software, San Diego, CA). Data were analyzed statistically by Student’s t-test, one-way ANOVA followed by Tukey’s post hoc test, and two-way repeated-measures ANOVA followed by Bonferroni’s post hoc test. Survival curves were compared by the log-rank (Mantel-Cox) test. The difference of P < 0.05 was considered statistically significant.
Results
Preparation of CD44-CAR mcDNA and Electroporation of Human T Cells
The humanized anti-CD44 scFv was synthesized according to previous research18 and linked to the third generation of CAR structure (Figure 1A). We cloned the anti-CD44 CAR structure into a parental plasmid and named as pMC.CMV-CD44-CAR. The recombinase ΦC31 separated pMC.CMV-CD44-CAR by mediating irreversible recombination at specific recognition sites of attB and attP. Then, the inducer L-arabinose was used for endonuclease reaction and the CD44-CAR mcDNA was successfully prepared. The bacterial backbone containing kanamycin was degraded (Figure 1B).
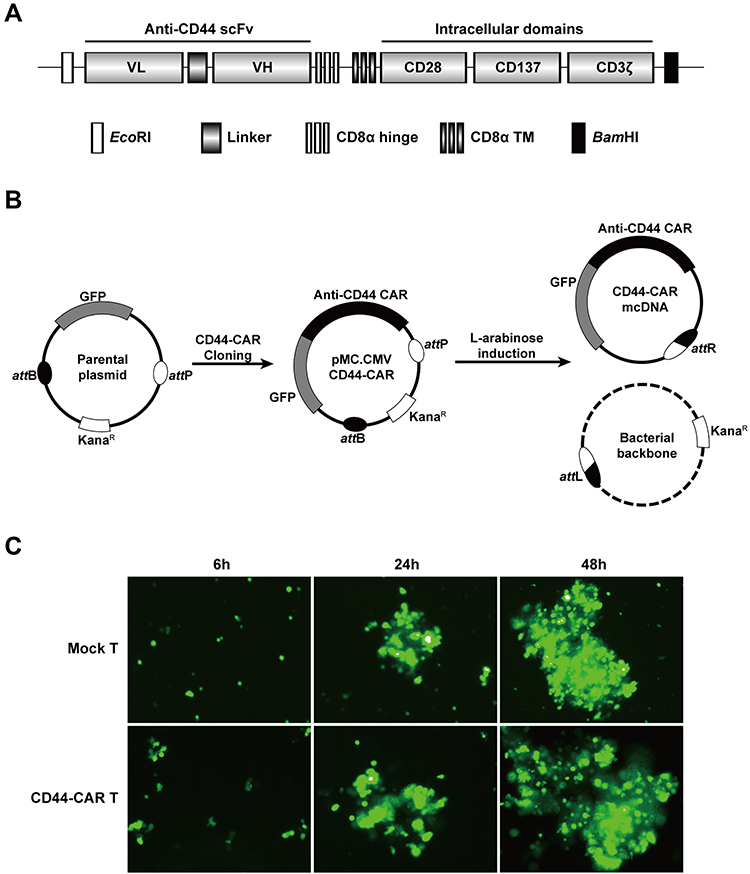 |
Figure 1 Construction of mcDNA and CD44-CAR T cells. (A) Schematic representation of anti-CD44 CAR structure. (B) Schematic diagram of CD44-CAR mcDNA generation. Anti-CD44 scFv was cloned into the parental plasmid to prepare pMC.CMV-CD44-CAR. L-arabinose was added to induce site-specific recombination. Bacterial backbone was digested for degradation and CD44-CAR mcDNA was generated. (C) Transfection efficacy demonstrated by fluorescence microscopy images within 48h at 400× magnification. |
We isolated human T cells from PBMCs and took 5×106 cells for each transfection. We obtained high-purity CD44-CAR mcDNAs (about 800ng/μL) after repeated extraction, and transfected them into human T cells via electroporation system. The products were CD44-CAR T cells. On the same conditions, we generated mock T cells by transfecting control plasmids containing GFP cassettes. Since both CD44-CAR T cells and mock T cells had GFP sequences, we evaluated transfection efficacy by observing the level of green fluorescence. The time points of the demonstration were set to 6 hours, 24 hours and 48 hours after transfection (Figure 1C). Because of CD44-CAR T cells had similar level of green fluorescence with mock T cells, the results of transfection by electroporation is preliminary satisfactory.
Identification and Proliferation of CD44-CAR T Cells
To illustrated the expression efficacy, we detected the GFP and the CD44-CAR expression on mock T and CD44-CAR T cells seven days after transfection. Normal T cells were used for control groups. Flow cytometry showed that by the FITC channel, the expression rate of GFP on mock T cells is 77.6% and on CD44-CAR T cells is 58.7%, by the PE channel, the expression rate of CD44-CAR on mock T cells is 4.51% and on CD44-CAR T cells is 54.2% (Figure 2A). For further demonstration, we repeated the above process three times and presented the statistics (Figure 2B). We added activating actors in culture medium for T cell proliferation (Described in materials and methods-Generation of CD44-CAR T Cells). The number of normal T, mock T and CD44-CAR T cells was respectively expanded 65, 60 and 50 times on day 14 (Figure 2C). We took 2×107 of each effector cells for protein extraction. Western blot analysis showed that the protein of exogenous CD3ζ was detected on 55kDa only in CD44-CAR T cells and the protein of endogenous CD3ζ was detected on 15kDa in all the three groups (Figure 2D). To summarized, the transfection strategy of CD44-CAR T cells, by electroporated non-viral mcDNA vectors, achieved an expression rate of more than 50% while expanding the number of cells to 50 times. The expression of exogenous CD3ζ protein confirmed the successful CAR transfection at the molecular level.
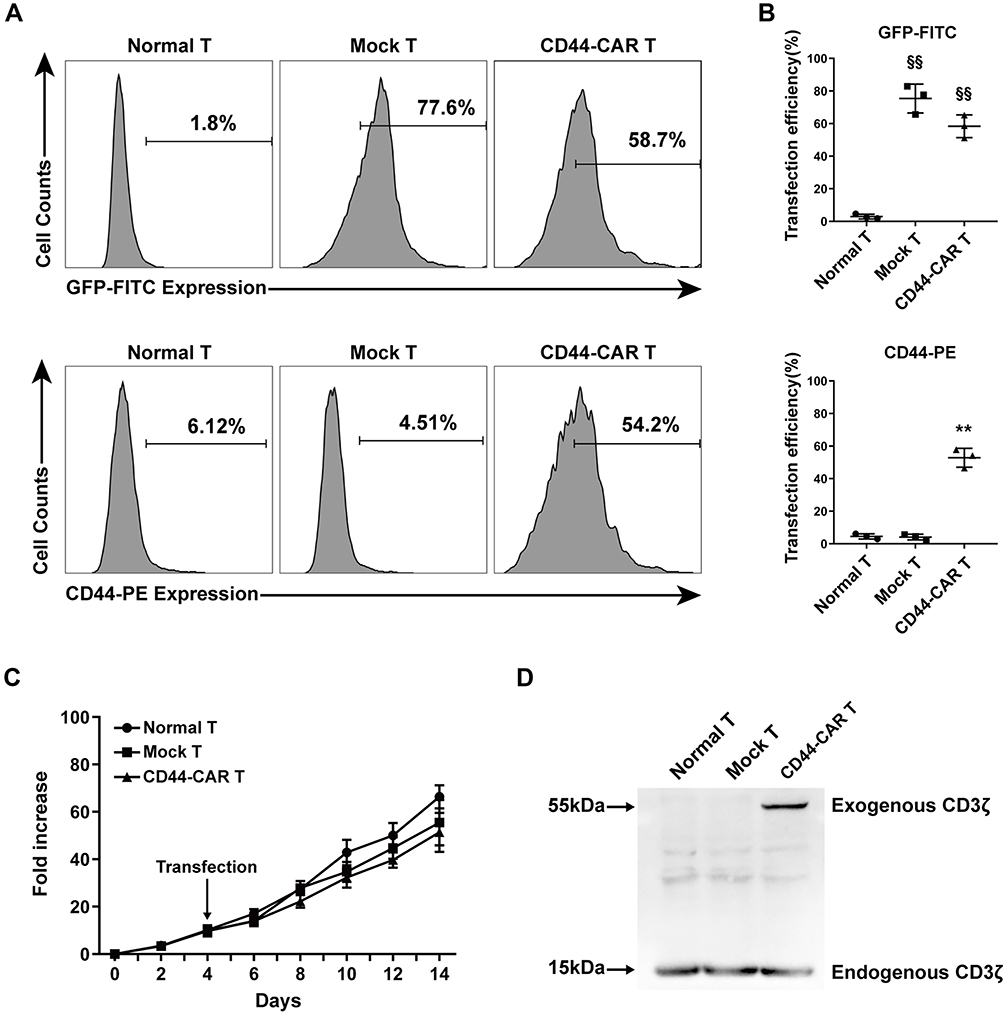 |
Figure 2 CD44-CAR expression analysis. (A) Flow cytometry showed CD44-CAR expressed in human T cells seven days after transfection. Normal T cells were used as control groups. Mock T cells were generated by electroporating plasmid encoding GFP under the same condition of CD44-CAR T generation. (B) Transfection efficiency of GFP gene or CD44-CAR in normal T, mock T and CD44-CAR T cells. Statistics are presented as the means ± SDs, n = 3 per group, §§ P<0.05 vs normal T group; ** P<0.05 vs normal T and mock T groups. (C) Fold expansion of normal T, mock T and CD44-CAR T cells, all cells were cultured in vitro and activated by anti-CD3/CD28 mAb, IL-2/IL-15 and IFN-γ. Statistics are presented as the means ± SDs. n = 3 per group. (D) The CAR protein expression was represented by Western blot analysis. The exogenous CD3ζ was detected to illustrate CD44-CAR expression. |
Phenotypic Characterization Detection of CD44-CAR T Cells
We used flow cytometry to detect CD3, CD4 and CD8 expression on CD44-CAR T cells, to know whether electroporation altered the phenotype of T cells. The single-staining method was used to detected CD3 expression by flow cytometry and the result showed similar expression rates on normal T and CD44-CAR T cells (Figure 3A). We obtained statistical data after three repeated detections (Figure 3B). For CD4/CD8 expression, we used double-staining method for flow cytometry. It showed similar expression rates on normal T and CD44-CAR T cells as well (Figure 3C). Statistics were presented for n=3 (Figure 3D). All statistics showed no significant difference, which indicated the phenotype of T cells did not alter after electroporation.
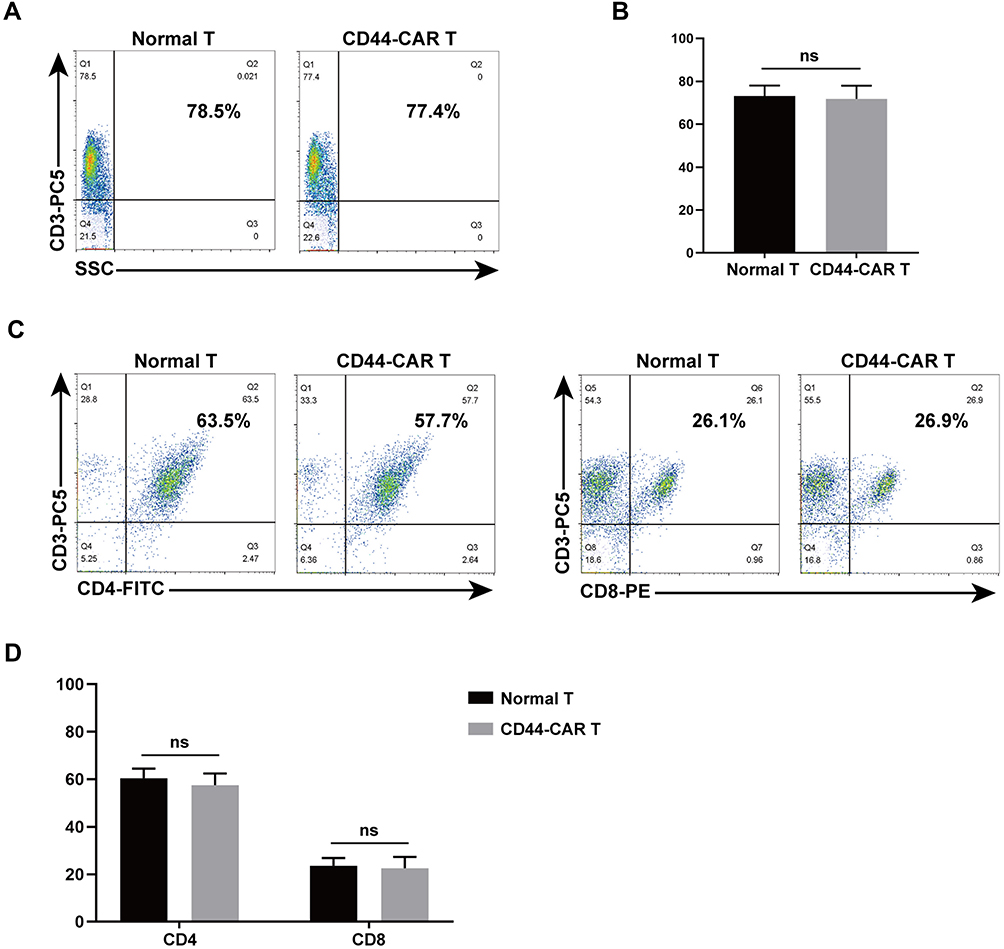 |
Figure 3 Phenotype analysis of normal T and CD44-CAR T cells. (A) After transfection, flow cytometry showed CD3 expression in normal T and CD44-CAR T cells on day 7. (B) No difference in normal T and CD44-CAR T cells of CD3 expression. Statistics are presented as the means ± SDs, n = 3 per group. ns, not significant. (C) Further analysis from CD3 positive cell populations, flow cytometry showed CD4 and CD8 expression in normal T and CD44-CAR T cells on day 7. (D) CD44-CAR T cells showed similar expression levels of CD4 and CD8 compared to normal T cells. Statistics are presented as the means ± SDs, n = 3 per group. ns, not significant. |
The in vitro Cytotoxicity of CD44-CAR T Cells Against HCC Cells
We selected five types of HCC cell lines for further experiments. First, we detected the expression of CD44 antigen on these cell lines by flow cytometry. The CD44 expression rate of Hep3B2, MHCC97H, SMMC-7721, HepG2 and PLC8024 was 79.5%, 97.6%, 88.1%, 9.5% and 3.7%, respectively (Figure 4A). Statistical data were obtained after repeating the detection of CD44 expression for three times (Figure 4B). Next, we extracted proteins from five types of tumor cells and detected them by Western blot. The results showed that Hep3B2, MHCC97H and SMMC-7721 were positively detected at 81kDa of CD44 protein, while HepG2 and PLC8024 were negatively detected (Figure 4C). Therefore, we concluded that CD44-positive cell lines were Hep3B2, MHCC97H and SMMC-7721; CD44-negative cell lines were HepG2 and PLC8024.
 |
Figure 4 CD44 expression in HCC cell lines and in vitro antitumor activity of CD44-CAR T cells. (A) Flow cytometry showed CD44 antigen expressed on tumor cell surface. The PE-conjugated anti-CD44 antibody used to stain five types of HCC cell lines and the anti-IgG1 antibody stained cells used as control groups. (B) Statistical analysis of CD44 expression on five types of HCC cell lines by flow cytometry. (C) Western blot analysis of CD44 protein expression on tumor cells. Anti-β-actin antibody used to stain control groups. (D) Cytotoxic activity analysis of CD44-CAR T cells. Five types of HCC cells cocultured with normal T, mock T and CD44-CAR T cells in increasing ratios (1:1, 5:1, 10:1 and 20:1) for 18 hours and CCK-8 assay used for detecting. (E) In vitro cytokine secretion (IL-2, IFN-γ and TNF-α) of CD44-CAR T cells analysis. Effector cells cocultured with target cells at a 10:1 ratio for 24 hours and ELISA assay used for detecting. All statistics are presented as the means ± SDs, n = 3 per group, ## P<0.05 vs HepG2 and PLC8024 groups; ** P<0.05 vs normal T and mock T groups. |
To evaluate the in vitro cytotoxic activity of CD44-CAR T cells, we cocultured normal T, mock T and CD44-CAR T cells with five types of HCC cells separately (Described in materials and methods-Cytotoxicity Assays). CD44-CAR T cells showed vigorous cytotoxic activity against Hep3B2, MHCC97H and SMMC-7721 but weak against HepG2 and PLC8024. Normal T and mock T cells had no significance on five types of tumor cell lines (Figure 4D). For the in vitro cytokine release assays, compared with other groups, CD44-CAR T cells secreted higher levels of IL-2, IFN-γ and TNF-α after cocultured with CD44-positive cell lines (Figure 4E). The above results demonstrated that CD44-CAR T cells initiated signaling pathways and activated costimulatory molecules through targeting and combining specific antigens.
Antitumor Activity of CD44-CAR T Cells Against CD44 Positive HCC in vivo
We inoculated 1×106 HCC cells into the subcutaneous back of mice and successfully obtained HCC xenograft mice. The xenograft mice received normal T, mock T and CD44-CAR T cells intravenous injection for treatment (Described in materials and methods-Xenograft Mouse Models). After CD44-CAR T cells treated mice were sacrificed, we stained tumor sections with anti-CD44 and anti-CD3 antibodies for immunofluorescence. Results indicated that, for Hep3B2, MHCC97H and SMMC-7721 cell lines, tumor tissues were positively-stained by CD44 and had rich infiltration by CD44-CAR T cells. But for HepG2 and PLC8024 cell lines, tumor tissues were negatively-stained by CD44 and had rare infiltration by CD44-CAR T cells (Figure 5A). The tumor volumes of Hep3B2, MHCC97H and SMMC-7721 xenograft mice treated with CD44-CAR T cells were significantly decreased than those with normal T and mock T cells treatments. While the tumor volumes of HepG2 and PLC8024 xenograft mice with normal T, mock T and CD44-CAR T cells treatments were no significant difference (Figure 5B). We detected the in vivo cytokine secretion on day 15 after the tumor volume reached 100mm3. The levels of IL-2, IFN-γ and TNF-α in Hep3B2, MHCC97H and SMMC-7721 xenograft mice were significantly higher than other groups (Figure 5C). CD44-CAR T cells exhibited strong in vivo antitumor activity on CD44-positive xenograft mice.
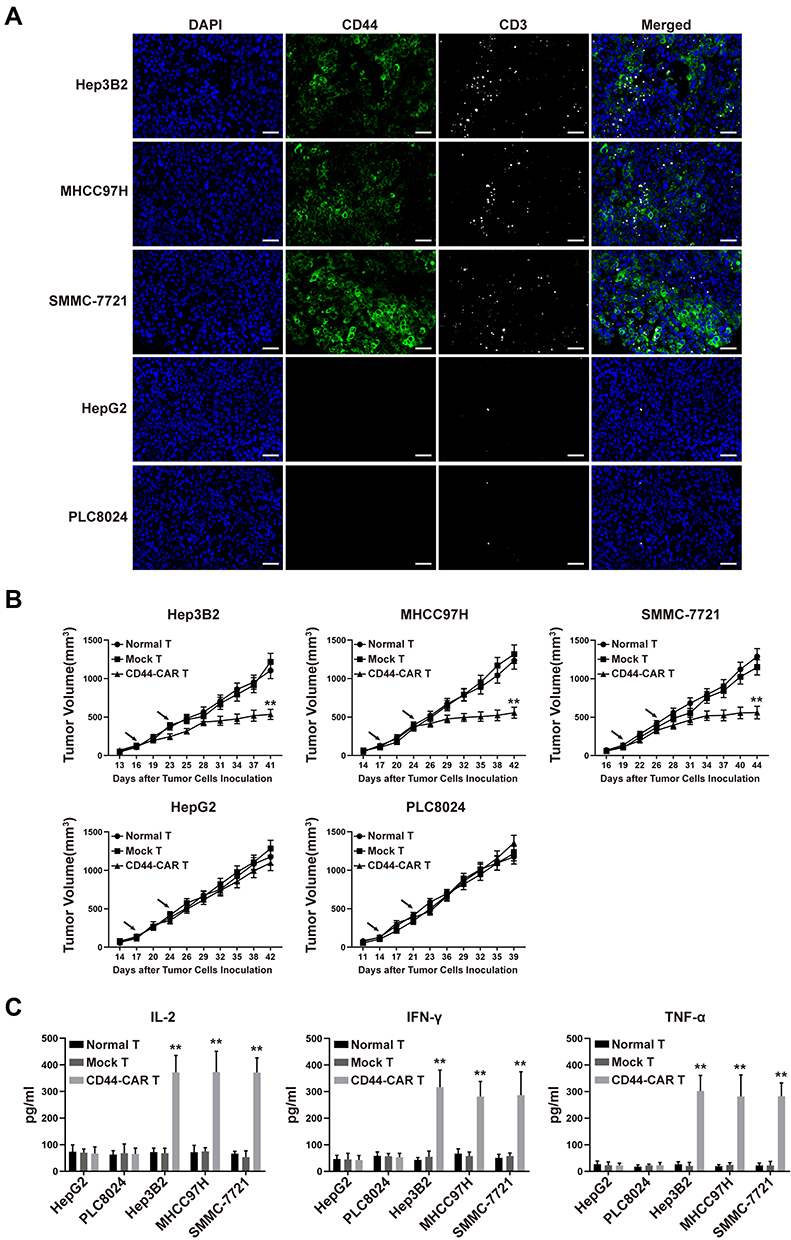 |
Figure 5 In vivo antitumor activity of CD44-CAR T cells against HCC. (A) HCC xenograft mice were given 1×107 effector cells treatment. After mice were sacrificed, tumor tissues of CD44-CAR T cells treated mice were harvested. Immunofluorescence images showed different CD44 expression and T cells infiltration (anti-CD3 staining) in five types of HCC cells. Scale bar = 50 μm. (B) After the tumor volume of xenograft mice reached about 100mm3, effector cells were injected twice (arrows marked). The volumes of xenograft mice tumors were recorded for statistical analysis. (C) One hundred μL of peripheral blood from treated mice on day 7 were obtained for the in vivo cytokine secretion assay according to the ELISA kit instrument. All statistics are presented as the means ± SDs, n = 5 for each group, ** P<0.05 vs normal T and mock T groups. |
Xenograft Mice with CD44-CAR T Cells Treatment Demonstrated Improved Vitality of Life
Each type of HCC cell inoculated into fifteen mice and randomly divided into three groups (n=5). Each group received normal T, mock T and CD44-CAR T cells treatments, respectively. After xenograft mice were sacrificed, we separated intact tumor tissues. We also fixed vital organs (heart, liver, lung, cerebrum and stomach) appropriately for the following steps. We photographed tumor tissues of xenograft mice to visualize the tumor size. The tumor sizes of Hep3B2, MHCC97H and SMMC-7721 xenograft mice with CD44-CAR T cells treatments were reduced and did not grow up before sacrificed (Figure 6A). We recorded the survival time of each mouse and exhibited survival curves of xenograft mice with CD44-CAR T treatments. There were respective three, two and four of Hep3B2, MHCC97H and SMMC-7721 xenograft mice survived more than 120 days. However, HepG2 and PLC8024 xenograft mice treated with CD44-CAR T cells all died before day 75 (Figure 6B). We measured the tumor weight of each mouse and made comparison (Figure 6C). CD44-CAR T cells significantly suppressed CD44-positive tumor growth in mice. We stained H&E with the vital organs of xenograft mice that were previously preserved. The organs of xenograft mice treated with CD44-CAR T cells were not different from normal T and mock T cells treated groups. The H&E staining photographs of Hep3B2 xenograft mice were exhibited only (Figure 6D). CD44-CAR T cells showed no side effects on xenograft mice after treatment.
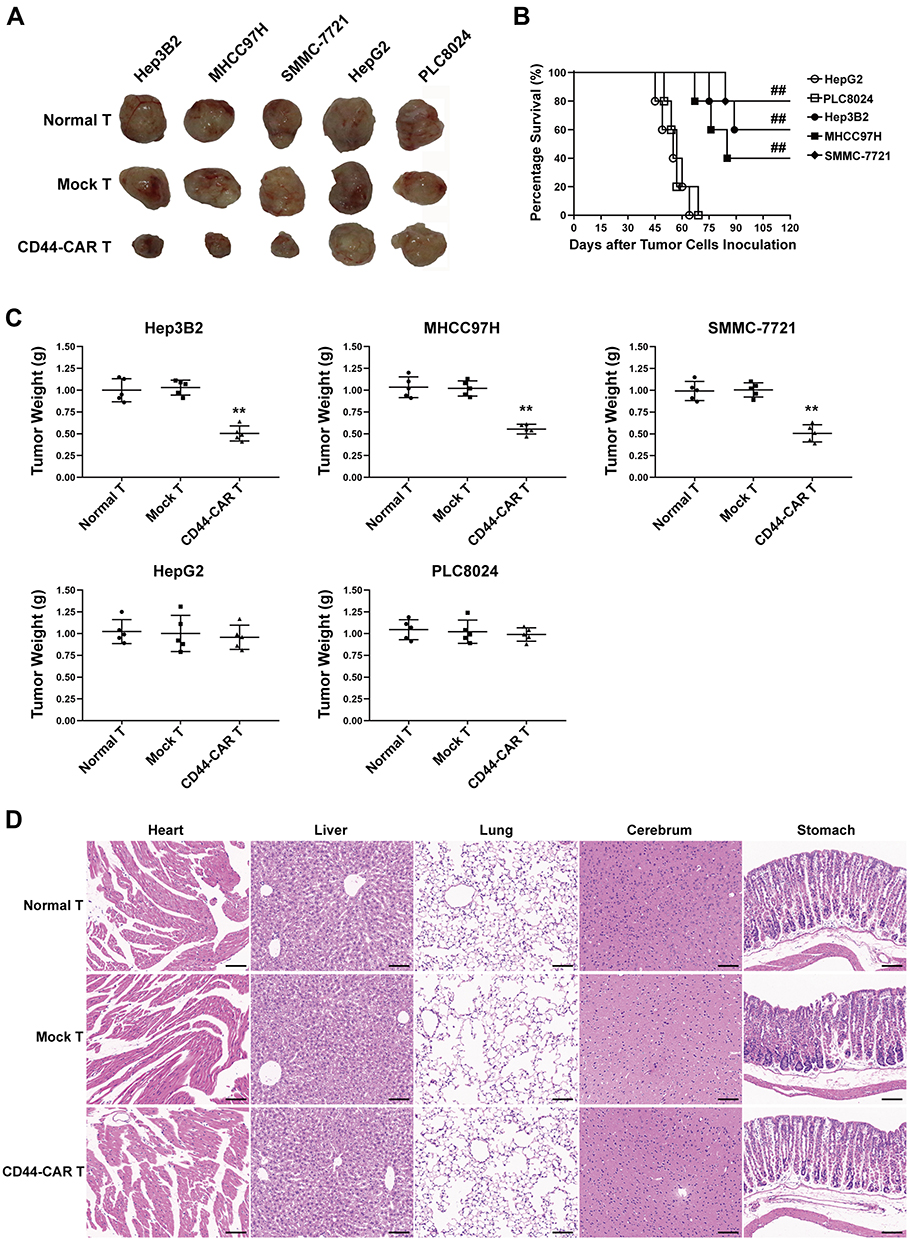 |
Figure 6 Tumor suppression capacity and safety of CD44-CAR T cells. (A) Tumor tissue images of sacrificed mice with different effector cell treatments had exhibited. (B) Survival time of xenograft mice with five types of HCC inoculation. Statistics were compared by the log-rank (Mantel-Cox) test, ## P<0.05 vs HepG2 and PLC8024 groups. (C) Statistical analysis showed the tumor weight of sacrificed mice. Statistics are presented as the means ± SDs, n = 5 for each group, ** P<0.05 vs normal T and mock T groups. (D) After sacrificing, the important organs of xenograft mice were obtained and cut into sections for H&E staining. Images of normal T, mock T and CD44-CAR T cells groups of sections were exhibited. Scale bar=100 µm. |
Discussion
Adoptive T cell immunotherapy has become a promising strategy for hematological malignancy,8,19 of which CD19-CAR T cell therapy is the most representative for acute lymphoblastic leukemia.20,21 After the extracellular domain, scFv, combines corresponding TAAs, signal pathways of CAR T cells are activated. Then, the intracellular domain of CAR T cells, costimulatory molecules, releases cytokines (IL-2, IFN-γ, TNF-α and so on) and cytotoxic granules to inhibit tumor progression.
The above mechanism has proven to be well-performed on certain solid tumors treatments,22 such as CEA-CAR T for colorectal cancer,23 EGFR/EGFRvIII-CAR T for Glioblastoma,24 GD2-CAR T for Neuroblastoma,25 MUC1-CAR T for triple-negative breast cancer26 and GPC3-CAR T for hepatocellular carcinoma.27 However, there is an obstruction persists on all efforts of CAR T cells against solid tumors. It is the restriction of tumor microenvironment (TME). The TME maintaining the state of immunosuppression attributes to three aspects: (1) network blood vessels that provide nutrition to tumor cells; (2) physical barriers composed of extracellular matrixes (ECMs); and (3) chemical barriers composed of inflammatory cytokines and tumor-derived chemokines.28–30 CSCs are a population of cells that exist on and inside the tumor, which keep the low oxygen of TME. Poor prognosis and immunotherapy resistance of tumors are closely related to hypoxic of TME.31,32 Decrease of mitochondria and low of oxidative phosphorylation caused by the hypoxic microenvironment enable tumor cells to have conditions that are more conducive to initiation, differentiation and metastasis.33,34 In this study, we engineered T cells to combine with CSC antigens of HCC. The outcomes were a large number of CAR T cells accumulated to the HCC cell surface and inside through the location of CSC antigens. CSC antigens activated the signaling pathway of CAR T cells, then the antitumor activity of CAR T cells strongly performed. After combining CSC antigens, CAR T cells infiltrated abundant other tumor cells and released heparanase to degrade components of the ECMs.35 The CSC-related potent antitumor responses proved to be critical factors in breaking the immunosuppression of hypoxic TME.
CD44, a single span transmembrane glycoprotein with multi-functions,36,37 encoded by the CD44 gene containing 20 exons, which located on chromosome 11 in humans.38,39 CD44 is highly expressed in HCC15 and is considered as a molecular marker of CSCs.40 Hyaluronic acid (HA), expressed by stromal and tumor cells and abundantly existed in ECM which is the main component of TME, was the main ligand of CD44.41 The humanized anti-CD44 scFv, derived from anti-CD44 mAbs (Figure 1A), was the exocellular domain of the third-generation CD44-CAR T cells we have prepared. It specifically combined with CD44 antigens and transited activated signals into T cells by the CAR structure, then cytokines and cytotoxic granules were released. This CD44-based antitumor mechanism pierced through the obstructs built by the TME. The superior antitumor activity of CD44-CAR T cells against CD44-positive HCC cells was exhibited both in vitro and in vivo. The electrical signals activated by CSC antigens conducted into T cells, increasing the levels of cytokines secretion and thus causing abundant T cells to expand and enhanced cytotoxicity (Figure 4D). After intravenously injecting CD44-CAR T cells into xenograft mice, different from TCR recognition system, CAR-based signal transduction pathway developed tumor suppressive effects in mice with an MHC-independent manner.
Compared with viral-based vectors, mcDNA is an ultimate small size nonviral DNA vector without a deleterious bacterial backbone.42 The nonviral mcDNA-mediated CAR T cell generation system we used in this study had advantages of improved transfection efficacy, prolonged transgene expression, uncomplicated extraction process and lower cost. After continuous attempts, we finalized the mcDNA extraction process using commercial kits for repeated purification to obtain products with clinical-grade purity. The third-generation of mcDNA-CD44-CAR structure (Figure 1A and 1B) was constructed and was transfected into human T cells by electroporation, then CD44-CAR T cells were successfully generated. We evaluated the transfection efficacy through green fluorescence exhibition (Figure 1C) and measured the expression efficacy through CD44-CAR detection. Until one week after transfection, the CD44-CAR expression efficacy detected by flow cytometry was 58.7% (Figure 2A). The HCC xenograft mice model we established received CD44-CAR T cells treatment and normal T cells injection. We compared vital organs in these two groups of mice by H&E staining and found no difference (Figure 6D). Before sacrificed, these two groups of alive mice were no hyperthermia, fatigue, anorexia, vomiting, diarrhea, irritability, twitch, or other severe cytokine release syndrome.
The mcDNA-mediated CAR T cells production system we established acquired satisfactory transfection efficiency while abandoning virus-related adverse effects. CD44-CAR T cells aggregated abundant antitumor factors to the CD44-positive HCC surroundings, and showed strong signal transmission capacity and cytokine secretion function both in vivo and in vitro. The CD44-positive HCC xenograft mice treated with CD44-CAR T cells received prolonged survival time and pronounced tumor reduction, which highlighted the promising prospect of mcDNA vectors and CSC-targeted CAR T cells for clinical application.
Conclusion
Our findings demonstrated that CD44-CAR-mcDNA was extracted with easier processes and transfected into T cells with higher efficiency. The results of in vitro and in vivo experiments showed the enhanced antitumor ability of CD44-CAR T cells, mainly because targeting CSC antigens produced TME breakthrough effects.
Abbreviations
HCC, hepatocellular carcinoma; CAR, chimeric antigen receptor; scFv, single-chain variable fragment; DC, dendritic cell; TILs, tumor infiltrating lymphocytes; CIK, cytokine induced killer; TCR, T cell receptor; TAAs, tumor-associated antigens; HLA, human leukocyte antigen; CSC, cancer stem cell; mcDNA, minicircle DNA; mAb, monoclonal antibody; PBMC, peripheral blood mononuclear cell; TME, tumor microenvironment; ECMs, extracellular matrixes.
Acknowledgments
The authors wish to thank Prof. Yi Guo (College of Life Sciences, Hebei Normal University) for technical support, Prof. Yunli Yan (Department of Cell Biology, Hebei Medical University) and Dr. Yanling Wang (Department of Cell Biology, Hebei Medical University) for facility support. This work was supported by the Graduate Innovation Fund of Hebei (grant numbers CXZZBS2019120).
Disclosure
The authors declare no potential conflicts of interest.
References
1. Villanueva A, Longo DL. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–1462.
2. Dawkins J, Webster RM. The hepatocellular carcinoma market. Nat Rev Drug Discov. 2018;18(1):13–14. doi:10.1038/nrd.2018.146
3. Galle PR, Forner A, Llovet JM, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236. doi:10.1016/j.jhep.2018.03.019
4. Kim YS, Shin SW. Hepatocellular carcinoma. N Engl J Med. 2019;381(1):e2.
5. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Cancer Immunother. 2018;359(6382):1361–1365.
6. Lv J, Zhao R, Wu D, et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J Hematol Oncol. 2019;12(1):18. doi:10.1186/s13045-019-0704-y
7. Zhao Z, Zheng L, Chen W, Weng W, Song J, Ji J. Delivery strategies of cancer immunotherapy: recent advances and future perspectives. J Hematol Oncol. 2019;12(1):126. doi:10.1186/s13045-019-0817-3
8. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the american society of clinical oncology. J clin oncol. 2018;36(10):1020–1044. doi:10.1200/JCO.2017.77.0446
9. Han J, Gao F, Geng S, et al. Minicircle DNA-engineered CAR T cells suppressed tumor growth in mice. Mol Cancer Ther. 2020;19(1):178–186. doi:10.1158/1535-7163.MCT-19-0204
10. Deng X, Gao F, Li N, et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res. 2019;9(5):945–958.
11. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. doi:10.1016/j.immuni.2019.12.011
12. Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66(19):9339–9344. doi:10.1158/0008-5472.CAN-06-3126
13. Ruiu R, Tarone L, Rolih V, et al. Cancer stem cell immunology and immunotherapy: harnessing the immune system against cancer’s source. 2019;164:119–188.
14. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. doi:10.1038/nrc3023
15. Hou Y, Zou Q, Ge R, Shen F, Wang Y. The critical role of CD133+CD44+/high tumor cells in hematogenous metastasis of liver cancers. Cell Res. 2011;22(1):259–272. doi:10.1038/cr.2011.139
16. Chen Z-Y, He C-Y, Ehrhardt A, Kay MA. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003;8(3):495–500. doi:10.1016/S1525-0016(03)00168-0
17. Zhao J, Huang P, Wang Z, et al. Synthesis of amphiphilic poly(β-amino ester) for efficiently minicircle DNA delivery in vivo. ACS Appl Mater Interfaces. 2016;8(30):19284–19290. doi:10.1021/acsami.6b04412
18. Wei X, Ma H ScFv against cancer stem cell specific protein CD44 and its applications. Chinese Patent. No. 103288958.
19. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
20. Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138–177ra138. doi:10.1126/scitranslmed.3005930
21. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase 1 dose-escalation trial. The Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
22. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Controlled Release. 2020;319:246–261. doi:10.1016/j.jconrel.2019.12.047
23. Zhang C, Wang Z, Yang Z, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther. 2017;25(5):1248–1258. doi:10.1016/j.ymthe.2017.03.010
24. Jiang H, Gao H, Kong J, et al. Selective targeting of glioblastoma with EGFRvIII/EGFR bitargeted chimeric antigen Receptor T cell. Cancer Immunol Res. 2018;6(11):1314–1326. doi:10.1158/2326-6066.CIR-18-0044
25. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. 2019;25(9):2915–2924. doi:10.1158/1078-0432.CCR-18-1811
26. Zhou R, Yazdanifar M, Roy LD, et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol. 2019;10:1149. doi:10.3389/fimmu.2019.01149
27. Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–6428. doi:10.1158/1078-0432.CCR-14-1170
28. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128. doi:10.3389/fimmu.2019.00128
29. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15(11):669–682. doi:10.1038/nri3902
30. Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm. 2011;8(3):635–641. doi:10.1021/mp1004228
31. van den Beucken T, Koch E, Chu K, et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat Commun. 2014;5(1):5203. doi:10.1038/ncomms6203
32. Jeong H, Kim S, Hong B-J, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79(4):795–806. doi:10.1158/0008-5472.CAN-18-2545
33. Najafi M, Farhood B, Mortezaee K, Kharazinejad E, Majidpoor J, Ahadi R. Hypoxia in solid tumors: a key promoter of cancer stem cell (CSC) resistance. J Cancer Res Clin Oncol. 2019;146(1):19–31. doi:10.1007/s00432-019-03080-1
34. Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15(6):501–513. doi:10.1016/j.ccr.2009.03.018
35. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. doi:10.1038/nm.3833
36. Xu H, Tian Y, Yuan X, et al. The role of CD44 in epithelial–mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–3792. doi:10.2147/OTT.S95470
37. Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11(1):64. doi:10.1186/s13045-018-0605-5
38. Rodrigo JP, Domínguez F, Alvarez C, González MV, Herrero A, Suárez C. Clinicopathologic significance of expression of CD44s and CD44v6 isoforms in squamous cell carcinoma of the supraglottic larynx. Am J Clin Pathol. 2002;118(1):67–72. doi:10.1309/F50H-6MLG-R7LM-2XFT
39. Spring FA, Dalchau R, Daniels GL, et al. The Ina and Inb blood group antigens are located on a glycoprotein of 80,000 MW (the CDw44 glycoprotein) whose expression is influenced by the In(Lu) gene. Immunology. 1988;64(1):37–43.
40. Yin T, Wang G, He S, Liu Q, Sun J, Wang Y. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41–45. doi:10.1016/j.cellimm.2015.11.009
41. Banerjee S, Modi S, McGinn O, et al. Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer. Clin Cancer Res. 2015;22(2):415–425. doi:10.1158/1078-0432.CCR-15-1155
42. Darquet AM, Cameron B, Wils P, Scherman D, Crouzet J. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther. 1997;4(12):1341–1349. doi:10.1038/sj.gt.3300540
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.