Back to Journals » OncoTargets and Therapy » Volume 15
Migfilin: Cell Adhesion Effect and Comorbidities
Authors Duan B, Qin Z, Gu X, Li Y
Received 6 January 2022
Accepted for publication 4 April 2022
Published 19 April 2022 Volume 2022:15 Pages 411—422
DOI https://doi.org/10.2147/OTT.S357355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Baoyu Duan,1,* Ziyao Qin,2,* Xuefeng Gu,1 Yanfei Li3
1Department of Pharmacy, Shanghai University of Medicine and Health Sciences, Shanghai, People’s Republic of China; 2Department of Research and Development, Shanghai Institute of Biological Products Co., Ltd., Shanghai, People’s Republic of China; 3Shanghai University of Medicine and Health Sciences Affiliated Zhoupu Hospital, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanfei Li, Shanghai University of Medicine and Health Sciences Affiliated Zhoupu Hospital, 1500 Zhouyuan Road, Shanghai, 201318, People’s Republic of China, Tel +86 21 6588 3180 Email [email protected] Xuefeng Gu, Department of Pharmacy, 279 Zhouzhu Road, Shanghai, 201318, People’s Republic of China, Tel +86 21 6588 3180, Email [email protected]
Abstract: Cell adhesion manifests as cell linkages to neighboring cells and/or the extracellular matrix (ECM). Migfilin is a widely expressed adhesion protein. It comprises three LIM domains in the C-terminal region and one proline-rich sequence in the N-terminal region. Through interplay with its various binding partners, such as Kindlin-2, Filamin, vasodilator-stimulated phosphoprotein (VASP) protein and the transcription factor CSX, Migfilin facilitates the dynamic association of connecting actomyosin fibers, orchestrating cell morphogenetic movement and cell adhesion, proliferation, migration, invasion, differentiation and signal transduction. In this review, to further elucidate the functional contributions of and pathogenesis induced by Migfilin, we focused on the structure of Migfilin and the targets which it directly binds with. We also summarized the role of Migfilin and its binding partners in the progression of different diseases and malignancies. As a possible candidate for coordinating various cellular processes and because of its association with both the pathogenesis and progression of certain tumors, Migfilin likely has utility as a therapeutic target against multiple diseases in the clinic.
Keywords: Migfilin, Filamin, Mig-2, Kindlin, cell–cell adhesions, cell-extracellular matrix adhesions
Introduction
In the cellular environment, adhesive relationships between a cell and its neighboring cells and the extracellular matrix (ECM) enable the formation of cell–cell and cell–ECM adhesion. Based on a combination of bioinformatic surveys, proteomic analysis and immunocytochemistry, over 200 adhesion-associated molecules, which are recruited to adhesion sites and include scaffolding and signaling proteins, have been previously identified.1 Notably, under normal homeostatic conditions, cell interactions with these adhesion-associated proteins are critical for two key biological functions. On the one hand, scaffolding proteins can physically anchor the actin cytoskeleton by either directly binding to the cytoplasmic domains of the main transmembrane cadherin or integrin receptors (such as Talin, Filamin and ɑ-actinin) or by indirectly connecting with these adhesion complexes through other scaffolding proteins (such as Vinculin and Zyxin).2–6 On the other hand, signaling proteins, including small G proteins of the Rho family, Src family kinases, the epidermal growth factor (EGF) receptor kinase and phosphatidylinositol 3-kinase (PI3K), can sensitize cells to chemical and mechanical properties in external environments and subsequently trigger intracellular signaling networks that orchestrate cell shape formation, viability, differentiation and metastasis.2–6 Thus, aberrant changes to cell adhesion framework lead to diverse pathologies, including the transition from benign tumors to malignant tumors.7
Notably, scientists attribute the dual functions of this adhesion complex-actin cytoskeleton module to certain domains, including the Src,8 pleckstrin,9 calponin homology domains10 and LIM11 domains. Among these domains, the LIM domain, named after its three core members (Lin-11, Isl-1 and Mec-3), is a highly conserved double-zinc finger motif with the following cysteine-rich sequence: CX2CX16-23HX2CX2CX2CX2CX2CX16-21CX2-3 (C/H/D; X represents any amino acid).12 The LIM domain acts as a key protein-binding interface within adhesion sites to mediate protein–protein interactions.
Migfilin, a LIM domain-containing protein, is reported to be activated during both cell–cell and cell–ECM adhesion in epithelial and endothelial cells.13,14 With a highly conserved structure, Migfilin is indispensable for connecting actomyosin fibers, maintaining cellular integrity and orchestrating actin-dependent cell morphogenetic movements, and it plays a functional role in modulating cell proliferation, migration and invasion.15–18 In addition, the role of Migfilin in shuttling from the cytoplasm to the nucleus, mediating transcriptional activity and affecting cell differentiation has also been investigated.19 According to recent studies, highly expressed Migfilin is involved in almost all types of cancers,20 which implies that Migfilin is linked to cancer predisposition and likely can be used as an important molecular marker for identifying different aspects of tumor behavior. Although some retrospective analyses of Migfilin structure, biological functions and relationship with certain serious diseases have been performed for years,14,21 breakthroughs in characterizing the biological role of Migfilin have been recently highlighted. It is important to summarize the most recent observations of the structure-dynamics and functional aspects of the adhesion complex-actin cytoskeletal network and oncogenic associations, with particular attention on Migfilin, to elucidate the physiological and pathological significance of Migfilin and inform the potential use of Migfilin as a therapeutic target for the prevention and treatment of various human diseases in the clinic.
The Structure of Migfilin
Known as a member of the LIM protein-containing Zyxin family, Migfilin directly binds with the actin-binding protein Filamin. Therefore, Migfilin has another name: Filamin binds LIM protein 1 (Figure 1A). Migfilin comprises three LIM domains in the C-terminal region and one proline-rich sequence in the N-terminal region, which occupy 51.8% and 48.2% of the full length, respectively (Figure 1B).20 Using multiple sequence comparison through log-expectation sequence alignment, Siddiqui et al observed that each C-terminal LIM domain of Migfilin contains a suite of four cysteine/histidine residues, which is an essential structure for coordinating the Zn2+ ions to form a zinc finger topology arranged in tandem and ultimately maintaining a high degree of sequence conservation.20 These highly conserved LIM regions in Migfilin are closely associated with protein–protein interactions. Notably, each LIM domain of Migfilin contains various hydrophobic amino acids linked to the zinc fingers within the LIM domain, and the LIM2 domain has been found to be relatively hydrophobic compared with the LIM1 and LIM3 domains. This hydrophobicity is one of the reasons that the LIM2 domain is regarded as an essential region for Migfilin targeting of its binding partners to the cell–cell and cell–ECM adhesion sites.14 In contrast, the proline-rich N-terminal sequence of Migfilin is reported to be highly variable, enabling a different set of partners that bind to this region. Thus, Migfilin can participate in diverse cellular processes, such as cell remodeling, cell growth, cell motility and oncogenesis, by binding to various proteins.
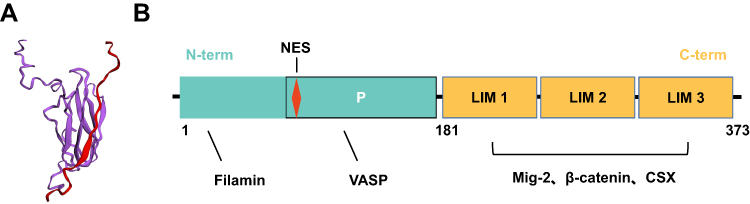 |
Figure 1 The Structure of Migfilin. (A) Solution NMR structure of the Filamin-Migfilin complex (PBD ID: 2K9U; Red, Migfilin; Purple, Filamin). (B) Schematic of domains and motifs of Migfilin protein. Migfilin comprises three LIM domains at the C-terminal region (C-term) and one proline-rich sequence at the N-terminal region (N-term), which occupy 51.8% and 48.2% of the full length, respectively. Migfilin is able to interact with Filamin and vasodilator-stimulated phosphoprotein (VASP) via its N-terminal region and proline-rich region (P), respectively. There also exists a nuclear export signal (NES) sequence in the proline-rich region, allowing Migfilin to shuttling between the cytoplasm and the nucleus. In addition, through the LIM 1/2/3 domains, Migfilin can interact with Mig-2, β-catenin and the cardiac-specific transcription factor CSX. |
Additionally, the proline-rich N-terminal sequence of Migfilin includes a nuclear export signal (NES) sequence that is not included in any of LIM domains (Figure 1B). The NES sequence can also be found in other Zyxin family members, such as Ajuba, Zyxin, WTIP and LPP. Recent work supports the hypothesis that the nuclear shuttling of Zyxin proteins may be an ancestral trait and that these proteins have retained this NES sequence evolutionally, resulting in not only their expression but also their shift in biological functions from nuclear roles to adhesion properties.22
Migfilin and Its Binding Partners
Migfilin is localized in both the ECM and the adjacent cells. With different binding regions, Migfilin is capable of coupling with distinct macromolecular complexes and forming cell–cell and cell–ECM adhesion structures. Migfilin has been reported to contact Kindlin-2, Filamin and VASP. Most scientists agree that the direct-binding partners of Migfilin are significant in regulating Migfilin dynamics and the cellular processes in which it is engaged, including actin-cytoskeleton orchestration, cell survival and cell mobility. In addition, an independent study demonstrated that Migfilin is able to facilitate cardiomyocyte differentiation by shuttling from the cytoplasm to the nucleus and regulating the activity of transcription factors. Herein, according to the unique sequences of Migfilin, we discuss the molecular activities and subcellular localizations of Migfilin with its binding partners and then further demonstrate the cellular adhesion effects of Migfilin in cell–cell and cell–ECM contact and in the nucleus and the diseases with which it is associated.
Migfilin and Kindlin
Kindlin-2, also named Mitogen inducible gene-2 (Mig-2), is known as a component of cell–ECM adhesion structures, especially in mammalian cells. Through interaction with an integrin cytoplasmic β tail, Mig-2 has the ability to modulate internal-to-external cell signaling and positively regulate integrin activation. This ability strengthens Mig-2 recruitment of integrin-mediated focal adhesions.23 The first imaging studies of Migfilin revealed that it physically interacted with Mig-2 via its C-terminal LIM domains.24 Migfilin can be recruited to the docking sites of Mig-2. In conclusion, Mig-2 is targeted to focal adhesion sites by binding with integrin,23 which in turn recruits Migfilin and Mig-2 to adhesion sites,24 showing that the molecular activities of integrin, Mig-2 and Migfilin play sequential roles.
The Migfilin fragment containing the proline-rich domain is reported to directly interact with Scr SH3 domains to a greater extent than with SH2 domains, which promotes Scr activation. This interaction is sufficient for protecting against anoikis. Therefore, at cell adhesion structures, the combination of Migfilin and Src seems to facilitate the suppression of cell apoptosis.25 In subsequent studies, Liu et al showed that Mig-2 phosphorylation by Src explains the mechanism of Migfilin and Src activation.26 Specifically, membrane-bound activated Src can couple with Mig-2 to phosphorylate Mig-2 at Y193, resulting in a strengthened Mig-2/Migfilin interaction and Migfilin recruitment to focal adhesion sites, which in turn promotes further Src activation.26 The finding of the Src-Mig-2-Migfilin-Src positive signaling cascade indicates that Scr activation may be required for Migfilin recruitment to cell–ECM adhesion structures (Figure 2).
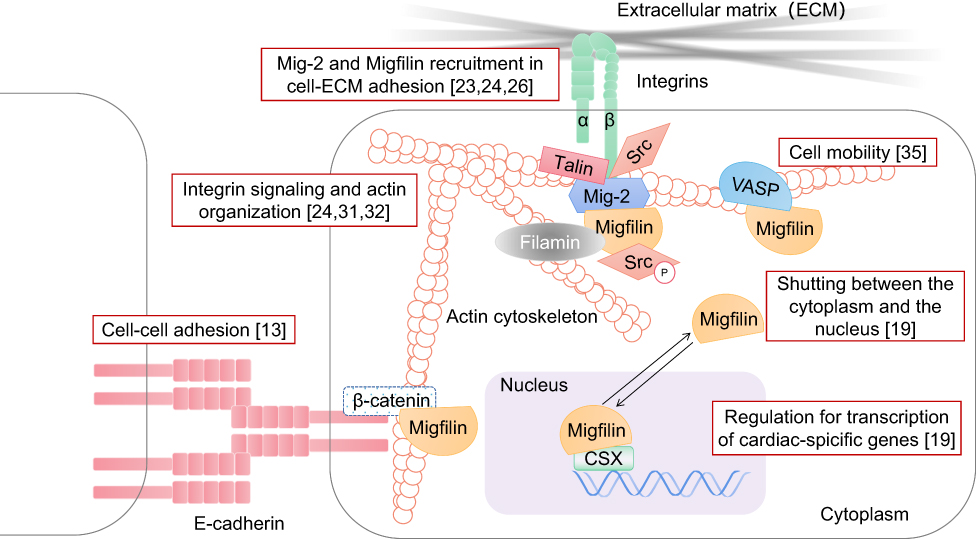 |
Figure 2 Migfilin and its binding partners. Firstly, in the integrin adhesome network, Kindlin-2 (Mig-2) directly binds with the integrin cytoplasmic β tail, which in turn strengthens Migfilin recruitment to the docking sites of Mig-2, mediating the cell–ECM adhesions. The finding of the Src-Mig-2-Migfilin-Src positive signaling cascade was also described, and Scr activation may be required for Migfilin recruitment to cell–ECM adhesion structures. Secondly, As a Filamin-binding LIM protein, Migfilin participates the formation of the Mig-2-Migfin-Filamin scaffolding complex, regulating integrin signaling and actin organization. Migfilin were also found to bind with β-catenin or the E-cadherin-β-catenin complex that are related to actin bundles, leading to Migfilin localization to the cell–cell adhesion structures. In addition, Migfilin participates the process of cell mobility through associating with VASP. Finally, Migfilin contains a functional NES sequence and shuttles from the cytoplasm into the nucleus in response to calcium. Migfilin itself also has a transcription-promoting role in cardiac development via interacting with CSX in the nucleus. |
Previously available data have shown that Migfilin recruitment and dynamics in cell adhesion are Mig-2 dependent. However, Kindlin-1, similar to the structure and function of Mig-2, has been implicated in focal adhesion together with Migfilin and Mig-2 in normal human keratinocytes and HaCaT cells and significantly affects cell proliferation and the cell migration rate.27 Intriguingly, through its C-terminal LIM domains, Migfilin was originally found to be coupled with Mig-2.24 In Mig-2-knockdown keratinocytes, Migfilin fragments containing three intact Migfilin LIM domains bound with Kindlin-1 and were recruited to focal adhesions, while Migfilin fragments lacking at least two Migfilin LIM domains did not couple with Kindlin-1.28 In summary, in the cellular Migfilin–Kindlin interaction, these three tandem LIM domains in Migfilin may dictate its cell–ECM adhesion localization capabilities. Equally important, silencing Kindlin-1 does not seem to affect the localization of either the Migfilin or Mig-2 protein at focal adhesions, indicating that Kindlin-1 may function independently of Migfilin and Mig-2.27 Together, these data support the idea that Kindlin is a crucial regulator of Migfilin dynamics and subcellular localization.
Migfilin and Filamin
Tu et al considered that in addition to the C-terminal LIM domains of Migfilin physically interacting with Mig-2, its N-terminal region is able to mediate interactions with the actin-binding protein Filamin,24 which plays a major role as an actin cross-linking protein in maintaining the actin network. The latest report from Ithychanda et al revealed that Migfilin binds to Filamin A/B/C repeat 21 and Filamin B repeat 11–13 constructs.29 Mig-2-Migfin-Filamin scaffolding complex formation may be the reason that depletion of Migfilin or Mig-2 impairs cell shape modulation.24 In subsequent studies, a critical pathway mediated by four key components, Talin, integrin, Filamin and Migfilin, in adhesive processes was identified, providing valuable information to examine integrin signaling and actin cytoskeleton reorganization. Specifically, there is considerable overlap between Talin (a mechanosensitive component of adhesion complexes such as Migfilin) and Filamin A-binding sites in integrin cytoplasmic β tails. Under certain circumstances, Filamin A cuts the talin-integrin interplay by competitively binding with integrin. Moreover, integrin cytoplasmic β tails and Migfilin overlap at the IgFLNa21 site of Filamin.30 Thus, Migfilin plays a role in competitively binding to the same site in Filamin as targeted by integrin and rescues the Filamin-induced inhibition of the talin–integrin interaction,31,32 reestablishing cell shape changes (Figure 2). In conclusion, Filamin seems to serve broadly as an inhibitor of integrin activation, and Migfilin seems to augment integrin activation. Most importantly, the balance between these different intracellular adaptor proteins plays a key role in regulating the kinetics of integrin signaling.
As the regulatory proteins of integrin signaling were discovered, the C-terminal LIM domains of Migfilin were also found to bind β-catenin or components of the E-cadherin-β-catenin complex that are related to actin bundles, leading to Migfilin localization to the junctions of adhering cells and connection with the actin cytoskeleton.13 Notably, the Mig-2-binding site in Migfilin that mediates Migfilin targeting to areas of cell–ECM adhesion partially overlaps with the β-catenin-binding site in Migfilin that mediates Migfilin targeting to cell–cell junctions. As a consequence, upregulation of Mig-2 expression may lead to a higher rate of Migfilin localization to cell–ECM adhesions, which translates into less Migfilin localized to cell–cell junctions (Figure 2).13
Migfilin and VASP
VASP has been discovered to be a regulator of the actin cytoskeleton and cell motility.33 It is a member of the conserved Ena/VASP protein family that consists of the EVH1 domain, EVH2 domain and a proline-rich central region.33,34 Among the multiple protein-binding regions of Migfilin, the single LPPPPP site (residues 104–109) in its proline-rich N-terminal domain is reported to mediate the interaction of Migfilin with the VASP EVH1 domain.35 Previous evidence indicated that knocking down Migfilin in HeLa, HT-1080 and MDA-MB-231 cells impaired the migration rate of these cells in Transwell chamber assays. Intriguingly, upregulation of Migfilin in HeLa, MDA-MB-231 and MDCK epithelial cells also led to a reduction in cell migration ability.35 In fact, the open VASP-binding site in the Migfilin proline-rich domain makes it possible for Migfilin to exert an inhibitory effect on cell motility (Figure 2).35 We can therefore attribute Migfilin suppression of cell migration to VASP.14,33 In addition, in recent years, compelling evidence has shown that silencing of VASP downregulates Migfilin expression levels in MDA-MB-231 breast cancer cells.36 In summary, VASP participation may be critical for Migfilin-mediated cell migration, and vice versa, which indicates that Migfilin and VASP serve as cofactors in the progression of tumor metastasis.
Notably, using pathway analysis, scientists have found that Migfilin, Zyxin and LPP can replace each other to engage VASP,20 indicating that three proteins might play similar roles and naturally compensate for each other under normal conditions.
Migfilin and Cancer Development
A recent study based on the GENEVESTIGATOR database demonstrated that the protein expression level of Migfilin and other Zyxin family members are upregulated in almost all cancer types.20 In contrast to identified family members, the Migfilin protein is mainly expressed in intraductal papillary-mucinous carcinoma in the pancreatic duct, mixed Signet ring cell adenocarcinoma in the digestive system, and premalignant esophageal cancer.20 Accordingly, Migfilin functions as a promising biomarker of tumor progression. In addition to Migfilin expression, changes in its binding partners exert some unusual effects, causing malignancies in the human body and leading to several other pathological characteristics.
Migfilin as a Biomarker for Cancer Diagnosis
Because of the involvement of Migfilin in regulating essential cellular functions affecting cell growth, survival, adhesion, motility, differentiation, and intracellular signaling, pathophysiological changes in the human body may be associated with altered Migfilin expression. Recently, a handful of studies investigated whether Migfilin can serve as a biomarker of physiological conditions in several human cancers.
Through tissue microarray techniques, scientists found that increased cytoplasmic levels of both Migfilin and its known binding partner Mig-2 are readily detected in more than one-half of human leiomyomas and leiomyosarcoma tissues, suggesting that twin adhesion proteins provide a necessary synergistic effect that results in the pathogenesis and progression of human leiomyosarcoma.37 In addition, Migfilin can orchestrate bone remodeling behaviors by balancing osteoclast differentiation and osteoblast properties.38 Accordingly, in examined osteoarthritis patient samples, both the mRNA and protein expression of Migfilin were dramatically elevated and correlated with the development and progression of disease.39 The findings showing positive expression of Migfilin in human leiomyosarcoma and osteoarthritis have been supported by similar statistical analyses of other human malignancies. Increased Migfilin expression can be detected in advanced glioma tissue samples and indicates a poor patient outcome.18,40 In vitro, the expression level of Migfilin also indicates the high invasive potential of HepG2 cells.17 Migfilin increases glioma cell migration and invasion, while its depletion halts human liver cancer cell motility.17,18,40 In summary, Migfilin may be a molecular indicator in the prognosis of several cancers and even their metastasis.
However, Migfilin is not readily expressed in all malignancies. For example, with regard to mesenchymal tumors, the expression of Migfilin can be positively related to higher leiomyosarcoma grades, while in examined primary tumors, Migfilin cannot be detected in central chondrosarcoma or enchondroma tissues,41 which indicates that Migfilin may have different functions even in certain tumors in the same category. The mRNA and protein levels of Migfilin and Mig-2 are disrupted in breast cancer patient tissues.42 In addition, He et al have also shown that Migfilin expression in esophageal squamous cell carcinoma patient samples with lymph node metastasis is lower than that in samples with no clinical metastatic potential, and Migfilin plays a suppressive role in esophageal cancer cell motility.43
Currently, no statistical analysis can explain why Migfilin can be expressed differently, showing even the opposite expression in human malignancies. Despite this lacking explanation, on the basis of its specific expression level, Migfilin is currently a prognostic biomarker candidate for identifying the pathogenesis and progression of certain tumors and for predicting the survival of patients with advanced cancer, which may indicate its role as a potential molecular therapeutic target against specific cancers.
The Roles of Migfilin and Kindlins in Human Cancers
Loss-of-function mutations in the KIND1 gene encoding Kindlin-1 are thought to be involved in the progressive human disorder Kindler syndrome (KS, a rare subtype of inherited epidermolysis bullosa) and are characterized by an aggressive phenotype of skin fragility, blistering and photosensitivity.44 KS places the patient at risk for cutaneous malignancies. A high risk of developing squamous cell carcinoma was evident in one case series of KS patients.45 Kindlin-1 plays a critical role in basal epidermal cells. A Kindlin-1-involved model in keratinocytes was extensively studied by Has et al, who suggested that, together with β1 integrin subunit, focal adhesion kinase (FAK), α-actinin and Migfilin, Kindlin-1 mediated the formation of focal adhesion complexes and induced Rho family GTPase Cdc42, Rac1 and RhoA signaling activation, further triggering the activation of multiple downstream molecular effectors and orchestrating filopodial extensions, lamellipodia protrusions and stress fibers.46 In addition, the absence of Kindlin-1 in epidermal cells is related to several abnormalities, such as cell polarity, cell adhesion, cell migration and plasma membrane protrusion activity.47 The lack of these Kindlin-1-related biological functions leads to a propensity for accelerated fragility, atrophy of keratinocytes and KS. Rognoni et al described a possible mechanistic role for Kindlin-1 in the development of KS, showing that mice with Kindlin-1-knockdown keratinocytes were inclined to suffer from cutaneous carcinoma (Figure 3).48 This finding suggests that Kindlin-1 expression may be related to maintained healthy epidermal cell function. To date, how Migfilin, an identified key-binding partner of Kindlin-1, regulates Kindlin-1 function in keratinocytes remains unknown; however, but it notable, that silencing of Kindlin-1 in HaCaT keratinocytes altered Migfilin distribution in focal adhesion complexes and greatly reduced Migfilin accumulation at early cell–ECM contact points.49 The interplay between Migfilin and Kindlin-1 in the pathology of KS, therefore, remains to be further explored.
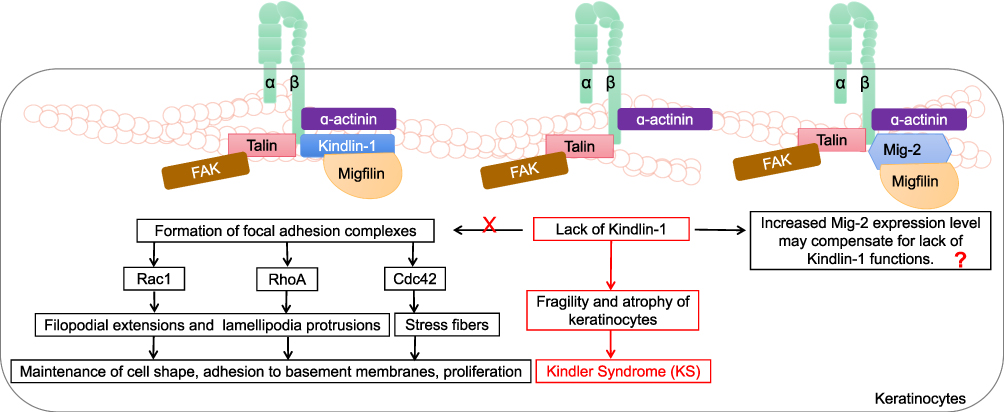 |
Figure 3 The roles of Migfilin and Kindlins in keratinocytes. Kindlin-1 forms focal adhesion complexes with integrin β1 subunit, focal adhesion kinase (FAK), α-actinin and Migfilin. Rho family GTPase Cdc42, Rac1 and RhoA signaling are activated and mediate filopodial extensions, lamellipodia protrusions, and stress fibers through multiple downstream molecular effectors, which plays an important role in maintaining cell shape, adhesion to basement membranes, proliferation of keratinocytes. The lack of these Kindlin-1-related biological functions forms the biological basis for fragility and atrophy of keratinocytes in the pathology of Kindler Syndrome (KS). It is possible that this process can proceed because an increase in Mig-2 expression level may compensate for lack of kindlin-1 functions. The interplay between Kindlin-1, Migfilin and Mig-2 in the pathology of KS remains to be further explored. |
Kindlin family members play distinctive roles in breast cancer. Upregulated Kindlin-1 expression in the tumor microenvironment is connected with more aggressive tumor progression and a poorer clinical prognosis in breast cancer.50 Moreover, decreased Mig-2 and Migfilin expression levels have also been detected in samples of human breast cancer.42 This finding implies that there may be a necessary synergistic reaction between Kindlin-1, Mig-2 and Migfilin that influences breast cancer development. At the cellular level, Kindlin-1 and Mig-2 have been extensively characterized with respect to their compensatory effects in breast cancer. Specifically, the depletion of Kindlin-1 led to an increase in Mig-2 expression level in several types of breast cancer cells, and vice versa.50 The compensatory role of Kindlin-1 and Mig-2 is reflected not only in their expression level but also in their ability to influence cell size, cell and subcellular localization, cell migration and cell invasion. Notably, Kindlin-1 silencing leads to overcompensation in metastasis through the upregulation of Mig-2 in breast cancer cells.50 In addition, compared to both Kindlins (Kindlin-1 and Mig-2) knockdown- or single-Mig-2-knockdown cells, single-Kindlin-1-knockdown cells exhibited a more significant decrease in invasive capability.50 Taken together, these findings indicate that Mig-2 may play a specific role in controlling cell-spreading ability and that Kindlin-1, in particular, may regulate cell invasion ability. However, as a binding partner of Mig-2, whether Migfilin is involved in this molecular compensation mechanism in breast cancer has not yet been determined.
In accordance with the observations of kindlins in breast cancer, other research groups have described the roles of kindlins in cell migration and invasion in glioma and pancreatic, gastric and colorectal carcinoma progression.51–54 The formation and maturation of invadopodia, which are actin-rich membranous protrusions, serve as key promoters of proteolytic degradation of the ECM and tumor metastasis. Liu et al identified IkappaB kinase subunit epsilon (IKKε) as a novel pivotal regulator of invadopodia formation and activation by phosphorylating Mig-2 at S159 and in turn causing colorectal cancer cell invasion.55 As already mentioned, Src activation mediates the interaction of Src with Mig-2 and phosphorylates Mig-2 at Y193, thereby resulting in Migfilin recruitment to cell–ECM adhesion structures and cell spreading orchestration.26 Considering these findings, there is a strong possibility that the recruitment of Migfilin may be followed by IKKε-mediated Mig-2-S159 phosphorylation to facilitate invadopodia formation. To determine whether Migfilin is associated with the IKKε-mediated signaling cascade in colorectal cancer metastasis, further study is required.
The Roles of Migfilin and Filamin A in Platelet Biology
Filamin A is currently the focus of considerable attention because its loss-of-function mutations likely result in a series of severe defects called Filaminopathies A, including tortuous and disorganized vasculature, abnormal blood vessels, defective adhesion junctions and misshapen vascular endothelial cells, suggesting a key role for Filamin A in the vasculature.56
In platelet circulation, idiosyncratically expressed integrin αIIbβ3 is maintained in resting conformation. As soon as the vasculature damages, integrin αIIbβ3 quickly shifts into active conformation, which mediates platelet aggregation.57 Mutations leading to the loss of Filamin A lead to the positive regulation of RhoA activity via integrin αIIbβ3 during proplatelet formation and thus give rise to macrothrombocytopenia.58 However, another previous study reported the opposite result: integrin αIIbβ3 activation occurs when Talin, in conjunction with Kindlin-3, binds to conserved motifs in the integrin β3 cytoplasmic domain.59 At this point, Filamin A participates in integrin β3 clustering, blocking the high affinity of Talin for the integrin β3 domain and impairing αIIbβ3 activation, which induces abnormalities in proplatelet formation.59 Migfilin can competitively bind to Filamin A, block its αIIbβ3-inhibiting effect and support long-term external-to-internal signaling (Figure 4).60 In other words, loss of Migfilin may enhance the Filamin A-mediated platelet αIIbβ3-inhibiting effect. However, Migfilin knockdown has been found to have no effect on platelet count or morphology.60 In summary, the loss of Filamin A activity induced by Filamin A mutations can lead to reduced αIIbβ3 activation, which results in abnormalities in hemostasis and thrombosis, indicating that, compared to changes in migfilin, changes in Filamin A seem to play a more profound role in platelet biology.
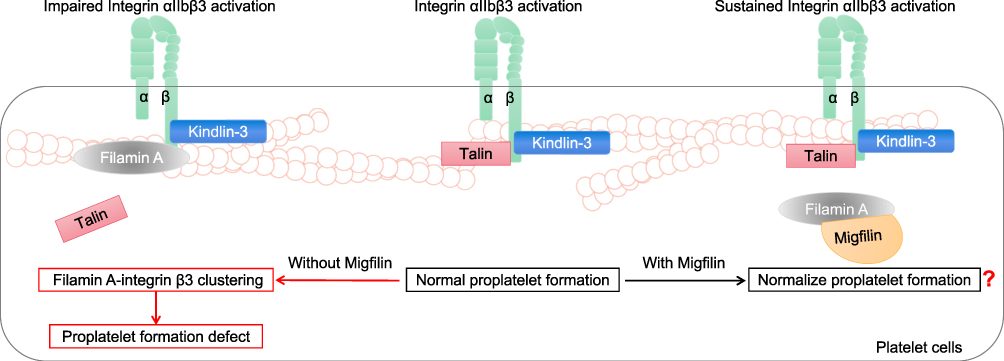 |
Figure 4 The roles of Migfilin and Filamin A in platelet biology. In platelet circulation, when the vasculature damages, integrin αIIbβ3 shifts into active conformation, which mediates platelet aggregation. During platelet activation, Talin, in conjunction with Kindlin-3, binds to conserved motifs in the integrin β3 cytoplasmic domain. That’s the normal proplatelet formation. At this point, Filamin A participates in integrin β3 clustering, blocking the high affinity of Talin for the integrin β3 and impairing αIIbβ3 activation, which induces proplatelet formation defect. Migfilin may bind to Filamin A, block its αIIbβ3-inhibiting effect and support a long-term Integrin αIIbβ3 activation. However, Migfilin knockdown has been found to have no effect on platelet count or morphology. A further elaboration of the role of Migfilin in normalizing the proplatelet formation and the Migfilin and Filamin A interaction network in platelets is warranted. |
The Role of Migfilin in Transcriptional Regulation
Migfilin is Involved in Transcriptional Regulation in Nucleus
In the VASP-binding proline-rich domain of Migfilin, there is also an NES sequence. Among Migfilin and other Zyxin family members, the sequence of the NES is considered to be conserved regardless of its origin and function, even though its position seems to vary from protein to protein,20 because of its ability to shuttle from the cytoplasm to the nucleus. Intriguingly, although Migfilin has been reported to localize to the nucleoli fibrillar center, its nuclear localization sequence (NLS) cannot be detected.20
One of the first studies on Migfilin provided evidence that its LIM domains associate with the homeodomain of the transcription factor CSX, which plays an essential role in normal heart development and heart functions and is involved in regulating cardiac differentiation in the P19CL6 cell line (Figure 2).19 The Migfilin-CSX interaction is required for the activation of the atrial natriuretic peptide (ANP) promoter, a downstream target gene of CSX. Akazawa et al discovered that Migfilin splicing variants containing only LIM2 and LIM3 domains have a higher likelihood of binding with CSX and exhibiting profound synergistic activation on the ANP promoter than full-length Migfilin,19 indicating that the LIM2 and LIM3 domains may function as promoter transactivation domains. These findings indicate that Migfilin itself has a transcription-promoting role in cardiac development, which simultaneously explains the pseudonym for Migfilin: CSX-associated LIM protein (Cal).
The Role of Migfilin and Other Cytoskeletal Proteins in Cardiac Hypertrophy
The cardiac hypertrophic response is an adaptive response for reducing increased myocardial wall stress and prevent cardiac dysfunction. However, sustained pressure and volume overload on the heart can trigger heart failure. Recent in vivo cardiovascular research confirmed that Migfilin has a special role and importance in the response to cardiac stress. Scientists have pointed out that the expression level of Migfilin transcripts and protein expression differences can be determined during the indicated times in wild-type mice with experimental chronic hypertensive stress.61 In addition, the loss of Migfilin drastically decreased hypertrophic remodeling of cardiomyocytes, reduced fibrosis and reduced cardiac failure under pressure overload in Migfilin-knockdown mice,19 indicating a role for Migfilin in cardiac hypertrophy.
Indeed, throughout the broad network of integrins linked to actin, the associated cytoskeletal proteins, including FAK, PYK2, vinculin, zyxin, VASP, and MENA, also have received attention because of their critical roles in heart hypertrophy. For example, phosphorylation of the adhesion protein FAK tyrosine is detected in cardiomyocytes in response not only to mechanical stretching but also to phenylephrine-mediated hypertrophic stimuli, showing a positive effect on cytoskeletal organization and cell spreading.62,63 Recent data have shown that vinculin was directly coupled with zonula occludens-1 (ZO-1) at cardiomyocyte–cardiomyocyte intercalated disc connections, stabilized gap junctions and modulated myocyte integrity.64 In addition, Zyxin, VASP, and MENA exert compensatory effects on cardiac structure and function.65 Although the molecular mechanism of several associated cytoskeletal proteins is indicated in heart development and cardiomyopathy, and although Zyxin, with a structure similar to that of Migfilin, is expressed in the heart and localized to the nucleus,20 these proteins are uniformly reported to work at the cytoplasmic face of the plasma membrane, not at the interaction sites of Migfilin. More importantly, based on the data analysis that Migfilin has an NES sequence and Mig-2 also bears an NLS sequence, enabling them to enter the nuclear compartment,20 it is thought that Migfilin and Mig-2 can localize not only at focal adhesions but also in the nucleus, where they coactivate CSX gene expression and promote cardiomyocyte differentiation.66 In summary, we speculate that Migfilin may be required for regulating heart hypertrophy, independent of other associated cytoskeleton proteins, except Mig-2, in the nucleus.
Conclusion
Based on the fact that the LIM domain plays an important role in actin cytoskeleton-based cell adhesion modules, an increasing number of experiments with Migfilin as the research target have demonstrated profound results.
With its highly conserved structure and interplay with partners to which it directly binds, such as Filamin, Mig-2, VASP and CSX, Migfilin has been found to move to different subcellular locations, ranging from focal adhesions and adhesion junctions to the cell nucleus, pointing to its capacity to regulate cell remodeling, proliferation, motility, differentiation and intracellular signaling, implicating Migfilin effects on multiple pathways and showing its significance in cell processes.
In addition, the indirect-binding mechanics of Migfilin need to be taken into account. For example, Migfilin can directly interact with Mig-2 through integrin-linked kinase (ILK).67 Through its direct binding to PINCH and Parvin, ILK is involved in integrin regulation of the actin cytoskeleton.68 Both Migfilin and ILK are common binding partners of Mig-2; however, whether Migfilin indirectly regulates ILK intracellular signaling to affect a wide variety of proteins with similar cellular functions is unknown. Similarly, VASP, a critical direct-binding target of Migfilin, also immediately binds to two other adhesion proteins, including Vinculin, establishing a physical link between integrin and the actin cytoskeleton.69 The dynamic network of Migfilin and the functionally related proteins (ILK, PINCH, Parvin and Vinculin) to which Migfilin is spatially and temporally related deserve further investigation, which may yield new insights into actin-based cell adhesion modules.
Since its discovery in 2002, Migfilin has been implicated as having an essential role in cancer and becoming a potential molecular target for therapy in the clinic. In regard to Migfilin expression, it seems to present different and even opposing level among human malignancies. In the cytoplasm, Migfilin expression is readily increased in leiomyosarcoma, glioma and osteoarthritis patient tissues59,60,67,68 but decreased in chondrosarcomas, enchondromas, breast cancer and metastatic esophageal squamous cell carcinoma patient samples.41–43 In addition, elevated Migfilin expression is associated with the high migratory and invasive potential of glioma cells,42,51 and similarly, our laboratory has established that Migfilin plays a role in inhibiting A375 melanoma cell proliferation and metastasis both in vivo and in vitro. However, considering the suppressive role of Migfilin in esophageal cancer cell motility as an example,55 Migfilin may have great but opposite effects on tumor metastasis. Thus, more prospective studies are required to determine whether Migfilin can be a significant novel target in cancer diagnosis and treatment.
Migfilin and its binding partners may exert an effect especially on skin disorders and platelet activation abnormalities. In normal human keratinocytes, Kindlin-1 forms focal adhesion complexes with Migfilin, integrin β1 subunit, FAK and α-actinin. The activated Rho family GTPase signaling mediates filopodial extensions, lamellipodia protrusions, and stress fibers, which plays an essential role in maintaining cell shape, adhesion to basement membranes, proliferation of keratinocytes. In the pathology of KS, the lack of these Kindlin-1-related biological functions forms the biological basis for fragility and atrophy of keratinocytes.46 It is possible that this process can proceed because an increase in Mig-2 expression level may compensate for lack of kindlin-1 functions. But the interplay between Kindlin-1, Migfilin and Mig-2 in the pathology of KS remains to be further explored.46 In normal proplatelet formation, integrin αIIbβ3 shifts into active conformation and meanwhile Talin and Kindlin-3 tend to bind with integrin β3 cytoplasmic domain. The absence of Migfilin promotes the Filamin A-integrin β3 binding, blocking the high affinity of Talin for the integrin β3 and impairing integrin activation, which induces proplatelet formation defect. There is a chance that Migfilin may bind to Filamin A, block its αIIbβ3-inhibiting effect and support a long-term Integrin αIIbβ3 activation. A further elaboration of the role of Migfilin in normalizing the proplatelet formation and the Migfilin-Filamin A interaction network in platelets is warranted.60
Because Migfilin possesses different protein–protein interaction domains,14 probing the detailed structure of Migfilin and the intricate Migfilin-containing adhesion network will undoubtedly help to explain the different expression levels and functions of Migfilin in different human diseases and help to provide a molecular and cellular basis for the generation of these comorbidities.
Data Sharing Statement
All data are available from Yanfei Li by request.
Acknowledgments
The authors thank Chuanyue Wu and Xudong Guo for helpful and constructive criticisms.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the establishment of title background, analysis, interpretation, or in all these areas; have drafted or written, or substantially revised or critically reviewed the article; have read and approved the final version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the matching project Fund of Shanghai University of Medicine and Health Sciences (E4-6200-20-201001), the Seed Fund of Shanghai University of Medicine and Health Sciences (E3-0200-20-201007-10), 2021 National Innovation and Entrepreneurship Training Program for College Students (A3-0200-21-309010-315) and Open Project Funding from the Department of Science and Technology (E4-6100-21-201048). This work has also been supported by the National Natural Science Foundation of China (31970599) and the clinical discipline project of Shanghai Pudong (PWYgy2018-03 and PWZzk2017-31).
Disclosure
The authors declare no competing interests in this work.
References
1. Winograd-Katz SE, Fassler R, Geiger B, et al. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol. 2014;15(4):273–288. doi:10.1038/nrm3769
2. Bachir AI, Horwitz AR, Nelson WJ, et al. Actin-based adhesion modules mediate cell interactions with the extracellular matrix and neighboring cells. Cold Spring Harb Perspect Biol. 2017;9(7):a023234. doi:10.1101/cshperspect.a023234
3. Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475(7356):316–323. doi:10.1038/nature10316
4. Nader GP, Ezratty EJ, Gundersen GG. FAK, talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat Cell Biol. 2016;18(5):491–503. doi:10.1038/ncb3333
5. Kobielak A, Fuchs E. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat Rev Mol Cell Biol. 2004;5(8):614–625. doi:10.1038/nrm1433
6. Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi:10.1146/annurev.cellbio.22.010305.104315
7. Harjunpaa H, Llort Asens M, Guenther C, et al. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol. 2019;10:1078. doi:10.3389/fimmu.2019.01078
8. Wu JC, Chen YC, Kuo CT, et al. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci Rep. 2015;5:18476. doi:10.1038/srep18476
9. Jorge R, Zarich N, Oliva JL, et al. hSos1 contains a new amino-terminal regulatory motif with specific binding affinity for its pleckstrin homology domain. J Biol Chem. 2018;293(29):11650. doi:10.1074/jbc.W118.004643
10. Yin LM, Schnoor M, Jun CD. Structural characteristics, binding partners and related diseases of the Calponin Homology (CH) domain. Front Cell Dev Biol. 2020;8:342. doi:10.3389/fcell.2020.00342
11. Winkelman JD, Anderson CA, Suarez C, et al. Evolutionarily diverse LIM domain-containing proteins bind stressed actin filaments through a conserved mechanism. PNAS. 2020;117(41):25532–25542. doi:10.1073/pnas.2004656117
12. Zheng Q, Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell. 2007;99(9):489–502. doi:10.1042/BC20060126
13. Gkretsi V, Zhang Y, Tu Y, et al. Physical and functional association of migfilin with cell-cell adhesions. J Cell Sci. 2005;118(Pt 4):697–710. doi:10.1242/jcs.01638
14. Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118(Pt 4):659–664. doi:10.1242/jcs.01639
15. Toeda Y, Kasamatsu A, Koike K, et al. FBLIM1 enhances oral cancer malignancy via modulation of the epidermal growth factor receptor pathway. Mol Carcinog. 2018;57(12):1690–1697. doi:10.1002/mc.22889
16. Hotta K, Kitamoto T, Kitamoto A, et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol Res. 2018;48(3):E320–E34. doi:10.1111/hepr.12992
17. Gkretsi V, Bogdanos DP. Experimental evidence of Migfilin as a new therapeutic target of hepatocellular carcinoma metastasis. Exp Cell Res. 2015;334(2):219–227. doi:10.1016/j.yexcr.2015.03.002
18. Ou Y, Ma L, Dong L, et al. Migfilin protein promotes migration and invasion in human glioma through epidermal growth factor receptor-mediated phospholipase C-gamma and STAT3 protein signaling pathways. J Biol Chem. 2012;287(39):32394–32405. doi:10.1074/jbc.M112.393900
19. Akazawa H, Kudoh S, Mochizuki N, et al. A novel LIM protein Cal promotes cardiac differentiation by association with CSX/NKX2-5. J Cell Biol. 2004;164(3):395–405. doi:10.1083/jcb.200309159
20. Siddiqui MQ, Badmalia MD, Patel TR. Bioinformatic analysis of structure and function of LIM domains of human zyxin family proteins. Int J Mol Sci. 2021;22(5):2647. doi:10.3390/ijms22052647
21. Larjava H, Plow EF, Wu C. Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 2008;9(12):1203–1208. doi:10.1038/embor.2008.202
22. Koch BJ, Ryan JF, Baxevanis AD. The diversification of the LIM superclass at the base of the metazoa increased subcellular complexity and promoted multicellular specialization. PLoS One. 2012;7(3):e33261. doi:10.1371/journal.pone.0033261
23. Shi X, Ma YQ, Tu Y, et al. The MIG-2/integrin interaction strengthens cell-matrix adhesion and modulates cell motility. J Biol Chem. 2007;282(28):20455–20466. doi:10.1074/jbc.M611680200
24. Tu Y, Wu S, Shi X, et al. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi:10.1016/s0092-8674(03)00163-6
25. Zhao J, Zhang Y, Ithychanda SS, et al. Migfilin interacts with Src and contributes to cell-matrix adhesion-mediated survival signaling. J Biol Chem. 2009;284(49):34308–34320. doi:10.1074/jbc.M109.045021
26. Liu Z, Lu D, Wang X, et al. Kindlin-2 phosphorylation by Src at Y193 enhances Src activity and is involved in Migfilin recruitment to the focal adhesions. FEBS Lett. 2015;589(15):2001–2010. doi:10.1016/j.febslet.2015.05.038
27. Lai-Cheong JE, Ussar S, Arita K, et al. Colocalization of kindlin-1, kindlin-2, and migfilin at keratinocyte focal adhesion and relevance to the pathophysiology of Kindler syndrome. J Invest Dermatol. 2008;128(9):2156–2165. doi:10.1038/jid.2008.58
28. Brahme NN, Harburger DS, Kemp-O’Brien K, et al. Kindlin binds migfilin tandem LIM domains and regulates migfilin focal adhesion localization and recruitment dynamics. J Biol Chem. 2013;288(49):35604–35616. doi:10.1074/jbc.M113.483016
29. Ithychanda SS, Hsu D, Li H, et al. Identification and characterization of multiple similar ligand-binding repeats in filamin: implication on filamin-mediated receptor clustering and cross-talk. J Biol Chem. 2009;284(50):35113–35121. doi:10.1074/jbc.M109.060954
30. Lad Y, Jiang P, Ruskamo S, et al. Structural basis of the migfilin-filamin interaction and competition with integrin beta tails. J Biol Chem. 2008;283(50):35154–35163. doi:10.1074/jbc.M802592200
31. Ithychanda SS, Das M, Ma YQ, et al. Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284(7):4713–4722. doi:10.1074/jbc.M807719200
32. Das M, Ithychanda SS, Qin J, et al. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PLoS One. 2011;6(10):e26355. doi:10.1371/journal.pone.0026355
33. Krause M, Dent EW, Bear JE, et al. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi:10.1146/annurev.cellbio.19.050103.103356
34. Barzik M, Kotova TI, Higgs HN, et al. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280(31):28653–28662. doi:10.1074/jbc.M503957200
35. Zhang Y, Tu Y, Gkretsi V, et al. Migfilin interacts with vasodilator-stimulated phosphoprotein (VASP) and regulates VASP localization to cell-matrix adhesions and migration. J Biol Chem. 2006;281(18):12397–12407. doi:10.1074/jbc.M512107200
36. Gkretsi V, Stylianou A, Stylianopoulos T. Vasodilator-Stimulated Phosphoprotein (VASP) depletion from breast cancer MDA-MB-231 cells inhibits tumor spheroid invasion through downregulation of Migfilin, beta-catenin and urokinase-plasminogen activator (uPA). Exp Cell Res. 2017;352(2):281–292. doi:10.1016/j.yexcr.2017.02.019
37. Papachristou DJ, Gkretsi V, Tu Y, et al. Increased cytoplasmic level of migfilin is associated with higher grades of human leiomyosarcoma. Histopathology. 2007;51(4):499–508. doi:10.1111/j.1365-2559.2007.02791.x
38. Xiao G, Cheng H, Cao H, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287(25):21450–21460. doi:10.1074/jbc.M111.331249
39. Gkretsi V, Papanikolaou V, Dubos S, et al. Migfilin’s elimination from osteoarthritic chondrocytes further promotes the osteoarthritic phenotype via beta-catenin upregulation. Biochem Biophys Res Commun. 2013;430(2):494–499. doi:10.1016/j.bbrc.2012.12.008
40. Ou Y, Wu Q, Wu C, et al. Migfilin promotes migration and invasion in glioma by driving EGFR and MMP-2 signalings: a positive feedback loop regulation. J Genet Genomics. 2017;44(12):557–565. doi:10.1016/j.jgg.2017.09.008
41. Papachristou DJ, Gkretsi V, Rao UN, et al. Expression of integrin-linked kinase and its binding partners in chondrosarcoma: association with prognostic significance. Eur J Cancer. 2008;44(16):2518–2525. doi:10.1016/j.ejca.2008.07.021
42. Gkretsi V, Papanikolaou V, Zacharia LC, et al. Mitogen-inducible Gene-2 (MIG2) and Migfilin expression is reduced in samples of human breast cancer. Anticancer Res. 2013;33(5):1977–1982.
43. He H, Ding F, Li Y, et al. Migfilin regulates esophageal cancer cell motility through promoting GSK-3beta-mediated degradation of beta-catenin. Mol Cancer Res. 2012;10(3):273–281. doi:10.1158/1541-7786.MCR-11-0419
44. Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. doi:10.1016/j.jaad.2014.01.903
45. Guerrero-Aspizua S, Conti CJ, Escamez MJ, et al. Assessment of the risk and characterization of non-melanoma skin cancer in Kindler syndrome: study of a series of 91 patients. Orphanet J Rare Dis. 2019;14(1):183. doi:10.1186/s13023-019-1158-6
46. Has C, Herz C, Zimina E, et al. Kindlin-1 Is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am J Pathol. 2009;175(4):1442–1452. doi:10.2353/ajpath.2009.090203
47. Herz C, Aumailley M, Schulte C, et al. Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J Biol Chem. 2006;281(47):36082–36090. doi:10.1074/jbc.M606259200
48. Rognoni E, Widmaier M, Jakobson M, et al. Kindlin-1 controls Wnt and TGF-beta availability to regulate cutaneous stem cell proliferation. Nat Med. 2014;20(4):350–359. doi:10.1038/nm.3490
49. Petricca G, Leppilampi M, Jiang G, et al. Localization and potential function of kindlin-1 in periodontal tissues. Eur J Oral Sci. 2009;117(5):518–527. doi:10.1111/j.1600-0722.2009.00651.x
50. Azorin P, Bonin F, Moukachar A, et al. Distinct expression profiles and functions of Kindlins in breast cancer. J Exp Clin Cancer Res. 2018;37(1):281. doi:10.1186/s13046-018-0955-4
51. Ou Y, Zhao Z, Zhang W, et al. Kindlin-2 interacts with β-catenin and YB-1 to enhance EGFR transcription during glioma progression. Oncotarget. 2016;7(46):74872–74885. doi:10.18632/oncotarget.12439
52. Mahawithitwong P, Ohuchida K, Ikenaga N, et al. Kindlin-1 expression is involved in migration and invasion of pancreatic cancer. Int J Oncol. 2013;42(4):1360–1366. doi:10.3892/ijo.2013.1838
53. Kong J, Du J, Wang Y, et al. Focal adhesion molecule Kindlin-1 mediates activation of TGF-β signaling by interacting with TGF-βRI, SARA and Smad3 in colorectal cancer cells. Oncotarget. 2016;7(46):76224–76237. doi:10.18632/oncotarget.12779
54. Wang Z, Yang Y, Cui Y, et al. Tumor-associated macrophages regulate gastric cancer cell invasion and metastasis through TGFbeta2/NF-kappaB/Kindlin-2 axis. Chin J Cancer Res. 2020;32(1):72–88. doi:10.21147/j.issn.1000-9604.2020.01.09
55. Liu G, Bao Y, Liu C, et al. IKKepsilon phosphorylates kindlin-2 to induce invadopodia formation and promote colorectal cancer metastasis. Theranostics. 2020;10(5):2358–2373. doi:10.7150/thno.40397
56. Kato K, Shiozawa T, Mitsushita J, et al. Expression of the mitogen-inducible gene-2 (mig-2) is elevated in human uterine leiomyomas but not in leiomyosarcomas. Hum Pathol. 2004;35(1):55–60. doi:10.1016/j.humpath.2003.08.019
57. Feng Y, Chen MH, Moskowitz IP. Filamin A (FLNA) is required for cell–cell contact in vascular development and cardiac morphogenesis. PANS. 2006;103(52):19836–19841. doi:10.1073/pnas.0609628104
58. Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115(12):3363–3369. doi:10.1172/JCI26989
59. Donada A, Balayn N, Sliwa D, et al. Disrupted filamin A/alphaIIbbeta3 interaction induces macrothrombocytopenia by increasing RhoA activity. Blood. 2019;133(16):1778–1788. doi:10.1182/blood-2018-07-861427
60. Metcalf DG, Moore DT, Wu Y, et al. NMR analysis of the alphaIIb beta3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc Natl Acad Sci U S A. 2010;107(52):22481–22486. doi:10.1073/pnas.1015545107
61. Zhou Y, Hu M, Chen X, et al. Migfilin supports hemostasis and thrombosis through regulating platelet alphaIIbbeta3 outside-in signaling. Haematologica. 2020;105(11):2608–2618. doi:10.3324/haematol.2019.232488
62. Haubner BJ, Moik D, Schuetz T, et al. In vivo cardiac role of migfilin during experimental pressure overload. Cardiovasc Res. 2015;106(3):398–407. doi:10.1093/cvr/cvv125
63. Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275(25):19250–19257. doi:10.1074/jbc.M909099199
64. Menashi EB, Loftus JC. Differential effects of Pyk2 and FAK on the hypertrophic response of cardiac myocytes. Cell Tissue Res. 2009;337(2):243–255. doi:10.1007/s00441-009-0807-9
65. Zemljic-Harpf AE, Godoy JC, Platoshyn O, et al. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J Cell Sci. 2014;127(Pt 5):1104–1116. doi:10.1242/jcs.143743
66. Brancaccio M, Hirsch E, Notte A, et al. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70(3):422–433. doi:10.1016/j.cardiores.2005.12.015
67. Ussar S, Wang HV, Linder S, et al. The Kindlins: subcellular localization and expression during murine development. Exp Cell Res. 2006;312(16):3142–3151. doi:10.1016/j.yexcr.2006.06.030
68. Horton ER, Humphries JD, James J, et al. The integrin adhesome network at a glance. J Cell Sci. 2016;129(22):4159–4163. doi:10.1242/jcs.192054
69. Legerstee K, Geverts B, Slotman JA, et al. Dynamics and distribution of paxillin, vinculin, zyxin and VASP depend on focal adhesion location and orientation. Sci Rep. 2019;9(1):10460. doi:10.1038/s41598-019-46905-2
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.