")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Milnacipran treatment and potential biomarkers in depressed patients following an initial SSRI treatment failure: a prospective, open-label, 24-week study
Authors Hashimoto T, Sakurai D, Oda Y, Hasegawa T, Kanahara N , Sasaki T, Komatsu H, Takahashi J, Oiwa T, Sekine Y, Watanabe H , Iyo M
Received 25 August 2015
Accepted for publication 9 November 2015
Published 10 December 2015 Volume 2015:11 Pages 3031—3040
DOI https://doi.org/10.2147/NDT.S95067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Tasuku Hashimoto,1,2 Daiji Sakurai,1 Yasunori Oda,1 Tadashi Hasegawa,3 Nobuhisa Kanahara,4 Tsuyoshi Sasaki,3 Hideki Komatsu,3,5 Junpei Takahashi,1,5 Takahiro Oiwa,6 Yoshimoto Sekine,4,5 Hiroyuki Watanabe,1 Masaomi Iyo1,3
1Department of Psychiatry, Chiba University Graduate School of Medicine, 2Sodegaura Satsukidai Hospital, 3Department of Psychiatry, Chiba University Hospital, 4Division of Medical Treatment and Rehabilitation, Centre for Forensic Mental Health, Chiba University, 5Choshi Kokoro Clinic, 6Mobara Shinkeika Hospital, Chiba, Japan
Background: We assessed the effect of switching patients with major depressive disorder to milnacipran following an initial selective serotonin reuptake inhibitor treatment failure, and explored potential biomarkers in their blood.
Methods: We conducted a prospective, open-label, 24-week trial. Depression was assessed with the 17-item Hamilton Depression Rating Scale. Patients showing a ≥50% reduction in Hamilton Depression Rating Scale scores from baseline to final visit were considered responders. Regarding adverse effects (AEs), moderate-to-severe AEs were specifically identified as effects that required any medical treatment or that induced treatment withdrawals. We also measured blood levels of various molecules including inflammatory cytokines.
Results: Of the 30 participants who enrolled, 17 completed this study. The responder rate was 30% (n=10). Baseline serum levels of interleukin-6 (Z=-2.155; P=0.031) and interleukin-8 (Z=-2.616; P=0.009) were significantly higher when moderate-to-severe AEs were present (n=13 patients with moderate-to-severe AEs). Serum levels of macrophage inflammatory protein-1β showed a significant continuous decrease from the baseline level (Friedman’s test: χ2=23.9, df=4, P<0.001) only in non-responders.
Conclusion: These results demonstrate that serum levels of interleukin-6, interleukin-8, and macrophage inflammatory protein-1β as potential blood biomarkers could be utilized to identify the responsiveness of patients to serotonin and norepinephrine reuptake inhibitor like milnacipran, or to identify those patients who may experience AEs strong enough to warrant discontinuation of treatment.
Keywords: major depressive disorder, milnacipran, biomarkers, adverse effects, cytokines
Introduction
Antidepressants are effective treatments for major depressive disorder (MDD). Selective serotonin reuptake inhibitors (SSRIs) are widely used to treat patients with MDD, and evidence suggests that several SSRIs are good first-line drugs for treating a current depressive episode.1,2 However, only approximately 30% of patients with MDD treated with the typical SSRI citalopram achieved symptomatic remission with the initial pharmacological treatment.3 Thus, it is extremely important to investigate alternative treatment strategies, such as switching to another antidepressant, for MDD patients for whom initial SSRI treatment failed.
Milnacipran is a serotonin and norepinephrine reuptake inhibitor (SNRI) that is used for the treatment of MDD in a number of countries. As the name suggests, milnacipran inhibits both serotonin and norepinephrine reuptake through high-affinity transporter binding, with potencies for both serotonin and norepinephrine equivalent to other SNRIs such as venlafaxine and duloxetine.4–6 Results of a previous meta-analysis suggest that switching to another class of antidepressants (eg, SNRIs) is better than switching to other SSRIs in the treatment of patients with SSRI-resistant MDD.7 Milnacipran’s pharmacological profile is different from other SNRIs with regard to its binding affinities, potencies of reuptake inhibition, and transporter selectivity. Specifically, milnacipran has a higher selectivity for the norepinephrine transporter compared to venlafaxine and duloxetine.4,8 There are few studies about the effects of switching patients with MDD to more noradrenergic-selective antidepressants following an initial SSRI treatment failure, in terms of not only clinical outcomes of efficacy and safety, but also the exploration of potential biomarkers that could indicate SSRI resistance, SNRI effectiveness, or the likelihood of adverse effects (AEs).
One method for identifying putative biomarkers related to treatments for MDD is to measure blood levels of molecules related to inflammation or neuronal function. We therefore measured the levels of various molecular species from peripheral blood samples, which included interleukin-6 (IL-6), interleukin-8 (IL-8), macrophage inflammatory protein (MIP)-1β, brain-derived neurotrophic factor (BDNF), serotonin, and the catecholamine metabolites homovanillic acid (HVA), 5-hydroxyindoleacetic acid (5-HIAA), and 3-methoxy-4-hydroxyphenylglycol (MHPG). These were chosen based on information from previous studies.9–14
The primary aim of this study was to assess the efficacy and safety of switching to milnacipran in patients with MDD following an initial SSRI treatment failure. The secondary aim of this study was to seek potential biomarkers by correlating clinical outcomes and blood levels of molecules that were presumably associated with MDD or its treatment.
Methods
Participants
Patients were recruited to the study at four sites in Japan. This study was conducted from April 2010 to March 2013. For inclusion criteria to be met, prospective subjects had to be 1) aged 20–74 years; 2) diagnosed according to the DSM-IV-TR criteria for MDD;15 3) unremitted patients treated by an SSRI (paroxetine, fluvoxamine, sertraline, or escitalopram) for at least 6 weeks prior to this study, and 4) with the score of the 17-item Hamilton Depression Rating Scale (HDRS)16 at the baseline was 14 or more. For exclusion criteria to be met, potential subjects had to be one of the following: 1) treated with selegiline hydrochloride; 2) allergic to milnacipran; 3) suffering from urinary retention; 4) pregnant or breast-feeding; 5) at significant risk of suicide; 6) diagnosed with a primary diagnosis including dementia, bipolar disorder, obsessive–compulsive disorder, eating disorders, or schizophrenia or other psychotic disorders; and 7) dealing with medical conditions judged to make the patient improper for use as a trial subject.
Ethics
The trial was approved by the Institutional Review Board of Chiba University Hospital (Chiba, Japan), and performed in accordance with the ethical standards laid down in the Helsinki Declaration of 1975, as revised in 1983. The trial was registered on the Clinical Trials Registry of the University Hospital Medical Information Network (UMIN, Tokyo, Japan) under registration number UMIN000003516. All subjects provided written informed consent for their participation in the study after the procedure had been fully explained to them.
Study design and procedure
We conducted a multicentre, prospective, open-label, 24-week trial. The starting dose of milnacipran was 25 mg in the first 1–2 weeks. Titration of the milnacipran dosage was flexible, from 100 to 200 mg, on the basis of the clinical judgment of the treating psychiatrists. However, the option to decrease the dosage for intolerance remained available. A given participant’s visit frequency was every 1–2 weeks in principle, to assess the safety of milnacipran, up to 8 weeks. Meanwhile, participants were tapered off their prior SSRIs for the first 4 weeks. The patients were evaluated at the baseline, and then after 4, 8, 12, and 24 weeks. Blood samples of the participants were collected from peripheral veins on the same day as the clinical evaluation.
Clinical assessments
To assess the severity of depressive symptoms, we used the HDRS. Response to milnacipran was defined as a ≥50% reduction in the HDRS score from baseline to each assessment day. Remission was defined by an HDRS score ≤7. We classified patients as responders, non-responders, and remitters at each point of the study, such that responders and remitters satisfied their respective definitions described earlier, and those patients who satisfied neither the responder nor remitter definitions were classified as non-responders. We also assessed patients using the Montgomery–Asberg Depression Rating Scale (MADRS),17 the 16-item Quick Inventory of Depressive Symptomatology-Self-Report – Japanese version (QIDS-SRJ),18,19 and the Clinical Global Impressions-Severity (CGI-S) scale.20 To assess social function, we used the Japanese version of the Social Adaptation Self-evaluation Scale (SASS-J),21 which is a validated self-evaluation scale for assessment of social functioning.22 The HDRS, MADRS, QIDS-SRJ, SASS, and CGI-S scores were measured at the baseline and 4, 8, 12, and 24 weeks.
In terms of safety, we collected information on all the AEs experienced by the participants during this study. We identified moderate-to-severe AEs as effects for which patients underwent medical treatments or were forced to withdraw from this study. We did not include moderate-to-severe AEs which patients experienced, but which required no intervention. We defined serious AEs as those causing death, any life-threatening conditions, hospitalization, or persistent disability.
Blood sample collection and measurements of molecules
All blood samples were collected between 10 am and 12 pm, at least 3 hours after breakfast, and were done at the same approximate time for each participant at every visit, although we did not strictly set a diet restriction. Serum samples were rapidly delivered to the Department of Psychiatry, Chiba University Graduate School of Medicine in anticoagulant tubes at 4°C, and stored at −80°C until use. The serum levels of IL-6, IL-8, MIP-1β, and BDNF were measured using the respective specific enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA). Plasma 5-HIAA, HVA, and MHPG concentrations, as well as whole blood serotonin concentration, were measured by SRL Inc. (Tokyo, Japan) using their standard laboratory protocols, including evaluation via high-performance liquid chromatography. Reference values provided by SRL on the basis of data from more than 100 healthy people were whole blood levels of serotonin, 57–230 (ng/mL); plasma levels of 5-HIAA, 1.0–6.0 (ng/mL); HVA, 4.4–15.1 (ng/mL); and MHPG 3.2–5.9 (ng/mL) (SRL, http://www.srl-group.co.jp/). Platelet counts were conducted at the Department of Molecular Diagnosis of Chiba University Hospital.23 Since platelets store serotonin,24 we calculated platelet serotonin as whole blood serotonin corrected with division by platelet number, to make the adjustment to whole blood serotonin measurements in participants that would be comparable to previous studies.13,25
Exploration of biomarkers from blood molecules on the basis of clinical outcomes
We first identified the clinical outcomes for participants in this study. We defined the short-term effectiveness of switching to milnacipran as the effect at the 8-week assessment, according to the convention used in Phase III clinical trials.26 The long-term effectiveness was defined by the result of the 24-week (final) assessment. Once the clinical outcomes and their distinct features were determined, we examined the relationships between these data and the data on the assayed blood chemicals to identify potential biomarkers.
Statistical analyses
We used parametric tests for the values of clinical measures because the data showed normal distribution. Data analyses were conducted on an intent-to-treat basis. Longitudinal efficacy outcomes (HDRS, MADRS, QIDS-SRJ, SASS-J, and CGI-S) were analyzed using a linear mixed effects model for repeated measures data with week as a fixed effect and subject as a random effect. Bonferroni’s adjustment was used for multiple comparisons.
We used non-parametric tests for the values of biological data from blood samples which did not follow a normal distribution. To analyze the changes of molecule levels in blood samples in the same individuals, we conducted Friedman’s test to screen for significance, followed by the Wilcoxon signed rank test. In comparisons between independent groups for biological values, we used the Mann–Whitney U-test. Two-tailed P-values below 0.05 were considered to indicate statistical significance in all analyses. All analyses were conducted using SPSS, version 20.0 (IBM Corporation, Armonk, NY, USA).
Results
Participants and clinical course outline
We screened 48 outpatients with MDD after initial SSRI treatment failure, of which 30 were enrolled in this study. Their demographic data are presented in Table 1.
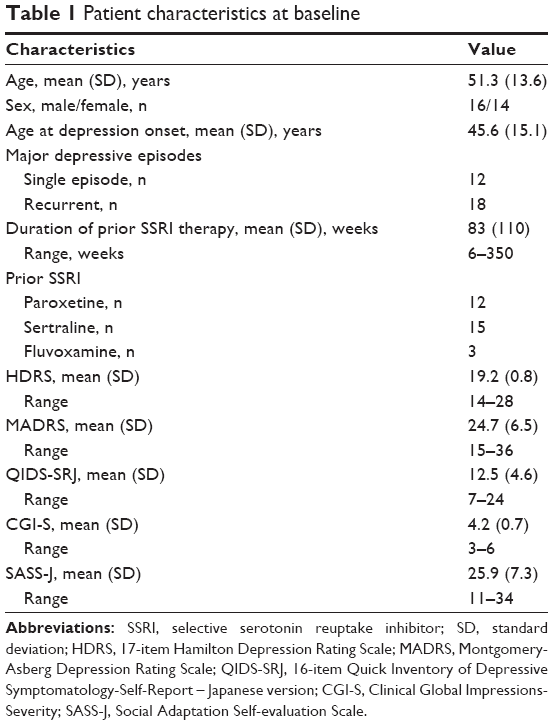 | Table 1 Patient characteristics at baseline |
By the first 8 weeks, ten participants withdrew due to AEs (n=8) or clinical deterioration (n=2) (Figure 1). Three more participants discontinued the study due to insufficient therapeutic response (Figure 1), so 17 participants ultimately completed this study (Figure 1). The daily mean peak dose of milnacipran was 108.8 mg (standard deviation [SD] 55.7), with a dose range of 12.5–200 mg. The average dose of milnacipran at the end of this study was 123.5 mg (SD 48.0).
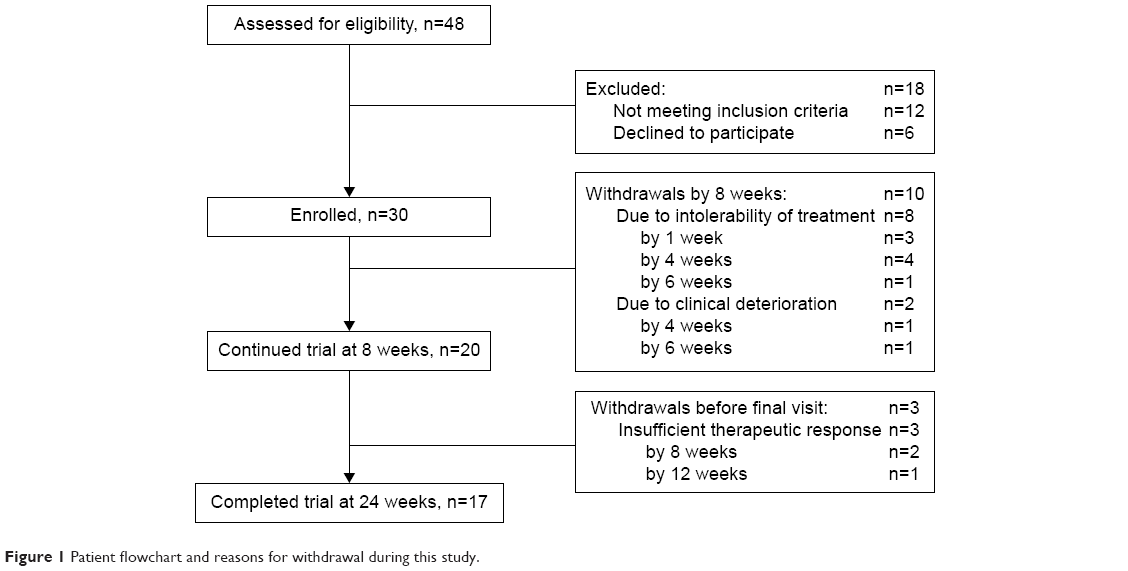 | Figure 1 Patient flowchart and reasons for withdrawal during this study. |
Efficacy
The clinical assessments at the baseline and each time point are presented in Table 2. As shown in Table 2, the HDRS scores were significantly reduced (ie, improved) vs baseline at 4, 8, 12, and 24 weeks. The other measurements to assess severity of depression, including the MADRS and the QIDS-SRJ also significantly decreased at each time point compared to baseline. The CGI-S and SASS-J scores increased (ie, improved) significantly at 8, 12, and 24 weeks, but not at 4 weeks, from baseline.
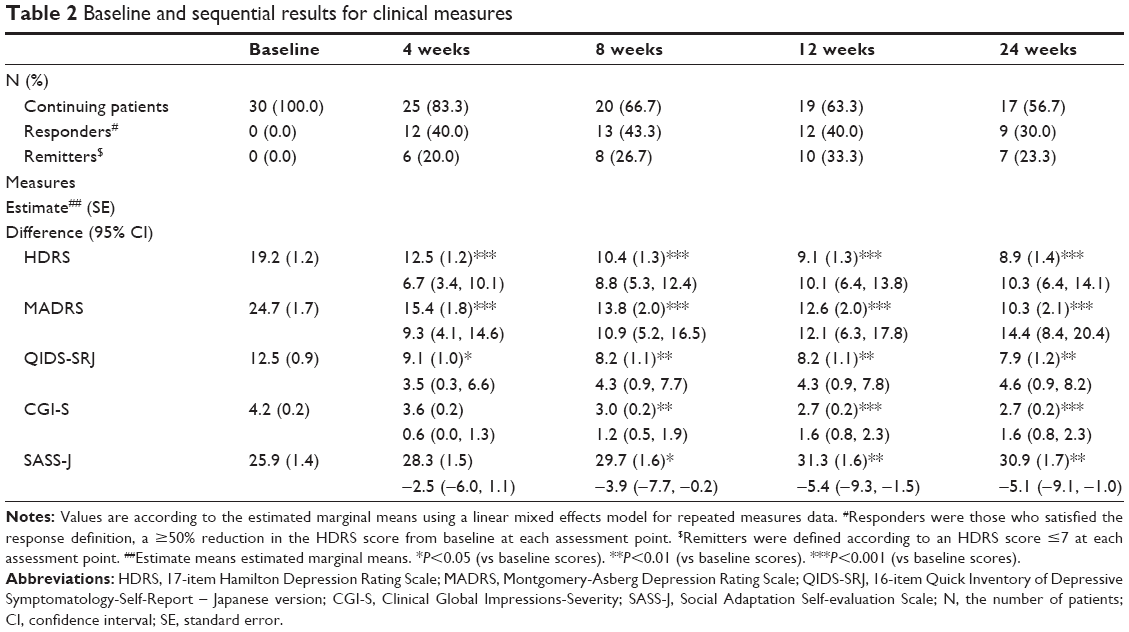 | Table 2 Baseline and sequential results for clinical measures |
Safety
The details of AEs in this study are shown in Table 3. A total of 23 AEs were reported by 16 patients over the study period. All AEs were considered to be related to milnacipran treatment and occurred within the first 4 weeks. No late-onset AEs (after the first 4 weeks) occurred. Table 3 provides the number and details of moderate-to-severe AEs. The rate of patients with moderate-to-severe AEs was 43.3% (n=13) in all 30 participants (Table 3). No serious AEs were observed in this study. Eight patients withdrew from this study because of AEs within 8 weeks, but no further withdrawals occurred (Figure 1).
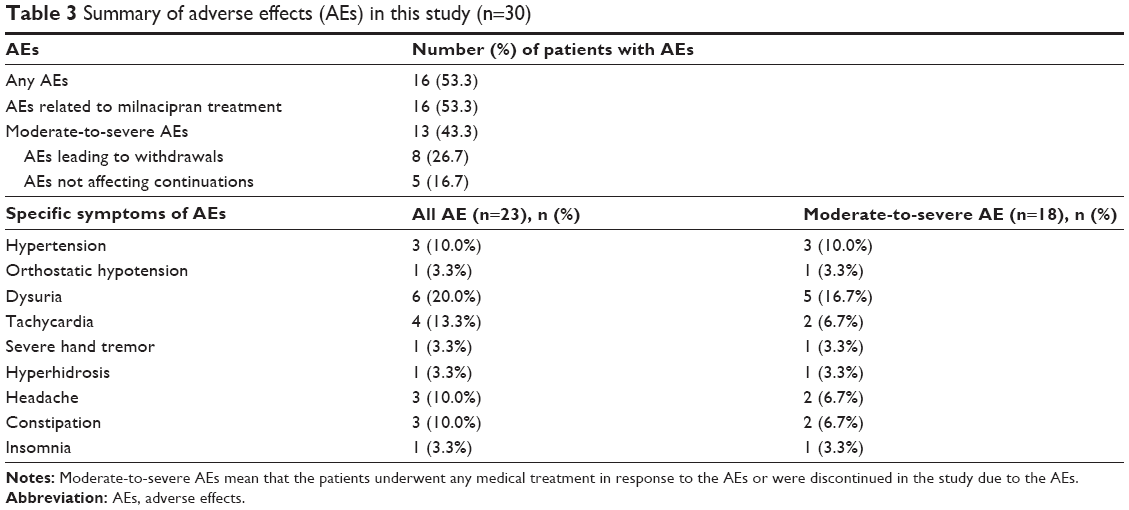 | Table 3 Summary of adverse effects (AEs) in this study (n=30) |
The relationships between clinical outcomes and possible blood biomarkers
As noted in Table 4, baseline serum levels of IL-6 and IL-8 were significantly higher in patients with moderate-to-severe AEs than in those patients without them. The levels of whole blood serotonin and platelet serotonin in patients with moderate-to-severe AEs were also significantly higher than in those patients without moderate-to-severe AEs (Table 4).
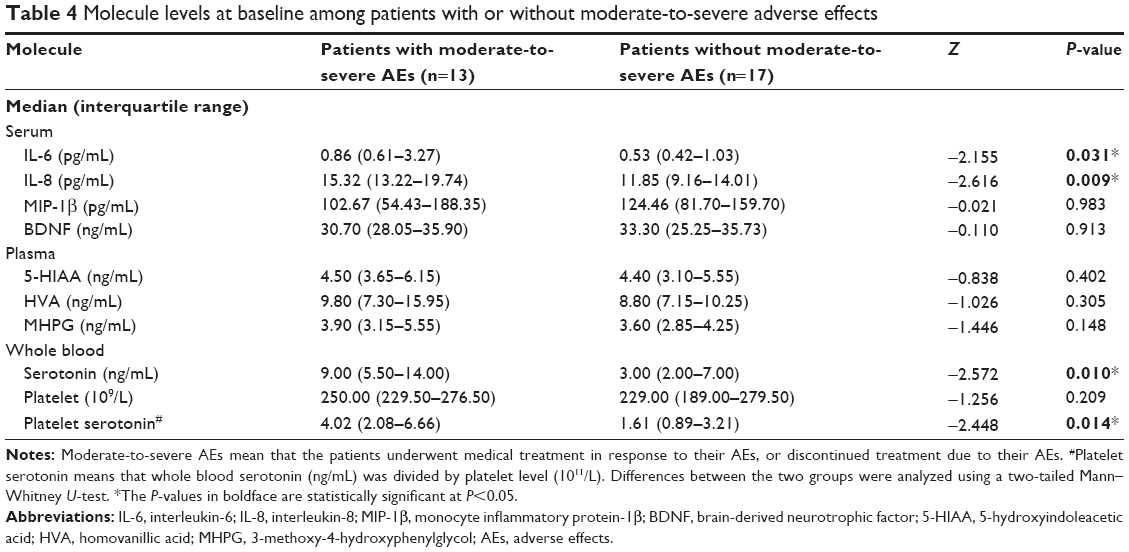 | Table 4 Molecule levels at baseline among patients with or without moderate-to-severe adverse effects |
Serum MIP-1β levels were assessed in the 17 participants who completed the full 24 weeks of the study, and were found to be significantly decreased from baseline (Friedman’s test, χ2=18.8, df=4, P<0.001). Moreover, comparing milnacipran responders to non-responders, serum levels of MIP-1β in non-responders (n=8) significantly decreased from baseline at all time points tested (Friedman’s test: χ2=23.9, df=4, P<0.001; the Wilcoxon signed rank test: at 4, 8, and 12 weeks, Z=−2.5, P<0.05; at 24 weeks, Z=−2.1, P<0.05, respectively), while those in responders (n=9) were not decreased at any time points (Figure 2). As shown in Figure 3, whole blood serotonin levels significantly increased above baseline over time in both responders (n=9) (Friedman’s test: χ2=21.8, df=4, P<0.001; the Wilcoxon signed rank test: at 4 weeks, Z=−2.3, P<0.05; at 8 weeks, Z=−2.5, P<0.05; at 12 and 24 weeks, Z=−2.7, P<0.01) and non-responders (n=8) (Friedman’s test: χ2=16.5, df=4, P<0.01; the Wilcoxon signed rank test: at 8, 12, and 24 weeks, Z=−2.4, P<0.05) (Figure 3). Platelet serotonin levels also significantly increased from baseline in both groups, responders (n=9) (Friedman’s test: χ2=21.8, df=4, P<0.001; the Wilcoxon signed rank test: at 4 weeks, Z=−2.2, P<0.05; at 8 weeks, Z=−2.5, P<0.05; at 12 and 24 weeks, Z=−2.7, P<0.01) and non-responders (n=8) (Friedman’s test: χ2=16.5, df=4, P<0.01; the Wilcoxon signed rank test: at 8, 12, and 24 weeks, Z=−2.4, P<0.05), while the levels of the other molecules were not statistically different (data not shown).
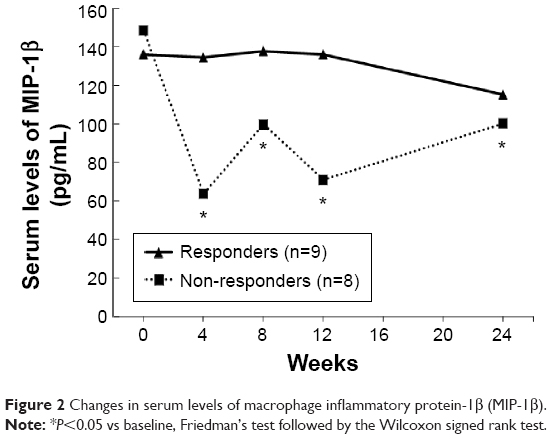 | Figure 2 Changes in serum levels of macrophage inflammatory protein-1β (MIP-1β). |
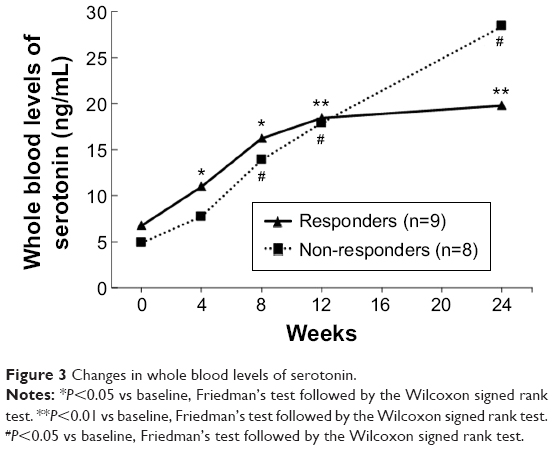 | Figure 3 Changes in whole blood levels of serotonin. |
Discussion
Five important results from this study deserve further mention here. First, in terms of efficacy, switching to milnacipran following an initial SSRI failure resulted in a 30.0% response rate, and a 23.3% remission rate, at the final assessment of patients with MDD. Second, in terms of safety, all AEs occurred in the early period (up to 4 weeks) after administration of milnacipran began, and the moderate-to-severe AEs were the main reason for withdrawal from this study. Third, this study showed that when moderate-to-severe AEs occurred after switching to milnacipran, the baseline serum levels of IL-6 and IL-8 in patients with them were significantly higher than in patients without them. Fourth, serum levels of MIP-1β decreased, and remained decreased throughout the 24-week trial period, in patients with MDD who did not respond to milnacipran. Fifth, whole blood levels of serotonin and platelet serotonin in patients, regardless of responsiveness condition, were increased after switching to milnacipran, and remained increased over the full study duration.
Switching to milnacipran following an initial SSRI failure resulted in a 30.0% response rate, and a 23.3% remission rate, at the final assessment of patients with MDD in this study. Other studies have examined the efficacy of switching pharmacotherapies following SSRI failure. In a representative large sample study, the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial,27 switching to other pharmacotherapies or cognitive therapy as the next step in patients with depression whose initial citalopram (SSRI) treatment failed produced a 27.3% rate of response and a 27.0% rate of remission. Moreover, a recent study reported that the efficacy of switching to the SNRI venlafaxine in patients whose first antidepressant treatment failed produced a 36.2% response rate, and a 19.9% remission rate.28 Our results herein are largely consistent with these studies, suggesting that switching to milnacipran could be an optional strategy for patients with MDD following failure of treatment with an SSRI or other type of antidepressant. Clearly, though, identifying predictors of treatment responsiveness in the pharmacotherapy of major depression would be a boon to clinical practice.
All AEs occurred in the first 4 weeks after administration of milnacipran had begun. Most AEs in this study were considered specific symptoms of inhibiting neuronal norepinephrine reuptake29 as SNRIs do by definition. Although previous reports described SNRIs such as milnacipran as comparatively well tolerated,29–31 minimizing side effects is important, especially for patients with depression who may have already experienced failure of their initial antidepressant treatments. Establishing means to prevent or prepare for the side effects related to switching pharmacotherapies is needed in clinical practice.
To the best of our knowledge, this is the first study to demonstrate that higher serum levels of IL-6 and IL-8 at the baseline could be related to adverse clinical effects of an antidepressant treatment in patients with major depression. However, several previous studies have described relationships between blood levels of cytokines or serotonin at the baseline and therapeutic responses in depressed patients.32–35 An inflammatory mechanism is thought to play an important role in the pathophysiology of depression.36–38 Hyperactivity of the sympathetic nervous system is associated with inflammatory dysregulation in patients with depression.38 Moreover, norepinephrine reuptake inhibitors such as milnacipran enhance noradrenergic action in the synaptic clefts.39 Therefore, it seems reasonable that patients with greater inflammation, as reflected by higher serum levels of IL-6 and IL-8, may be more likely to suffer from adverse side effects due to administration of noradrenergic antidepressants such as milnacipran. Measurements of serum IL-6 and IL-8 levels before administration of noradrenergic antidepressants might therefore be useful as predictive biomarkers to guide the direction of further treatment.
This study is also the first to report that serum levels of MIP-1β are continuously decreased in non-responsive, but not in responsive, patients with depression after administration of a noradrenergic antidepressant. MIP-1β is a chemokine, or chemotactic cytokine, which is related to the movement of leukocytes to sites of inflammation in injury or inflammatory diseases.40 Two in vitro studies have demonstrated that norepinephrine modulates the release of some cytokines from dendritic cells.41,42 Although the mechanism behind the decrease in serum MIP-1β here is still unknown, switching to milnacipran might directly affect serum MIP-1β through its noradrenergic activity. With regard to clinical practice, Lehto et al reported lower serum levels of MIP-1β in patients with depression than in healthy people, a result they ascribed to prolonged depressive symptoms.12 Given that our non-responders would have residual and persistent depressive symptoms, our results are compatible with those of the above report. Thus, a change in serum MIP-1β levels may represent a monitorable biomarker for responsiveness to treatment with noradrenergic antidepressants such as milnacipran.
Serotonin levels were also significantly different following the switch to milnacipran, regardless of responsiveness. Serotonin in the blood is mainly stored in platelets, which acquire the serotonin through cell-surface serotonin transporters.24 SSRIs and related antidepressants such as tricyclics inhibit serotonin reuptake, and can decrease blood serotonin levels by blocking platelet-mediated storage.32,43–46 Because milnacipran has lower potency and binding affinity for the serotonin transporter compared to SSRIs,4,8 it would also increase blood serotonin levels relative to an SSRI, as both our data and the abovementioned studies confirmed. It is possible for SSRIs to predispose patients to a risk of bleeding through the suppression of platelet function.47,48
Moreover, in comparison to other SNRIs, given that the selectivity for the serotonin transporter is relatively higher compared to other SNRIs such as venlafaxine and duloxetine than milnacipran,4,8 milnacipran might not affect bleeding risk better than other SNRIs. Our findings here indicate that milnacipran might be a suitable antidepressant for the treatment of patients with depression who may also be at risk for bleeding problems.
In addition, the data herein also demonstrated that the baseline whole blood levels of serotonin were different between patients with and without moderate-to-severe AEs after switching to milnacipran treatment. However, this result is difficult to interpret, because both the baseline levels in patients with and without moderate-to-severe AEs were remarkably lower than the reference values of whole blood serotonin related to prior SSRI treatments.32,43–46 To clarify this relationship in milnacipran treatment, studies excluding patients treated with serotonin reuptake inhibitor-based antidepressants will need to be designed.
Three main limitations to this study must be noted. First, this study was not rigorously designed. To confirm our findings, especially those regarding efficacy and safety in switching to milnacipran treatment, a rigorous study including control groups (placebo and/or other antidepressants), randomized assignments, and double-blind assessments must be performed. Second, the sample size of this study was small. Further studies with a larger sample are needed to establish not only the clinical effects, but also to verify the current potential biomarkers and identify new ones. Third, the relationships between our putative biomarkers (IL-6, IL-8, and MIP-1β) and the clinical outcomes are thus far limited to those related to switching to an SNRI in patients with depression who were treated with an SSRI first. Whether these biomarkers can be generalized to other SNRIs or other treatment regimens remains to be seen. Indeed, the utility of these biomarkers is a key question requiring further studies, such as those including other noradrenergic antidepressants and/or a patient cohort with no prior SSRI exposure.
Conclusion
This study showed that switching to milnacipran is a potentially effective strategy in the pharmacological treatment of patients with depression following the failure to respond to an initial SSRI. Moreover, our study identified serum levels of IL-6, IL-8, and MIP-1β as potential blood biomarkers that could be utilized to identify the responsiveness of patients to SNRIs such as milnacipran, or to identify those patients who may experience AEs strong enough to warrant discontinuation of treatment.
Acknowledgments
We would like to thank Editage (www.editage.jp) for English language editing. We declare that this work was financially supported by Chiba University Psychiatry Doumonkai (Chiba, Japan) and Chiba University Graduate School of Medicine (Chiba, Japan). There was no financial support by any other companies. This study was materially supported by medical information and printing services from Asahi Kasei Pharma (Tokyo, Japan). Asahi Kasei Pharma had no role in the study design, patient recruitment, analysis, data interpretation, and writing of the paper. We thank all patients and doctors for their participation in this study. We also thank the doctors and medical staff members of Choshi Kokoro Clinic (Tsuneo Senba, Rumiko Ishigami), Sodegaura Satsukidai Hospital (Syuichi Kikuchi), and Mobara Shinkeika Hospital (Masatoshi Suzuki, Kiyotoshi Sakashita, Naoko Takase) for recruiting the patients. We also thank the researchers at the Division of Clinical Neuroscience Chiba University Centre for Forensic Mental Health (Tamaki Ishima, Yuko Fujita, Kenji Hashimoto) and the Department of Psychiatry, Chiba University Graduate School of Medicine (Miwako Nakamura), for assisting in this work. We further thank the clinical research nurses for supporting this work at Chiba University Hospital (Junko Goto, Kaoru Ikeda, Komako Ito, Chisako Fujishiro).
Disclosure
Dr Tasuku Hashimoto has received honoraria as speaker/consultant from Astellas, GlaxoSmithkline, Meiji Seika, Mochida, Otsuka, Tanabe Mitsubishi, Yoshitomiyakuhin, and received research/grant support from Astellas, Chugai, Otsuka, Pfizer, Shionogi. Dr Tadashi Hasegawa has received honoraria as speaker/consultant from Astellas, Meiji Seika, Mochida, Otsuka, Yoshitomiyakuhin, Shionogi, Eli Lilly, Dainippon-Sumitomo, and received research/grant support from Astellas, Novartis. Dr Nobuhisa Kanahara reported honoraria from Otsuka, Janssen and Eli Lilly. Dr Tsuyoshi Sasaki received honoraria as speaker/consultant from Astellas, Daiichi Sankyo, Dainippon-Sumitomo, Eli Lilly, Janssen, Mochida, Shionogi, Otsuka, Taisho, Yoshitomi, and received research/grant support from Novartis, Otsuka and Taisho. Dr Hideki Komatsu has received research/grant support from Dainippon-Sumitomo, Novartis and Eli Lilly. Dr Hiroyuki Watanabe has received honoraria as speaker/consultant from Eli Lilly, Janssen, Astellas, Meiji Seika, Mochida, Otsuka, Tanabe Mitsubishi, Yoshitomiyakuhin, Shionogi, Dainihon-Sumitomo. Dr Masaomi Iyo has received a consultant fee from Eli Lilly, Dainippon-Sumitomo, Pfizer and Abbott and honoraria from Janssen, Eli Lilly, Otsuka, Meiji Seika, Astellas, Dainippon Sumitomo, Ono, GlaxoSmithKline, Takeda, Mochida, Kyowa Hakko, MSD, Eisai, Daiichi-Sankyo, Novartis, Teijin, Shionogi, Hisamitsu and Asahi Kasei. Drs Daiji Sakurai, Yasunori Oda, Takahiro Oiwa, Junpei Takahashi and Yoshimoto Sekine declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Lam RW, Kennedy SH, Grigoriadis S, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. III. Pharmacotherapy. J Affect Disord. 2009;117 Suppl 1:S26–S43. | ||
Bauer M, Pfennig A, Severus E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 1: update 2013 on the acute and continuation treatment of unipolar depressive disorders. World J Biol Psychiatry. 2013;14(5):334–385. | ||
Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. | ||
Koch S, Koch S, Hemrick-Luecke SK, et al. Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology. 2003;45(7):935–944. | ||
Takano A, Halldin C, Farde L. SERT and NET occupancy by venlafaxine and milnacipran in nonhuman primates: a PET study. Psychopharmacology (Berl). 2013;226(1):147–153. | ||
Nogami T, Takano H, Arakawa R, et al. Occupancy of serotonin and norepinephrine transporter by milnacipran in patients with major depressive disorder: a positron emission tomography study with [(11)C]DASB and (S,S)-[(18)F]FMeNER-D(2). Int J Neuropsychopharmacol. 2013;16(5):937–943. | ||
Papakostas GI, Fava M, Thase ME. Treatment of SSRI-resistant depression: a meta-analysis comparing within-versus across-class switches. Biol Psychiatry. 2008;63(7):699–704. | ||
Vaishnavi SN, Nemeroff CB, Plott SJ, Rao SG, Kranzler J, Owens MJ. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biol Psychiatry. 2004;55(3):320–322. | ||
Roy A, Pickar D, De Jong J, Karoum F, Linnoila M. Norepinephrine and its metabolites in cerebrospinal fluid, plasma, and urine. Relationship to hypothalamic–pituitary–adrenal axis function in depression. Arch Gen Psychiatry. 1988;45(9):849–857. | ||
Mitani H, Shirayama Y, Yamada T, Kawahara R. Plasma levels of homovanillic acid, 5-hydroxyindoleacetic acid and cortisol, and serotonin turnover in depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(3):531–534. | ||
Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169–1180. | ||
Lehto SM, Niskanen L, Herzig KH, et al. Serum chemokine levels in major depressive disorder. Psychoneuroendocrinology. 2010;35(2):226–232. | ||
Wulsin LR, Musselman D, Otte C, Bruce E, Ali S, Whooley MA. Depression and whole blood serotonin in patients with coronary heart disease from the Heart and Soul Study. Psychosom Med. 2009;71(3):260–265. | ||
Yoshimura R, Hori H, Ikenouchi-Sugita A, Umene-Nakano W, Ueda N, Nakamura J. Higher plasma interleukin-6 (IL-6) level is associated with SSRI- or SNRI-refractory depression. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(4):722–726. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Text revision. Washington, DC; 2000. | ||
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. | ||
Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. | ||
Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item quick inventory of depressive symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54(5):573–583. | ||
Fujisawa D, Nakagawa A, Tajima M, et al. Cross-cultural adaptation of the Quick inventory of depressive symptomatology, self-report (QIDS-SR). Jpn J Stress Sci. 2010;25:43–52 [in Japanese]. | ||
Guy W. Clinical Global Impression Scale. In: ECDEU Assessment Manual for Psychopharmacology–Revised. Rockville, MD: National Institute of Mental Health; 1976:218–222. | ||
Ueda N, Suda A, Nakagawa M, et al. Reliability, validity and clinical utility of a Japanese version of the Social Adaptation Self-evaluation Scale as calibrated using the Beck Depression Inventory. Psychiatry Clin Neurosci. 2011;65(7):624–629. | ||
Bosc M. Assessment of social functioning in depression. Compr Psychiatry. 2000;41(1):63–69. | ||
Shimada H, Oohira G, Okazumi S, et al. Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma. J Am Coll Surg. 2004;198(5):737–741. | ||
Mohammad-Zadeh LF, Moses L, Gwaltney-Brant SM. Serotonin: a review. J Vet Pharmacol Ther. 2008;31(3):187–199. | ||
Schins A, Hamulyak K, Scharpe S, et al. Whole blood serotonin and platelet activation in depressed post-myocardial infarction patients. Life Sci. 2004;76(6):637–650. | ||
Wisniewski SR, Rush AJ, Nierenberg AA, et al. Can phase III trial results of antidepressant medications be generalized to clinical practice? A STAR*D report. Am J Psychiatry. 2009;166(5):599–607. | ||
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. | ||
Souery D, Calati R, Papageorgiou K, et al. What to expect from a third step in treatment resistant depression: A prospective open study on escitalopram. World J Biol Psychiatry. 2014:1–11. | ||
Whiskey E, Taylor D. A review of the adverse effects and safety of noradrenergic antidepressants. J Psychopharmacol. 2013;27(8):732–739. | ||
Nakagawa A, Watanabe N, Omori IM, et al. Milnacipran versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2009;(3):CD006529. | ||
Kasper S, Pail G. Milnacipran: a unique antidepressant? Neuropsychiatr Dis Treat. 2010;6:23–31. | ||
Alvarez JC, Gluck N, Fallet A, et al. Plasma serotonin level after 1 day of fluoxetine treatment: a biological predictor for antidepressant response? Psychopharmacology (Berl). 1999;143(1):97–101. | ||
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370–379. | ||
Eller T, Vasar V, Shlik J, Maron E. Pro-inflammatory cytokines and treatment response to escitalopram in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):445–450. | ||
Rethorst CD, Toups MS, Greer TL, et al. Pro-inflammatory cytokines as predictors of antidepressant effects of exercise in major depressive disorder. Mol Psychiatry. 2013;18(10):1119–1124. | ||
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. | ||
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–741. | ||
Raison CL, Lowry CA, Rook GA. Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry. 2010;67(12):1211–1224. | ||
Dell’Osso B, Palazzo MC, Oldani L, Altamura AC. The noradrenergic action in antidepressant treatments: pharmacological and clinical aspects. CNS Neurosci Ther. 2011;17(6):723–732. | ||
Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. | ||
Goyarts E, Matsui M, Mammone T, et al. Norepinephrine modulates human dendritic cell activation by altering cytokine release. Exp Dermatol. 2008;17(3):188–196. | ||
Yanagawa Y, Matsumoto M, Togashi H. Enhanced dendritic cell antigen uptake via alpha2 adrenoceptor-mediated PI3K activation following brief exposure to noradrenaline. J Immunol. 2010;185(10):5762–5768. | ||
Marshall EF, Stirling GS, Tait AC, Todrick A. The effect of iproniazid and imipramine on the blood platelet 5-hydroxytrptamine level in man. Br J Pharmacol Chemother. 1960;15:35–41. | ||
Ross SB, Aperia B, Beck-Friis J, Jansa S, Wetterberg L, Aberg A. Inhibition of 5-hydroxytryptamine uptake in human platelets by antidepressant agents in vivo. Psychopharmacology (Berl). 1980;67(1):1–7. | ||
Poirier MF, Le Quan-Bui KH, Loo H, Meyer P. Changes in human platelet 5 HT level after 1 week of chlorimipramine treatment. Psychopharmacology (Berl). 1985;86(1–2):194–196. | ||
Reikvam AG, Hustad S, Reikvam H, Apelseth TO, Nepstad I, Hervig TA. The effects of selective serotonin reuptake inhibitors on platelet function in whole blood and platelet concentrates. Platelets. 2012;23(4):299–308. | ||
Serebruany VL. Selective serotonin reuptake inhibitors and increased bleeding risk: are we missing something? Am J Med. 2006;119(2):113–116. | ||
Halperin D, Reber G. Influence of antidepressants on hemostasis. Dialogues Clin Neurosci. 2007;9(1):47–59. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.