Back to Journals » OncoTargets and Therapy » Volume 7
Microtubule-targeting agents in oncology and therapeutic potential in hepatocellular carcinoma
Received 11 November 2013
Accepted for publication 28 January 2014
Published 16 April 2014 Volume 2014:7 Pages 575—585
DOI https://doi.org/10.2147/OTT.S46019
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Herbert H Loong, Winnie Yeo
Department of Clinical Oncology, Sir YK Pao Centre for Cancer, Hong Kong Cancer Institute, State Key Laboratory in Oncology in South China, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, NT, Hong Kong
Abstract: In mammalian cells, microtubules are present both in interphase and dividing cells. In the latter, microtubules forming the mitotic spindle are highly dynamic and exquisitely sensitive to therapeutic inhibitors. Developed to alter microtubule function, microtubule-binding agents have been proven to be highly active as an anticancer treatment. Significant development of microtubule-binding agents has taken place in recent years, with newer anti-tubulin agents now showing novel properties of enhanced tumor specificity, reduced neurotoxicity, and insensitivity to chemoresistance mechanisms. Hepatocellular carcinoma remains one of the most difficult cancers to treat, with chemotherapies being relatively ineffective. There is now evidence to suggest that microtubule-binding agents may be effective in the treatment of hepatocellular carcinoma, especially when used in combination with mammalian target of rapamycin inhibitors. Preclinical models have suggested that the latter may be able to overcome resistance to microtubule binding agents. In this review article, recent developments of novel microtubule binding agents and their relevance to the treatment of hepatocellular carcinoma will be discussed.
Keywords: chemotherapy, microtubule-binding agents, microtubular stabilization and destabilization, mTOR inhibition
Introduction
Hepatocellular carcinoma (HCC) is the third commonest cause of cancer-related deaths worldwide.1,2 While surgical resection and liver transplantation are the two mainstays of treatment with a potential for cure, these approaches can only be applied to patients with early-stage HCC.3 The majority of patients diagnosed with HCC are not amenable to a curative resection, or eventually progress despite locoregional treatments at which point systemic treatment, with a palliative intent, is considered. Cytotoxic chemotherapies have not been effective. To date, the only systemic agent that has been shown to provide survival benefit over best supportive care is sorafenib, a multikinase inhibitor of vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR) and Raf.4–6 However, the overall prognosis of HCC patients remains poor. There is thus a need to identify better therapies for patients with advanced disease.
Gene expression profiling studies have recently shown that microtubule-related cellular assembly and organization is the most crucial event in HCC development,7–9 suggesting microtubules to be an important target for therapeutic intervention in HCC. Microtubule-targeting agents are compounds that bind to soluble and/or polymerized tubulin in the microtubules, thus affecting microtubule function. Microtubules, constituting the mitotic spindle in dividing mammalian cells, are exquisitely sensitive to therapeutic inhibitors. For this reason, several microtubule-binding agents (MBAs) have been developed with different aims, including for use as pesticides, antiparasitics, and cytotoxic chemotherapeutics. These agents exert antimitotic activity and inhibit cell proliferation by inducing cell cycle arrest and apoptosis.
In the age of small-molecule and monoclonal antibody-based targeted therapies in cancer treatment, it is noteworthy that significant resources are still being devoted to the identification and evaluation of novel MBAs. This is partly due to the large untapped reservoir of potential therapeutic natural compounds, which influence microtubule dynamics, and also our growing understanding of the role of microtubule cytoskeleton in cancer cells.
This review will serve to highlight recent findings and to speculate on the potential and future directions of microtubule targeting, alone or in combination with other compounds, with focused discussion on the potential role of this class of agent in HCC.
Biology of the microtubule
The microtubule has a critical regulatory function that affects not only mitosis, but also cytoskeletal shape, cell motility, and intracellular protein and organelle transport.10 Microtubules are composed of α- and β-tubulin subunits, each consisting of approximately 450 amino acids with a molecular weight of 50,000 Da, and are compacted into very complex monomeric structures.11 The unique functions of microtubules and the principal actions of anti-microtubule agents relate to the dynamic equilibrium between α- and β-tubulin heterodimer subunits and the microtubule polymer. Tubulin polymerization occurs by a nucleation–elongation mechanism, in which the slow formation of a short microtubule “nucleus” is followed by rapid elongation of the microtubule at its ends by the reversible, noncovalent addition of α- and β-tubulin heterodimers. In essence, the microtubule polymer is in a complex and dynamic equilibrium with the intracellular pool of α- and β-tubulin heterodimers, which incorporates free heterodimers into the polymerized structure and simultaneously releases heterodimers into the soluble tubulin pool. Microtubule assembly and disassembly occur simultaneously at both ends of the microtubule, and the net effect, elongation versus shortening, is determined by several factors, including the concentration of free tubulin and various chemical mediators such as magnesium and calcium ions and guanosine triphosphate.11
Two fundamental processes govern microtubule dynamics in vivo. The first, known as treadmilling, is the net growth at one end of the microtubule and the net shortening at the opposite end.12,13 Treadmilling plays a role in many microtubule functions, most notably the polar movement of the chromosomes during the anaphase stage of mitosis. The second dynamic behavior, termed dynamic instability, occurs when the plus ends of individual microtubules switch spontaneously between states of slow sustained growth and rapid shortening.11 The rate of dynamic instability is accelerated during mitosis, which results in the formation and attachment of the mitotic spindles to the chromosomes. The rate and magnitude of both dynamic instability and treadmilling can be altered by microtubule-associated proteins and other regulatory proteins, variable expression of tubulin isotypes, posttranslational tubulin modifications, and tubulin mutations.14,15
MBAs disrupt microtubule dynamics that involve the mitotic spindle. Suppression of the two major dynamic processes of the mitotic spindle, namely treadmilling and dynamic instability, are the principal means by which MBAs disrupt cellular function and induce cell death.
Mechanisms of action of MBAs
Having been first isolated half a century ago, the vinca alkaloids and, subsequently, the taxanes are two families of MBAs currently administered in various indications including solid tumors and hematological malignancies. Table 1 highlights a selection of MBAs currently in clinical practice or undergoing clinical trials.
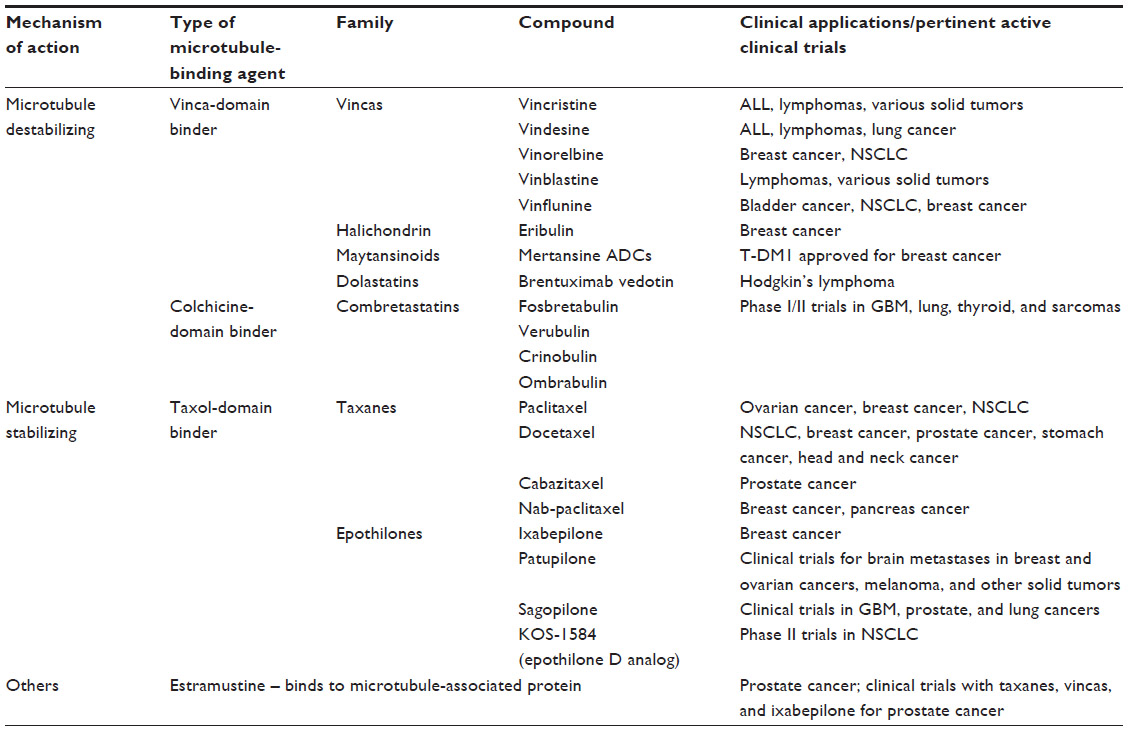 | Table 1 Microtubule-binding agents currently in clinical practice or in development in clinical trials |
While vinca alkaloids are often given in combination regimens with other cytotoxics, taxanes have established their role both as single agents and as an essential partner in doublet chemotherapies, often in combination with platinum-based agents. The armamentarium of MBAs also consists of newer agents, including epothilones and halichondrins, together with a resurgence of interest in maytansinoids when incorporated into antibody–drug conjugates (ADCs). MBAs are peculiar in their structural diversity and complexity. Significant difficulties have been encountered in their development, as many agents were isolated from marine organisms or flora, which were not cultivated and only present in minute amounts. Demand for these agents to be available in the clinic has also led to improvements in partial and total synthesis techniques.
Destabilization and stabilization of microtubules
MBAs are often classified into two major groups: microtubule-destabilizing and microtubule-stabilizing agents. Both classes of drug potently suppress microtubule dynamics. Microtubule-destabilizing agents inhibit microtubule polymerization when present at high concentrations, typically binding to the vinca or colchicine domains, of which the former is more readily available in clinical practice. Vinca-domain binders include the vinca alkaloids (vincristine, vinblastine, vindesine, and vinorelbine), originally isolated from the periwinkle plant Catharanthus rosea, which represent the oldest and most diverse family of MBAs and have helped improve treatment outcomes in both hematological and solid malignancies. Their oncolytic activity is due to inhibition of formation and possibly the disruption of mitotic spindle microtubules, thereby arresting cell division in metaphase.16 Vinflunine is a novel fluorinated vinca alkaloid obtained by semi-synthesis using superacid chemistry to selectively introduce two fluorine atoms at the 20′ position of catharanthine moiety of vinorelbine.17 Vinflunine is currently approved in Europe by the European Medicines Agency (EMEA), as a second-line agent for inoperable or metastatic urothelial carcinoma, in combination with cisplatin as doublet chemotherapy.18 It has also seen clinical activity in advanced non-small-cell lung cancer19 and in breast cancers,20 even in patients refractory to vinorelbine treatment.20
Eribulin, a synthetic derivative of the marine sponge natural product halichondrin B, is a naturally occurring mitotic inhibitor with a unique mechanism of action of binding predominantly to a small number of high-affinity sites at the plus ends of existing microtubules.21 Eribulin exerts its anticancer effects by triggering apoptosis of cancer cells following prolonged irreversible mitotic blockade. It has been shown to be active in patients with metastatic breast cancer relapsing after anthracyclines and taxanes.22 In a Phase III open-label randomized study22 that investigated eribulin versus the physician’s choice of treatment, overall survival was significantly improved in women assigned to eribulin compared with the physician’s choice (13.1 versus 10.6 months, respectively; P=0.041). Neutropenia was commoner in patients receiving eribulin (52% versus 30% at all grades) and peripheral neuropathy was the commonest adverse event ([AE] (5%) leading to discontinuation of eribulin.
Recent development of ADCs utilizes antibodies to deliver cytotoxic agents specifically to antigen-expressing tumors. ADCs with potent MBAs as payloads have created a new and exciting role for MBAs in anticancer therapy. Maytansinoids and auristatins both bind to the vinca-binding domain of tubulin and have similar cytotoxic potency in the picomolar range. The ADCs made with these tubulin-binding agents release the active payload after internalization and processing within endosomes or lysosomes.23–25 Brentuximab vedotin comprises the anti-CD30 monoclonal antibody cAC10 conjugated to the cytotoxic agent monomethyl auristatin E, a synthetic analog of the tubulin polymerization inhibitor dolastatin 10, via linkage to a chimeric immunoglobulin (Ig)G1 antibody formed by partial reduction of interchain disulfide bonds of Ig. Brentuximab vedotin has been granted accelerated conditional approval by the US Food and Drug Administration (FDA) after its efficacy was proven in a single-arm Phase II trial of Hodgkin’s lymphoma patients who had progressed after an autologous stem cell transplant, or in patients who were ineligible for transplant, having progressed on at least two chemotherapy regimens.26 Tumor reductions were seen in 94% of the over 100 patients recruited in the trial.
Trastuzumab emtansine (T-DM1) combines the humanized IgG1 anti-human epidermal growth factor 2 (HER2) antibody, trastuzumab, with the maytansinoid DM1 utilizing an uncleavable cross-linking agent. This particular conjugate design was selected for clinical development based on potent antiproliferative activity in vitro and antitumor activity with reduced toxicity in vivo in preclinical models including HER2-overexpressing models resistant to trastuzumab.27 Its clinical development has been promising. The landmark EMILIA Phase III trial28 that compared T-DM1 with lapatinib and capecitabine in metastatic HER2-positive breast cancer patients who had previously received trastuzumab treatment showed significant improvement in efficacy endpoints in the treatment arm, as well as fewer grade 3 or 4 AEs. The robust antitumor activity and good tolerability of T-DM1 has led to a significant paradigm shift in the treatment in HER2-positive metastatic breast cancer, and the study has led to clinical approval for its use in HER2-positive metastatic breast cancer in the second-line setting.28 A recent Phase II study of T-DM1 versus trastuzumab in combination with docetaxel in trastuzumab-naïve HER2-positive metastatic breast cancer patients as first-line treatment29 showed significantly better progression-free survival in the T-DM1 arm (14.2 months versus 9.2 months; hazard ratio 0.59; 95% confidence interval: 0.36–0.97). Fewer grade 3 or more severe AEs were also documented in the T-DM1 arm, and a significantly lower number of AEs leading to discontinuation of treatment was noted. In addition, a randomized Phase III study involving T-DM1 seeks to confirm the benefit of this agent in the first-line setting; enrollment has been completed and analysis of results is pending (NCT01120184).30
Colchicine-site binders include colchicine and its analogs, combretastatins, podophyllotoxin, 2-methoxyestradiol, steganacins, and curacins.31 Combretastatins in particular have been shown to target and disrupt vasculature, possibly by their effects on the microtubule cytoskeleton of endothelial cells. In Phase I trials of combretastatin A4, unusual toxicities such as tumor pain, ataxia, and cardiovascular modifications, including prolonged corrected QT interval and electrocardiography changes consistent with acute coronary syndrome,32–34 have been observed. A key issue for the approval of this family of agents in clinical use will be the ability to limit its toxicities in the normal vasculature.
Microtubule-stabilizing agents enhance microtubule polymerization at high drug concentrations. The most representative group in this category is taxol-domain binding agents, including paclitaxel and docetaxel, which are commonly used in the treatment of breast and non-small-cell lung cancers, and cabazitaxel, which has recently received approval for use in metastatic prostate cancer that has failed prior docetaxel therapy. Unlike other tubulin-targeting drugs such as colchicine that inhibit microtubule assembly, taxanes stabilize the microtubule polymer and protect it from disassembly.35 Chromosomes are thus unable to achieve a metaphase spindle configuration. This blocks progression of mitosis, and prolonged activation of the mitotic checkpoint triggers apoptosis or reversion to the G phases of the cell cycle without cell division.
Issues with currently available taxanes include the fact that their water solubility is poor and the use of surfactants, such as Cremophor EL® - BASF SE (Limburgerhof, Germany) in the case of paclitaxel, or ethanol in the cases of docetaxel and cabazitaxel, is required for intravenous administration, thus increasing the risk of hypersensitivity reactions.36 Novel taxane formulations have also been developed with the intent of reducing issues associated with poor solubility or hypersensitivity.37 Nanoparticle albumin-bound (nab)-paclitaxel was associated with better outcome, a lower rate of severe neutropenia, and a similar rate of reversible sensory neuropathy when pitched against conventional docetaxel therapy in patients with advanced metastatic breast cancer.38
The other class of microtubule-stabilizing agents under the taxane-domain-binder category tested and in clinical practice includes epothilones, which were originally isolated from the myxobacterium Sorangium cellulosum. Although taxanes and epothilones occupy the same binding site on β-tubulin, electron crystallography studies and molecular modeling suggest that epothilones do not share a common pharmacophore with taxanes.39 They have shown promise as a novel family of agents for cancer treatment as they may retain activity against taxane-resistant tumors.
Ixabepilone is a semi-synthetic analog of epothilone B.40 Ixabepilone was developed to overcome the narrow therapeutic index of epothilone B and is thought to be more metabolically stable. The increased stability of ixabepilone may be due to the substitution of the lactone with a lactam, which reduces its metabolism by carboxylesterase.40 Compared with paclitaxel, ixabepilone showed similar antitumor activity in paclitaxel-sensitive tumors and three- to fivefold higher activity in paclitaxel-resistant tumors that overexpress P-glycoprotein or contain a tubulin mutation.41 Moreover, ixabepilone had better antitumor activity against a panel of pediatric tumor xenograft models compared with paclitaxel, which was either ineffective or effective only at toxic doses.42 Ixabepilone is approved as monotherapy for the treatment of locally advanced or metastatic breast cancer (MBC) resistant or refractory to anthracyclines, taxanes, and capecitabine in the United States. Additionally, it is indicated in combination with capecitabine in patients with locally advanced or MBC that is resistant to anthracyclines and taxanes or resistant to taxanes with contraindications to anthracycline therapy. However, in a randomized Phase II study as first-line therapy for metastatic breast cancer in which ixabepilone was compared to weekly paclitaxel, both combined with bevacizumab, ixabepilone was reported to have similar efficacy relative to weekly paclitaxel but was associated with significantly greater risk of grade 3 or 4 neutropenia.43
Patupilone is several times more potent than taxanes in its ability to induce tubulin dimerization and stabilizing microtubules in vitro and appears to have a different β-tubulin binding site than taxanes.44,45 Importantly, patupilone is also not affected by common tumor resistance mechanisms, including β-tubulin mutation and overexpression of drug efflux pumps.46–49 Its antitumor activity has been shown in several clinical trials with various tumor types, including prostate, ovarian, and colon cancers.50–53
In addition, estramustine is a stable conjugate of estradiol and nornitrogen mustard with antimitotic properties. Binding of the drug to microtubule-associated proteins, tubulin, and proteins of the nuclear matrix are presently considered to be the most likely mechanisms underlying the cytotoxicity of estramustine.54 The agent is used in androgen-independent prostatic carcinoma,55 and ongoing trials55–57 are testing its role in combination with taxanes, vincas, and ixabepilone for prostate cancer.
Further to the above, the family of serine/threonine kinases called Aurora is emerging as an extremely important controller of cell mitosis, which is essential to maintaining genomic stability. The Aurora kinase inhibitors, although not strictly belonging to microtubule-targeted agents, are another class of agents that control cell mitoses by regulating the formation of the mitotic spindle in cell division. This is particularly important because aurora kinase is the first defense against the possible development of an aneuploid clone and is the controller of correct chromosome segregation. Aberrant expression of one or more of the three members of the Aurora family (Aurora A, B, or C) has been observed in many solid and hematological malignancies.59
Antiangiogenic effects and associated vascular disruption
Formation of new blood vessels is an integral part of migration of endothelial cells and tumor progression. Two approaches to inhibit vascular formation are to inhibit angiogenesis and to destroy the integrity of existing tumor vasculature using vascular-disruptive agents,60 with processes appearing to be sensitive to microtubule-targeted drugs.31 Vinca alkaloids have been shown to damage tumor vasculature in animal models. For example, in vitro studies have shown that vinflunine can induce a rapid change in the morphology of endothelial cells and disrupt the network of capillary-like structures, indicating potential vascular-disrupting activity.61 Furthermore, vinflunine also shows antiangiogenic properties, since it inhibits endothelial cell migration and the capacity of these cells to organize into a network of capillary-like structures. While it is unclear whether antitumor activity manifested by these compounds is due to classical antimitotic effects on microtubule dynamics or vascular disruption or both, MBAs should be regarded as having antiangiogenic activities. This belief has also prompted novel methods aiming to assess changes in tumor perfusion, such as dynamic contrast-enhanced magnetic resonance imaging measurements of gadolinium uptake and washout as well as positron emission tomography.
MBAs in HCC
Throughout the last 2 decades, extensive preclinical studies with various classes of cytotoxics have reconfirmed the fact that HCC is a relatively chemoresistant tumor. Doxorubicin is a cytotoxic agent that has been tested in various settings over the past few decades.62 Aside from intravenous administration, prior studies have also investigated different methods of drug delivery in the form of doxorubicin drug-eluting beads given via a transarterial approach63–65 or the use of thermosensitive liposomal doxorubicin66,67 as adjunctive treatment on top of radiofrequency ablation of individual tumors. These different studies showed varying degrees of success.
A recent advancement for systemic therapy in HCC was made when two large Phase III randomized controlled trials showed that sorafenib, a multikinase antiangiogenic small molecule tyrosine kinase inhibitor, can lead to an improvement in overall survival in both Western and Asian patients with inoperable HCC when compared to best supportive care.4,6,68,69 There has been a continuing desire amongst investigators to identify other systemic agents that may also bear therapeutic potential. Microtubule assembly is a potential therapeutic target in HCC, and a number of systemic agents that target this have been investigated.
Vinca alkaloids
Surprisingly, although often considered as the most mature group of MBAs, and with several of these agents currently approved for other oncological indications that are in routine clinical practice, vinca alkaloids as single agents have not been extensively studied in HCC.
Vindesine was tested in a Phase II study in 16 patients with advanced HCC but did not show a therapeutic effect.70 Vinflunine has been studied in a Phase I dose-escalation trial in patients with liver dysfunction.71 A high number of patients with HCC and concomitant liver cirrhosis (Child-Pugh grades A and B) were included in the study (18/30 patients). A subgroup analysis was carried out in these patients to estimate vinflunine’s antitumor activity. All 18 patients were outside Milan criteria and ineligible for liver transplantation. Thirteen patients had previously been treated with chemotherapy. The World Health Organization performance status scores of these patients were predominantly 0–1. One partial response (PR) was observed while eleven patients (61.1%) had stable disease yielding a disease control rate (PR + stable disease) of 66.7%. The median progression-free survival was 3.4 months (95% confidence interval: 2.6–6.0). Myelosuppression was one of the most common toxicities. However, there was no occurence of febrile neutropenia. Fatigue and abdominal pain were other less common adverse events.71 Although the disease control rate appeared promising, at the time of preparation of this manuscript, we were not aware of any dedicated clinical trials of vinflunine for HCC.
Taxanes
Taxanes, specifically docetaxel and paclitaxel, have been shown72,73 to have tumor suppressive effects in preclinical models on HCC cell lines. As drug delivery by nanoparticles has been shown to allow for improved efficacy with reduced side effects in other tumors, nab-paclitaxel was also studied in a panel of HCC cell lines.9 Nab-paclitaxel showed an effective half maximal inhibitory concentration dose that was 15-fold lower than that of paclitaxel or docetaxel. When compared with doxorubicin, the half maximal inhibitory concentration dose (IC50) was 450-fold lower. Flow cytometry confirmed that nab-paclitaxel induced a cell cycle blockade at G2/M phase, thus leading to increased apoptosis following treatment.9 In vivo animal models also showed that nab-paclitaxel readily inhibited xenograft growth with less toxicity to host cells compared to other MBAs and doxorubicin.9 In addition, it has been reported that STMN1 is a key factor in controlling microtubule dynamics; this microtubule regulatory gene is critical to cellular processes such as cell motility and cell division during mitosis.74,75 A direct synergistic effect has been reported when nab-paclitaxel is administered with gene silencing of STMN1.9
Clinical investigations of taxanes, however, have been less promising. In a Phase I clinical trial, a weekly 1-hour infusion of paclitaxel (70 mg/m2 starting dose escalated to a maximum of 100 mg/m2) in patients with unresectable HCC resulted in a 63% disease stabilization rate.76 However, in a Phase II clinical trial, paclitaxel (175 mg/m2) administered every 3 weeks failed to show anticancer effects in patients with HCC.77 It is unclear whether this discrepancy in clinical activity between the weekly versus 3-weekly regimen is associated with the fact that activity of paclitaxel is directly related to the cell cycle, and whether it is for this reason that more frequent administration might improve efficacy. In breast cancer, however, paclitaxel administered in a more continuous low-dose manner has been shown to exhibit more proapoptotic and antiangiogenic properties, increasing its antineoplastic effects.78,79
Docosahexaenoic acid (DHA)–paclitaxel (Taxoprexin®, Protarga Pharmaceuticals, Philadelphia, USA), a conjugate formed by the covalent linkage of the fatty acid DHA with paclitaxel, was developed with an aim of increasing drug exposure without worsening toxicity. A single-center Phase II trial of DHA-paclitaxel in advanced primary cancers of the liver, including HCC as well as gallbladder and biliary tract cancers (NCT00422877),80 was registered as having been completed in December 2008. However, results of this study do not appear to be readily available in published literature.
Epothilones
Six epothilones have been studied in preclinical and clinical trials,81 with patupilone (epothilone B) being the most investigated in HCC. Specifically to HCC, preclinical studies with patupilone as a single agent have shown it to be more potent than docetaxel, paclitaxel, or doxorubicin in HCC cell lines.44 This approach has also been taken to a two-stage Phase II clinical trial,82 which demonstrated an acceptable safety profile but with only modest clinical activity. Of the 24 evaluable patients recruited into the first stage, only one patient had a confirmed partial response through four cycles. Median progression-free survival was 3 months, and ten patients (40%) progressed within the first two cycles. Initial results of this study did not meet the predetermined efficacy criteria to proceed onto the second stage of the study.
Aurora kinase inhibitors
The overexpression of Aurora kinase B, which specifically regulates chromosome segregation, cytokinesis, and protein localization to the centromere and kinetochore during mitosis, is correlated with genetic instability in HCC.83–85 It has been identified as an independent predictor of recurrence.84,86 Small molecule inhibitors of Aurora kinases are undergoing Phase I and II trials87–89 in various malignancies, and these may potentially be ideal candidates for treatment of HCC.
MBAs in combination with PI3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) inhibition
Prior studies have shown that activation of the upstream signaling molecule of the mTOR, namely Akt, results in marked increase in resistance against microtubule-directed agents, while mTOR inhibitors were able to inhibit this PI3K/Akt/mTOR-associated resistance.90 As both microtubules and the mTOR pathway are considered druggable targets with therapeutic compounds for other indications already developed and available, the combination of these agents in the treatment of HCC has also been investigated. mTOR is a key effector in the PI3K/Akt/mTOR pathway and plays a critical role in regulating cell proliferation, survival, and angiogenesis;91 as such, it is a prominent potential therapeutic target for HCC. Aberrant activation of mTOR is common in HCC, accounting for ~45% of cases, and its overexpression has been associated with poor prognosis in HCC patients.92–94 The downstream targets of mTOR include ribosomal p70S6 kinase and the eukaryotic initiation factor binding protein (4E-BP1), which regulates cell cycle, cell growth, and protein synthesis.95 Recently, it has been shown that the mTOR inhibitor everolimus specifically impaired cell-cycle progression in non-transformed hepatocytes with DNA damage in vivo, and long-term treatment with everolimus markedly delayed liver tumor development that is induced by DNA damage, indicating that mTOR activation has a substantial effect on hepatocarcinogenesis.96
Aside from the mTOR inhibitor everolimus, the antitumor effects of the PI3K inhibitor BEZ235 and BKM120 were also studied preclinically in seven hepatoma cell lines as monotherapy and combination therapy with doxorubicin, cisplatin, irinotecan, or fluorouracil in vitro and in xenografts.97 Targeting the PI3-kinase/Akt/mTOR pathway at multiple levels may therefore provide more effective antitumor activity than selective inhibition of only one component of the pathway. Furthermore, an upstream inhibition of the pathway by blocking PI3K not only prevents feedback activation, but also inhibits the activation of several other kinases, transcription factors, and proteins that promote survival and growth of cancer cells. Treatment with these compounds reduced tumor growth mainly by inhibiting cell-cycle progression rather than induction of apoptosis. Clinically, mTOR targeting with everolimus has shown a modest antitumor effect in HCC, with a disease control rate of 44% in a Phase I/II study.98 A global Phase III study of everolimus in patients with HCC after progression on sorafenib (EVerOlimus for LiVer cancer Evaluation-1 [EVOLVE-1]) NCT0103522999 has recently been reported to have failed to reach its primary endpoint in improving OS with everolimus.100
Temsirolimus has also undergone a Phase I/II clinical trial as a single agent in patients with unresectable HCC.101 The Phase I portion identified 25 mg weekly every 3 weeks to be the maximum tolerated dose. In the study population, over half of whom had had prior systemic therapy for advanced HCC, temsirolimus demonstrated tumor stabilization of more than 12 weeks in 39% of patients. Temsirolimus has also been studied in combination with sorafenib.102 The Phase I dose-finding trial established a lower maximum tolerated dose of this combination in HCC patients than in those with other malignancies; the recommended Phase II dose for HCC is 10 mg intravenous temsirolimus weekly and 200 mg sorafenib by mouth twice daily.102 The Phase II trial for the combination is currently ongoing (NCT01687673).103
It is believed that, in most cancers, the effect of mTOR targeting is indeed largely cytostatic, which may limit its potential as a single agent in cancer therapy.104 Combination treatment with a cytotoxic agent may represent a more effective therapeutic direction.105 Prior studies have demonstrated involvement of the mTOR pathway in resistance to MBAs;90 activation of Akt has been shown to result in a marked increase in resistance against microtubule-directed agents, while rapamycin (an mTOR inhibitor) treatment was able to inhibit this PI3K/Akt/mTOR-associated resistance.90 This has led to the hypothesis that co-targeting mTOR and microtubules may be a more potent treatment strategy for HCC. There is now renewed enthusiasm about the potential activity of patupilone in combination with mTOR inhibitors. Indeed, recent in vivo and in vitro studies have shown that a combination of mTOR inhibitor temsirolimus and low-dose vinblastine had a marked antitumor effect.106 Similarly, everolimus was also investigated with low-dose patupilone, with promising results45 reconfirming a synergistic effect between mTOR inhibitors and MBAs in HCC cells. More interestingly, accompanying mechanistic studies demonstrated that the marked antitumor activity of the combination was not affected by further suppression of the mTOR signaling pathway compared with everolimus alone, but may be due to possible induction of apoptosis in these HCC models mediated by the combination treatment.45 The combination was also found to induce a significant reduction in microvessel density in tumors when compared to those treated with vehicle control, indicating potential antiangiogenic activity.45
With the hypotheses of the potential advantages in combination treatment of mTOR inhibition and microtubule targeting having generated exciting preclinical results, there is now a need to translate these into clinical investigations and early-phase human trials.
MBA toxicities and potential implications for the treatment of HCC
While there have been few dedicated clinical investigations of MBAs in patients with HCC, common toxicities of MBAs may pose a challenge – either when MBAs are used as single agents or in combination treatment. Treatment of inoperable HCC is often complicated by medical comorbidities. The commonest and most obvious comorbid condition is liver cirrhosis, which in itself can have significant implications for a patient’s functional condition and body reserve.
Neuropathy induced by established MBAs is potentially a severe and dose-limiting side effect,107 which is also dose-cumulative. It usually manifests as a painful and debilitating peripheral axonal neuropathy for which there is currently no effective symptomatic treatment.108 Patients with preexisting neuropathies, such as in cases of chronic alcoholism, which is also a risk factor for HCC, may be more severely affected. A major difficulty in the screening of novel MBAs for neuropathy is the lack of adequate preclinical models. Myeloid toxicity is also frequently observed with MBAs, with subtle differences between compounds within the same family.31 Neutropenia is often the most frequent and/or severe side effect, but thrombocytopenia remains a concern. This may be particularly relevant to HCC patients with low platelet and white blood cell levels due to preexisting liver cirrhosis and portal hypertension. Bleeding risks associated with thrombocytopenia may be magnified by potential coagulopathies in this subset of patients. On the other hand, some toxicities are relatively compound specific, such as fluid retention observed in patients receiving docetaxel31 or diarrhea after patupilone therapy.109,110
Given the complexity of physiological states of patients with HCC and its comorbidities, prior dose-determining studies in other patient populations may not necessarily apply to patients with HCC.
Conclusion
As most patients with HCC present with advanced and inoperable disease, there is still an unmet need for better systemic treatment. While sorafenib has made some headway in this regard, other lines of systemic therapy need to be developed to provide options for patients who become refractory to or who are ineligible for sorafenib.
Preclinical studies have suggested that MBAs appear to be a class of agents that may fit into this armamentarium. There is a need, however, to establish whether a single-agent sequential approach or a combination approach, possibly at low doses in combination with mTOR inhibition, may lead to better results. Given the fact that patients with advanced/inoperable HCC can be physiologically distinct from other populations, there is also a need to embark on early-phase dose-finding trials, preferably in the setting of early-phase studies, to establish toxicity and tolerability profiles in this unique patient population.
Disclosure
The authors report no conflicts of interest in this work.
References
He J, Gu D, Wu X, et al. Major causes of death among men and women in China. N Engl J Med. 2005;353:1124–1134. | |
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. | |
Poon RT, Fan ST, Lo CM, et al. Improving survival results after resection of hepatocellular carcinoma: a prospective study of 377 patients over 10 years. Ann Surg. 2001;234:63–70. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. | |
Cheng AL, Guan Z, Chen Z, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: subset analyses of the phase III Sorafenib Asia-Pacific trial. Eur J Cancer. 2012;48:1452–1465. | |
Patil MA, Chua MS, Pan KH, et al. An integrated data analysis approach to characterize genes highly expressed in hepatocellular carcinoma. Oncogene. 2005;24:3737–3747. | |
Tung CY, Jen CH, Hsu MT, Wang HW, Lin CH. A novel regulatory event-based gene set analysis method for exploring global functional changes in heterogeneous genomic data sets. BMC Genomics. 2009;10:26. | |
Zhou Q, Ching AK, Leung WK, et al. Novel therapeutic potential in targeting microtubules by nanoparticle albumin-bound paclitaxel in hepatocellular carcinoma. Int J Oncol. 2011;38:721–731. | |
Higa GM. The microtubule as a breast cancer target. Breast Cancer. 2011;18:103–119. | |
Rowinsky EK, Calvo E. Novel agents that target tublin and related elements. Semin Oncol. 2006;33:421–435. | |
Farrell KW, Jordan MA, Miller HP, Wilson L. Phase dynamics at microtubule ends: the coexistence of microtubule length changes and treadmilling. J Cell Biol. 1987;104:1035–1046. | |
Margolis RL, Wilson L. Microtubule treadmilling: what goes around comes around. Bioessays. 1998;20:830–836. | |
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. | |
Kavallaris M, Verrills NM, Hill BT. Anticancer therapy with novel tubulin-interacting drugs. Drug Resist Updat. 2001;4:392–401. | |
Himes RH, Kersey RN, Heller-Bettinger I, Samson FE. Action of the vinca alkaloids vincristine, vinblastine, and desacetyl vinblastine amide on microtubules in vitro. Cancer Res. 1976;36:3798–3802. | |
Bennouna J, Campone M, Delord JP, Pinel MC. Vinflunine: a novel antitubulin agent in solid malignancies. Expert Opin Investig Drugs. 2005;14:1259–1267. | |
Ortmann CA, Mazhar D. Second-line systemic therapy for metastatic urothelial carcinoma of the bladder. Future Oncol. 2013;9:1637–1651. | |
Bennouna J, Breton JL, Tourani JM, et al. Vinflunine – an active chemotherapy for treatment of advanced non-small-cell lung cancer previously treated with a platinum-based regimen: results of a phase II study. Br J Cancer. 2006;94:1383–1388. | |
Blasinska-Morawiec M, Tubiana-Mathieu N, Fougeray R, Pinel MC, Bougnoux P. Phase II study of intravenous vinflunine after failure of first-line vinorelbine based regimen for advanced breast cancer. Breast. 2013;22:58–63. | |
Kuznetsov G, Towle MJ, Cheng H, et al. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389. Cancer Res. 2004;64:5760–5766. | |
Cortes J, O’Shaughnessy J, Loesch D, et al; EMBRACE (Eisai Metastatic Breast Cancer Study Assessing Physician’s Choice Versus E7389) investigators. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377:914–923. | |
Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30:631–637. | |
Erickson HK, Lambert JM. ADME of antibody-maytansinoid conjugates. AAPS J. 2012;14:799–805. | |
Erickson HK, Lewis Phillips GD, Leipold DD, et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther. 2012;11:1133–1142. | |
Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30:2183–2189. | |
Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–9290. | |
Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. | |
Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J et al. Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2013:20;31(9):1157–1163. | |
Hoffmann-La Roche. A Study of Trastuzumab Emtansine (T-DM1) Plus Pertuzumab/Pertuzumab Placebo Versus Trastuzumab [Herceptin] Plus a Taxane in Patients With Metastatic Breast Cancer (MARIANNE). Available from: http://www.clinicaltrials.gov/ct2/show/NCT01120184. | |
Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. | |
Rustin GJ, Galbraith SM, Anderson H, et al. Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. J Clin Oncol. 2003;21:2815–2822. | |
Stevenson JP, Rosen M, Sun W, et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol. 2003;21:4428–4438. | |
Cooney MM, Radivoyevitch T, Dowlati A, et al. Cardiovascular safety profile of combretastatin a4 phosphate in a single-dose phase I study in patients with advanced cancer. Clin Cancer Res. 2004;10: 96–100. | |
Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994;5 Suppl 6:S3–S6. | |
Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–1598. | |
Terwogt JM, Nuijen B, Huinink WW, Beijnen JH. Alternative formulations of paclitaxel. Cancer Treat Rev. 1997;23:87–95. | |
Gradishar WJ, Krasnojon D, Cheporov S, et al. Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J Clin Oncol. 2009;27:3611–3619. | |
Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. | |
Wartmann M, Altmann KH. The biology and medicinal chemistry of epothilones. Curr Med Chem Anticancer Agents. 2002;2:123–148. | |
Lee FY, Smykla R, Johnston K, et al. Preclinical efficacy spectrum and pharmacokinetics of ixabepilone. Cancer Chemother Pharmacol. 2009;63:201–212. | |
Peterson JK, Tucker C, Favours E, et al. In vivo evaluation of ixabepilone (BMS247550), a novel epothilone B derivative, against pediatric cancer models. Clin Cancer Res. 2005;11:6950–6958. | |
Rugo HS, Campone M, Amadori D, et al. A randomized, phase II, three-arm study of two schedules of ixabepilone or paclitaxel plus bevacizumab as first-line therapy for metastatic breast cancer. Breast Cancer Res Treat. 2013;139:411–419. | |
Mok TS, Choi E, Yau D, et al. Effects of patupilone (epothilone B; EPO906), a novel chemotherapeutic agent, in hepatocellular carcinoma: an in vitro study. Oncology. 2006;71:292–296. | |
Zhou Q, Wong CH, Lau CP, et al. Enhanced antitumor activity with combining effect of mTOR inhibition and microtubule stabilization in hepatocellular carcinoma. Int J Hepatol. 2013;2013:103830. | |
Altmann KH, Wartmann M, O’Reilly T. Epothilones and related structures – a new class of microtubule inhibitors with potent in vivo antitumor activity. Biochim Biophys Acta. 2000;1470:M79–M91. | |
Kowalski RJ, Giannakakou P, Hamel E. Activities of the microtubule-stabilizing agents epothilones A and B with purified tubulin and in cells resistant to paclitaxel (Taxol(R)). J Biol Chem. 1997;272: 2534–2541. | |
Tsimberidou AM, Lewis N, Reid T, et al. Pharmacokinetics and antitumor activity of patupilone combined with midazolam or omeprazole in patients with advanced cancer. Cancer Chemother Pharmacol. 2011;68:1507–1516. | |
Tsimberidou AM, Takimoto CH, Moulder S, et al. Effects of patupilone on the pharmacokinetics and pharmacodynamics of warfarin in patients with advanced malignancies: a phase I clinical trial. Mol Cancer Ther. 2011;10:209–217. | |
Chi KN, Beardsley E, Eigl BJ, et al. A phase 2 study of patupilone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel: Canadian Urologic Oncology Group study P07a. Ann Oncol. 2012;23:53–58. | |
Melichar B, Casado E, Bridgewater J, et al. Clinical activity of patupilone in patients with pretreated advanced/metastatic colon cancer: results of a phase I dose escalation trial. Br J Cancer. 2011;105: 1646–1653. | |
Forster M, Kaye S, Oza A, et al. A phase Ib and pharmacokinetic trial of patupilone combined with carboplatin in patients with advanced cancer. Clin Cancer Res. 2007;13:4178–4184. | |
Ten Bokkel Huinink WW, Sufliarsky J, Smit WM, et al. Safety and efficacy of patupilone in patients with advanced ovarian, primary fallopian, or primary peritoneal cancer: a phase I, open-label, dose-escalation study. J Clin Oncol. 2009;27:3097–3103. | |
Hudes G. Estramustine-based chemotherapy. Semin Urol Oncol. 1997;15:13–19. | |
Memorial Sloan-Kettering Cancer Center. Ixabepilone With or Without Estramustine in Treating Patients With Progressive Prostate Cancer. NCT00025194. http://www.clinicaltrials.gov/ct2/show/NCT00025194. | |
Clinques universitaires Saint-Luc-Universite Catholique de Louvain. Safety and Efficacy Study of of Docetaxel vs Docetaxel Estramustine in Hormone Refractory Prostatic Cancer. NCT00541281. http://www.clinicaltrials.gov/ct2/show/NCT00541281. | |
MD Anderson Cancer Center. Combination Chemotherapy With Ketoconazole in Treating Patients With Prostate Cancer. NCT00003084. http://www.clinicaltrials.gov/ct2/show/NCT00003084. | |
Loblaw DA, Walker-Dilks C, Winquist E, Hotte S; Genitourinary Cancer Disease Site Group of Cancer Care Ontario’s Program in Evidence-Based Care: Systemic therapy in men with metastatic castration-resistant prostate cancer: a systematic review. Clin Oncol (R Coll Radiol). 2013;25:406–430. | |
Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. | |
Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–435. | |
Kruczynski A, Poli M, Dossi R, et al. Anti-angiogenic, vascular-disrupting and anti-metastatic activities of vinflunine, the latest vinca alkaloid in clinical development. Eur J Cancer. 2006;42:2821–2832. | |
Carr BI. Hepatocellular carcinoma: current management and future trends. Gastroenterology. 2004;127:S218–S224. | |
Song MJ, Chun HJ, Song do S, et al. Comparative study between doxorubicin-eluting beads and conventional transarterial chemoembolization for treatment of hepatocellular carcinoma. J Hepatol. 2012;57: 1244–1250. | |
Prajapati HJ, Dhanasekaran R, El-Rayes BF, et al. Safety and efficacy of doxorubicin drug-eluting bead transarterial chemoembolization in patients with advanced hepatocellular carcinoma. J Vasc Interv Radiol. 2013;24:307–315. | |
Martin R, Geller D, Espat J, et al. Safety and efficacy of trans arterial chemoembolization with drug-eluting beads in hepatocellular cancer: a systematic review. Hepatogastroenterology. 2012;59:255–260. | |
Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: an adjuvant to increase the cure rate of radiofrequency ablation in liver cancer. Future Oncol. 2011;7:937–945. | |
Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother. 2009;10:333–343. | |
Abou-Alfa GK, Amadori D, Santoro A, et al. Safety and efficacy of sorafenib in patients with hepatocellular carcinoma (HCC) and Child-Pugh A versus B cirrhosis. Gastrointest Cancer Res. 2011;4:40–44. | |
Bruix J, Raoul JL, Sherman M, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol. 2012;57:821–829. | |
Falkson G, Burger W. A phase II trial of vindesine in hepatocellular cancer. Oncology. 1995;52:86–87. | |
Saliba F, Paule B, Adam R, et al. Vinflunine in patients with advanced unresectable hepatocellular carcinoma and liver impairment. J Clin Oncol. 2007;25:15023. | |
Gagandeep S, Novikoff PM, Ott M, Gupta S. Paclitaxel shows cytotoxic activity in human hepatocellular carcinoma cell lines. Cancer Lett. 1999;136:109–118. | |
Geng C-X, Zeng Z-C, Wang J-Y. Docetaxel inhibits SMMC-7721 human hepatocellular carcinoma cell growth and induces apoptosis. World J Gastroenterol. 2003;9:696–700. | |
Belletti B, Nicoloso MS, Schiappacassi M, et al. Stathmin activity influences sarcoma cell shape, motility, and metastatic potential. Mol Biol Cell. 2008;19:2003–2013. | |
Chen J, Abi-Daoud M, Wang A, et al. Stathmin 1 is a potential novel oncogene in melanoma. Oncogene. 2013;32:1330–1337. | |
Strumberg D, Erhard J, Harstrick A, et al. Phase I study of a weekly 1 h infusion of paclitaxel in patients with unresectable hepatocellular carcinoma. Eur J Cancer. 1998;34:1290–1292. | |
Chao Y, Chan WK, Birkhofer MJ, et al. Phase II and pharmacokinetic study of paclitaxel therapy for unresectable hepatocellular carcinoma patients. Br J Cancer. 1998;78:34–39. | |
Milross CG, Mason KA, Hunter NR, Chung WK, Peters LJ, Milas L. Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. J Natl Cancer Inst. 1996;88:1308–1314. | |
Belotti D, Vergani V, Drudis T, et al. The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res. 1996;2:1843–1849. | |
Luitpold Pharmaceuticals. Taxoprexin® Treatment for Advanced Primary Cancers of the Liver. Availiable from http://www.clinicaltrials.gov/ct2/show/NCT00422877 as accessed on 25 Feb 2014. | |
Cheng KL, Bradley T, Budman DR. Novel microtubule-targeting agents – the epothilones. Biologics. 2008;2:789–811. | |
Venook AP, Poon R, Kang YK, et al. Evaluation of patupilpone as monotherapy in patients with advanced hepatoceullar carcinoma (HCC). J Clin Oncol. 2007;25:15055. | |
Aihara A, Tanaka S, Yasen M, et al. The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma. J Hepatol. 2010;52:63–71. | |
Tanaka S, Arii S, Yasen M, et al. Aurora kinase B is a predictive factor for the aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg. 2008;95:611–619. | |
Tanaka S, Noguchi N, Ochiai T, et al. Outcomes and recurrence of initially resectable hepatocellular carcinoma meeting milan criteria: rationale for partial hepatectomy as first strategy. J Am Coll Surg. 2007;204:1–6. | |
Girdler F, Gascoigne KE, Eyers PA, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–3675. | |
Mita M, Gordon M, Rejeb N, et al. A phase l study of three different dosing schedules of the oral aurora kinase inhibitor MSC1992371A in patients with solid tumors. Target Oncol. Epub July 6, 2013. | |
Foran J, Ravandi F, Wierda W, et al. A Phase I and pharmacodynamic study of AT9283, a small-molecule inhibitor of Aurora kinases in patients with relapsed/refractory leukemia or myelofibrosis. Clin Lymphoma Myeloma Leuk. Epub November 14, 2013. | |
Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32: 44–50. | |
VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther. 2004;3:1605–1613. | |
Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. | |
Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–8425. | |
Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27:255–261. | |
Buitrago-Molina LE, Vogel A. mTor as a potential target for the prevention and treatment of hepatocellular carcinoma. Curr Cancer Drug Targets. 2012;12:1045–1061. | |
Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. | |
Buitrago-Molina LE, Pothiraju D, Lamle J, et al. Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology. 2009;50:500–509. | |
Kirstein MM, Boukouris AE, Pothiraju D, et al. Activity of the mTOR inhibitor RAD001, the dual mTOR and PI3-kinase inhibitor BEZ235 and the PI3-kinase inhibitor BKM120 in hepatocellular carcinoma. Liver Int. 2013;33:780–793. | |
Zhu AX, Abrams TA, Miksad R, et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer. 2011;117:5094–5102. | |
Novartis. Global Study Looking at the Combination of RAD001 Plus Best Supportive Care (BSC) and Placebo Plus BSC to Treat Patients With Advanced Hepatocellular Carcinoma. (EVOLVE-1). http://www.clinicaltrials.gov/ct2/show/NCT01035229 - As accessed on 25 Feb 2014. | |
Zhu AX, Kudo M, Assenat E, et al. EVOLVE-1: Phase 3 study of everolimus for advanced HCC that progressed during or after sorafenib. J Clin Oncol. 2014;32(Suppl 3). In: ASCO Gastrointestinal Cancers Symposium 2014: January 16–18, 2014; San Francisco, CA. Abstract 172. | |
Yeo W, Chan SC, Mo F, et al. A phase I/II study of mTOR inhibitor temsirolimus in patients with unresectable hepatocellular carcinoma (HCC). ESMO 15th World Congress on Gastrointestinal Cancer; Barcelona. 3–6 July 2013;iv32. | |
Kelley RK, Nimeiri HS, Munster PN, et al. Temsirolimus combined with sorafenib in hepatocellular carcinoma: a phase I dose-finding trial with pharmacokinetic and biomarker correlates. Ann Oncol. 2013;24:1900–1907. | |
University of California, San Francisco. Phase II Combination of Temsirolimus and Sorafenib in Advanced Hepatocellular Carcinoma. http://www.clinicaltrials.gov/ct2/show/NCT01687673 - As accessed on Feb 25, 2014 | |
Mabuchi S, Altomare DA, Connolly DC, et al. RAD001 (Everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007;67:2408–2413. | |
Tam KH, Yang ZF, Lau CK, Lam CT, Pang RW, Poon RT. Inhibition of mTOR enhances chemosensitivity in hepatocellular carcinoma. Cancer Lett. 2009;273:201–209. | |
Zhou Q, Lui VW, Lau CP, et al. Sustained antitumor activity by co-targeting mTOR and the microtubule with temsirolimus/vinblastine combination in hepatocellular carcinoma. Biochem Pharmacol. 2012;83:1146–1158. | |
Canta A, Chiorazzi A, Cavaletti G. Tubulin: a target for antineoplastic drugs into the cancer cells but also in the peripheral nervous system. Curr Med Chem. 2009;16:1315–1324. | |
Argyriou AA, Koltzenburg M, Polychronopoulos P, Papapetropoulos S, Kalofonos HP. Peripheral nerve damage associated with administration of taxanes in patients with cancer. Crit Rev Oncol Hematol. 2008;66:218–228. | |
Larkin JM, Kaye SB. Potential clinical applications of epothilones: a review of phase II studies. Ann Oncol. 2007;18 Suppl 5:v28–v34. | |
Larkin JM, Kaye SB. Epothilones in the treatment of cancer. Expert Opin Investig Drugs. 2006;15:691–702. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.