Back to Journals » OncoTargets and Therapy » Volume 13
Microsatellite Instability-Related ACVR2A Mutations Partially Account for Decreased Lymph Node Metastasis in MSI-H Gastric Cancers
Authors Zhao L, Zhang J, Qu X, Yang Y, Gong Z, Yang Y, Wu Z, Guo W
Received 30 January 2020
Accepted for publication 15 April 2020
Published 5 May 2020 Volume 2020:13 Pages 3809—3821
DOI https://doi.org/10.2147/OTT.S247757
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Liqin Zhao,1,2,* Jieyun Zhang,1,2,* Xiaofei Qu,2,3,* Ya’nan Yang,1,2 Zhe Gong,1,2 Yue Yang,1,2 Zhenhua Wu,1,2 Weijian Guo1,2
1Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, People’s Republic of China; 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, People’s Republic of China; 3Cancer Institute, Fudan University Shanghai Cancer Center, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weijian Guo
Department of Medical Oncology, Fudan University Shanghai Cancer Center, 270 Dong’an Road, Shanghai 200032, People’s Republic of China
Tel +86-21-64175590
Fax +86-21-64170366
Email [email protected]
Purpose: Gene mutations play important roles in tumour metastasis, which significantly affect the prognosis of gastric cancer (GC) patients. This study aimed to compare lymph node (LN) metastasis of GCs with different microsatellite instability (MSI) statuses and explore the effect of ACVR2A mutations on GC LN metastasis.
Materials and Methods: The association between clinicopathologic characteristics and MSI status or ACVR2A mutational status was analysed based on a GC dataset from The Cancer Genome Atlas (TCGA). The association of ACVR2A mutations with MSI status was assessed. Whole-exome sequencing data of 157 GCs from Chinese patients at Fudan University Shanghai Cancer Center were used to validate the association of mutated ACVR2A and MSI status. Survival plots were obtained from the KMPlot and cBioPortal databases. The roles of ACVR2A and its common mutants in GC cell migration and proliferation were assayed in vitro.
Results: LN metastasis was significantly decreased in MSI-H GCs compared with microsatellite instability-low or microsatellite stable (MSI-L/MSS) GCs (P=0.016). As the most frequently mutated gene in MSI-H GCs, mutated ACVR2A was significantly associated with MSI-H (P< 0.001) and a higher mutation frequency (P< 0.001). Additionally, a tendency toward decreased LN metastasis was observed in GCs with mutated ACVR2A, although the P value was not statistically significant (P=0.052). Higher expression of ACVR2A predicted a poor prognosis, but patients with ACVR2A mutations had slightly better disease-free survival. Two polyadenine microsatellite loci in the ACVR2A coding region were hotspot mutation sites. In vitro experiments demonstrated that wild-type ACVR2A promoted GC cell migration probably via the Snail/Slug-EMT pathway, while ACVR2A truncated mutants lost this function.
Conclusion: MSI-H GCs had lower LN metastasis partially due to ACVR2A mutations. Mutated ACVR2A was significantly associated with MSI-H in GC, making it a potential biomarker that could be useful in choosing candidates for immunotherapy.
Keywords: microsatellite instability, activin A receptor type 2A, gene mutation, stomach neoplasms, neoplasm metastasis
Introduction
Gastric cancer (GC), as the 6th most commonly diagnosed cancer and the 3rd most common cause of cancer death worldwide; GC led to almost 0.8 million cancer deaths and over one million new patients in 2018, making it one of the deadliest cancers worldwide.1 The direct cause of death in advanced GC patients is a series of complications caused by tumour metastasis.2 Therefore, an improved understanding of the process of tumour metastasis is essential for the treatment of GC patients.
Tumour metastasis is a very complex process involving both the intrinsic properties of tumour cells and the host response.3 Traditional thought holds that the more gene mutations accumulate in cancer cells, the more heterogeneous and divergent the cancer cells are, hence the increased possible emergence of metastatic cancer cell clones.4,5 Impairment of mismatch repair genes, which are important for maintaining human genome stability, leads to microsatellite instability (MSI) in various types of cancers.6 Microsatellites are short tandem repeats of nucleotides that are mostly located in the noncoding regions of genes or near the telomeric regions of chromosomes, and loss or gain of nucleotide repeats is called MSI, which is a powerful predictive biomarker for immunotherapy.6,7 Depending on the degree, MSI can be divided into three groups: MSI-H (microsatellite instability-high), MSI-L (microsatellite instability-low), and MSS (microsatellite stable).8 As previously reported, the tumour mutation burden (TMB) of MSI-H-related tumours is significantly higher than that of their MSI-L/MSS counterparts,9 which suggests the possible emergence of metastatic cancer cell clones in MSI-H cancers according to the traditional thinking mentioned above. However, previous reports revealed that MSI-H GCs showed a lower tendency of lymph node (LN) metastasis and better survival than MSI-L/LMSS GCs,10 which contradicted the speculation that MSI-H tumours are more likely to migrate to other sites.
GC is one of the malignancies with the highest proportion of MSI-H. A previous study reported that activin A receptor type 2A (ACVR2A) was one of the most frequently mutated genes in MSI-H gastrointestinal cancers.11 ACVR2A contains two polyadenine (A8) microsatellite loci, which are located in exon 3 and exon 10; it is there that most mutations occur in MSI-H tumours, and the mutation types are truncations because of frameshifts induced by nucleotide deletion.12
ACVR2A encodes a transmembrane type 2 receptor that mediates the functions of activin, which is a member of the transforming growth factor-beta (TGF-β) superfamily involved in diverse biological processes, including epithelial–mesenchymal transition (EMT).13 In addition to major ligand activin, inhibin A and some bone morphogenetic proteins (BMPs) are potential ligands for ACVR2A. The activin signalling pathway has been reported to play important roles in regulating cell differentiation, proliferation, and apoptosis in various cancer cells.14,15 Previous studies showed that mutated ACVR2A lost the function of promoting migration mediated by wild-type ACVR2A through activin signalling in colon cancer and attenuated activin signalling in prostate cancer cells.16,17
Hence, we speculate that a lower tendency of LN metastasis in MSI-H GCs may be associated with the ACVR2A mutation. In the present study, we compared clinicopathological features and gene mutations in GCs with different MSI statuses, analysed the association of ACVR2A mutations with MSI status, mutation frequency, and LN metastasis using data downloaded from a dataset from The Cancer Genome Atlas (TCGA), and detected gene mutations in Chinese GC patients by whole-exome sequencing (WES) technology. We also generated stable GC cell lines overexpressing wild-type ACVR2A and three ACVR2A mutants and explored the function and possible mechanism of wild-type ACVR2A and its mutants in regulating GC cell migration and proliferation.
Materials and Methods
Clinical Samples
A total of 157 fresh frozen primary site specimens of GC were obtained from the tissue bank of Fudan University Shanghai Cancer Center (FUSCC). Genomic DNA was extracted from these specimens, and WES was applied to detect gene mutations. The MSI status of these patients was derived from tumour-normal paired sequence data using MSIsensor,18 which calculated MSI scores using a C++ program that detected somatic microsatellite changes, and samples with MSI score >3.5 were considered MSI-H, while MSI score <3.5 were considered MSS. Tumour mutation burden was defined as the number of somatic mutations per mega-base (Mb). Our study was approved by the Institutional Medical Ethics Committee of FUSCC. All samples were collected with written informed consent from patients, and our study protocol was conducted in accordance with the Declaration of Helsinki.
Data Acquisition and Analysis
The mutation data (n=395) of TCGA GC samples were downloaded from the cBioPortal database (https://www.cbioportal.org/), and gene expression data, clinical information and MSI status information (n=443) of TCGA samples were downloaded from the FireBrowse database (https://www.firebrowse.org/). The survival analysis of 593 GC patients from the Gene Expression Omnibus (GEO) dataset (GSE14210, GSE15459, GSE22377, GSE 29272 and GSE51105; n=593) and 375 GC patients from the TCGA dataset were obtained from the KMPlot database (https://kmplot.com). Survival analysis of ACVR2A-mutated GCs and wildtype GCs was obtained from the cBioPortal database based on TCGA data (https://www.cbioportal.org/).
Gastric Cancer Cell Lines and Culture
Two strains of human GC cell lines (MKN28 and AGS) were obtained from the Surgical Institution of Ruijin Hospital Affiliated to Medical College of Shanghai Jiaotong University. Another two strains of human GC cell lines (HGC27 and MGC803) and a normal human immortalized gastric mucosal cell line (GES-1) were obtained from the Chinese Academy of Sciences, Shanghai Cell Institution. The use of MKN28 and AGS cell lines was approved by the Institutional Medical Ethics Committee of FUSCC. The MKN28 cell line, which was chosen for the in vitro experiments, was authenticated by the STR profile. All the cell lines were cultured in DMEM with high glucose (HyClone, Utah, USA) except GES-1, which was cultured in RPMI-1640 medium (HyClone, Utah, USA). The two kinds of culture medium were both supplemented with 10% foetal bovine serum (FBS) and antibiotics.
Cell Line Genomic DNA Extraction and Sequencing
Genomic DNA from the five cell lines mentioned above was extracted using a DNA extraction kit (Tiangen, Beijing, China). Then, exon 3 and exon 10 of ACVR2A were sequenced at TsingKe Biological Technology Company (Nanjing, China). Exon-specific primers for ACVR2A are as follows: Exon 3F 5ʹ-TAGTAACAGGAATCCAGGAAC-3ʹ, Exon 3R 5ʹ- CCATCTTGTGATGCCTGTAC-3ʹ; Exon 10F 5ʹ-GTTCCTTATGTCCTCTGTGC-3ʹ, Exon 10R 5ʹ-GAATATCCTGTGTGAAGATCAC-3ʹ.
Plasmids, Lentivirus, and Infection
Lentiviral vectors carrying wild-type ACVR2A and three mutated versions of ACVR2A (c.1309-1310delAA, c.1310delA, and c.285delA) as well as a puromycin resistance cassette were obtained from Hanyin Biotechnology Company (Shanghai, China). Stable cell lines overexpressing ACVR2A and the ACVR2A mutants were generated by infection with the lentiviruses. Successfully infected cells were selected by adding puromycin to the culture medium. Overexpression efficiency was assayed by RT-PCR and Western blotting.
RNA Extraction and Quantitative Real-Time PCR Analysis
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). A PrimeScript reagent Kit with gDNA Eraser (Takara, Dalian, China) was used to reverse transcribe RNA into cDNA, and then quantitative real-time PCR was performed using TB Green Ex TaqTM (Takara, Dalian, China) with an Applied Biosystems Prism 7900 system (Life Technologies, Carlsbad, CA, USA). The sequences of the primers were as follows: ACVR2A-Forward: AGTTGGCGTTTGCCGTCTT, ACVR2A-Reverse: GCCGCCGTTTATCTTTGTCAC; GAPDH-Forward: ACCCAGAAGACTGTGGATGG, and GAPDH-Reverse: TTCTAGACGGCAGGTCAGGT.
Western Blotting
Total cellular proteins were extracted using RIPA cell lysis buffer (Beyotime, Shanghai, China) supplemented with protease inhibitors and phosphatase inhibitors (Bimake, Shanghai, China), and protein concentrations were quantified by a BCA protein assay kit (Beyotime, Shanghai, China). Then, Western blot analysis was performed as described previously,19 and a total of 30 µg of protein from each sample was used to detect proteins of interest. The primary antibodies for Western blotting were as follows: anti-ACVR2A (Abcam, Cambridge, UK), anti-Snail (CST, Danvers, MA, USA), anti-Slug (CST, Danvers, MA, USA), anti-Vimentin (CST, Danvers, MA, USA), anti-Smad2/3 (CST, Danvers, MA, USA), anti-phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) (CST, Danvers, MA, USA), anti-E-cadherin (BD Biosciences, NJ, USA), anti-Flag (MBL, Japan), anti-GAPDH (Proteintech, Wuhan, China) and anti-β-actin (Proteintech, Wuhan, China).
Cell Migration Assay
A transwell chamber assay was used to detect the migration of gastric cancer cells. Briefly, 2x104 cells were suspended in 200 μL serum-free medium and plated on the membrane of the upper chamber (24-well insert, pore size: 8 μm; BD FalconTM, NJ, USA), and 600 μL medium with 20% FBS was added to the lower chamber. Twenty-four hours later, the membranes of the upper chamber were fixed in 4% paraformaldehyde for 30 minutes and then were stained with 1% crystal violet for 30 minutes. Cells in the upper chamber were gently wiped off with cotton swabs, and cells that migrated to the opposite side of the membrane were counted.
Cell Proliferation Assay
Cell Counting Kit-8 (CCK8) assays, plate colony formation assays and cell cycle analyses were used to detect cell proliferation. For the CCK8 assay, cells were plated into 96-well plates (2000 cells per well), and relative cell numbers were detected by CCK8 reagent according to the manufacturer’s instructions on day 1, day 2, day 3, and day 4. For the plate colony formation assay, cells were plated into 6-well plates (1000 cells per well) and then were cultured for two weeks. Colonies were washed with PBS twice, fixed with 4% paraformaldehyde for 30 minutes and stained with 1% crystal violet for 30 minutes. Finally, crystal violet was washed away, and colonies were photographed and counted. For cell cycle analysis, cells were grown in 6-cm dishes until they reached 50% confluence. After 24 hours of serum starvation, cells were cultured with complete medium containing 10% FBS for 48 hours. Then, the cells were harvested and washed twice with PBS and fixed with 70% ethanol overnight at 4°C. The next day, the cells were washed with PBS twice and then were stained with propidium iodide containing ribonuclease A at room temperature for 30 minutes. Finally, cells were analysed with flow cytometry for cell cycle analysis.
Statistical Analysis
All statistical analyses were performed using SPSS Statistics software version 20.0 (IBM Corp., NY, USA) or R language (version 3.5.1). Analysis of differentially mutated genes in MSI-H GCs and MSI-L/MSS GCs was processed by R language and assayed by chi-square tests. The correlation between MSI status and LN metastasis was analysed by chi-square or Fisher’s exact tests, and chi-square tests were also used to analyse the correlation between ACVR2A mutation and MSI status or clinicopathological variables. The difference in MSI score or TMB between the ACVR2A-mutated group and the ACVR2A wild-type group of our sequenced samples was assayed by an unpaired T-test. One-way ANOVA was applied to test the migration, colony formation, proliferation ability and cell cycle of ACVR2A wild-type or mutant-overexpressing cells. Unless stated otherwise, all p values were two-sided, and p<0.05 was considered statistically significant.
Results
Mutated ACVR2A is Strongly Associated with MSI-H and High TMB, and MSI-H GCs Had Lower LN Metastasis, Probably Due to ACVR2A Mutation
Analysis of TCGA data revealed that ACVR2A is the gene with the most dramatically different mutation rate between the MSI-H group of GCs and MSI-L/MSS group of GCs (75.34% VS 1.24%, p<0.001) (Table 1). The MSI-H GCs less commonly had LN metastasis than their MSI-L/MSS counterparts (n=443, p=0.016), and it was more likely to occur in elderly and female gastric cancer patients (Table 2). We also analysed the association between MSI status and clinical variables in our sequenced patients (Supplemental Table 1) and validated that MSI-H was more likely to occur in elderly patients. However, the association of N staging with MSI status was not observed, which we thought was due to the relatively smaller sample size and to having fewer MSI-H patients. In the cohort from TCGA, further analysis revealed that the rate of MSI-H in the ACVR2A mutation group was significantly higher than that in the ACVR2A wild-type group (93.22% vs 5.36%, p<0.001) (Figure 1A). In addition, we found that patients with mutations of c.1310delA/1309-1310delAA or c.285delA were all MSI-H GCs. Our own sequence data also verified that most patients with mutated ACVR2A were MSI-H (80% vs 4.76%, p<0.001) (Figure 1B), and ACVR2A mutations were correlated with high mutation frequency and a high MSI score or TMB (Figure 1C–E). In TCGA cohort, only two patients carried silent mutations, but they simultaneously carried nonsilent mutations, so we classified them as patients with nonsynonymous mutations.
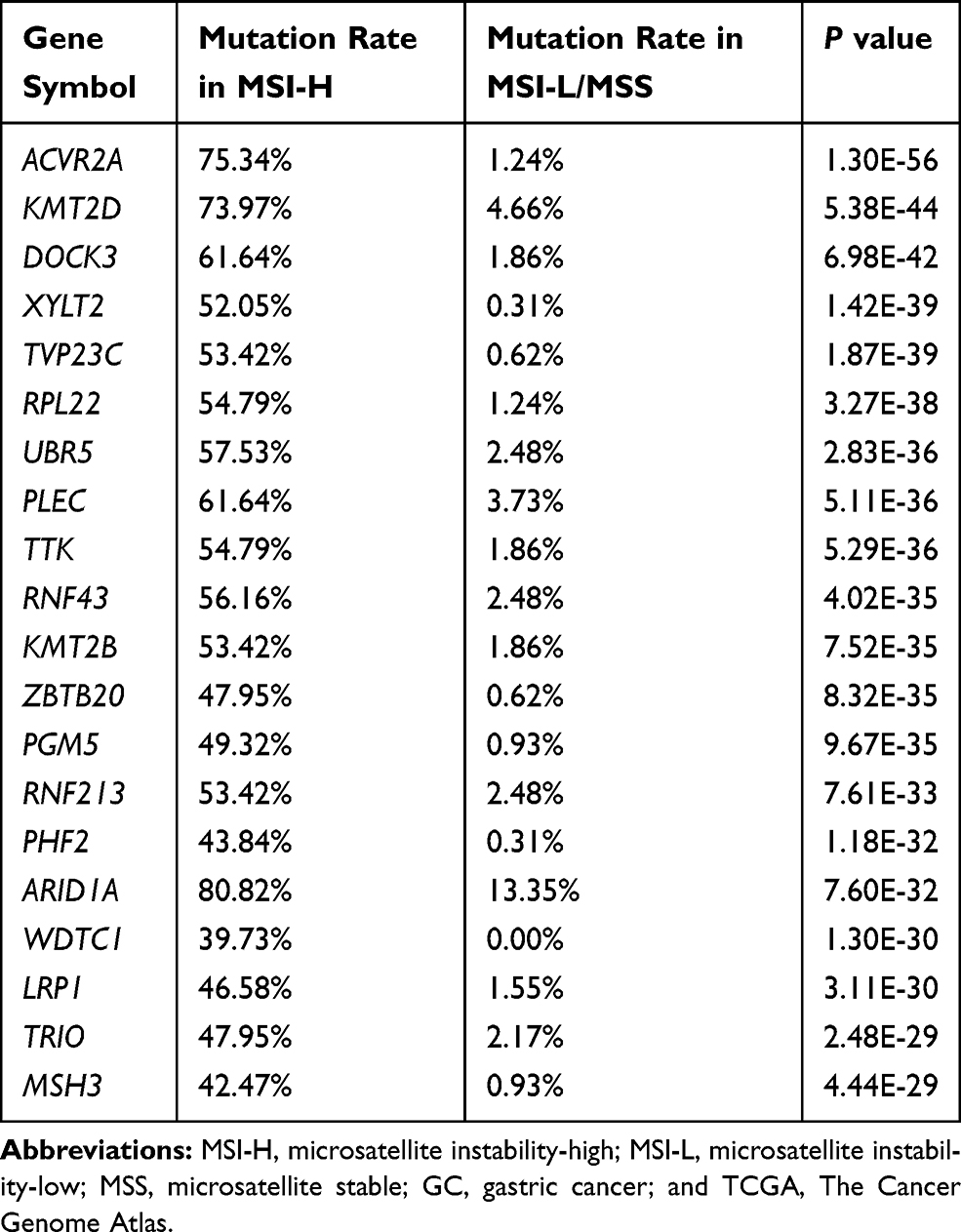 |
Table 1 Top 20 Genes Frequently Mutated in MSI-H GCs vs MSI-L/MSS GCs in TCGA Dataset |
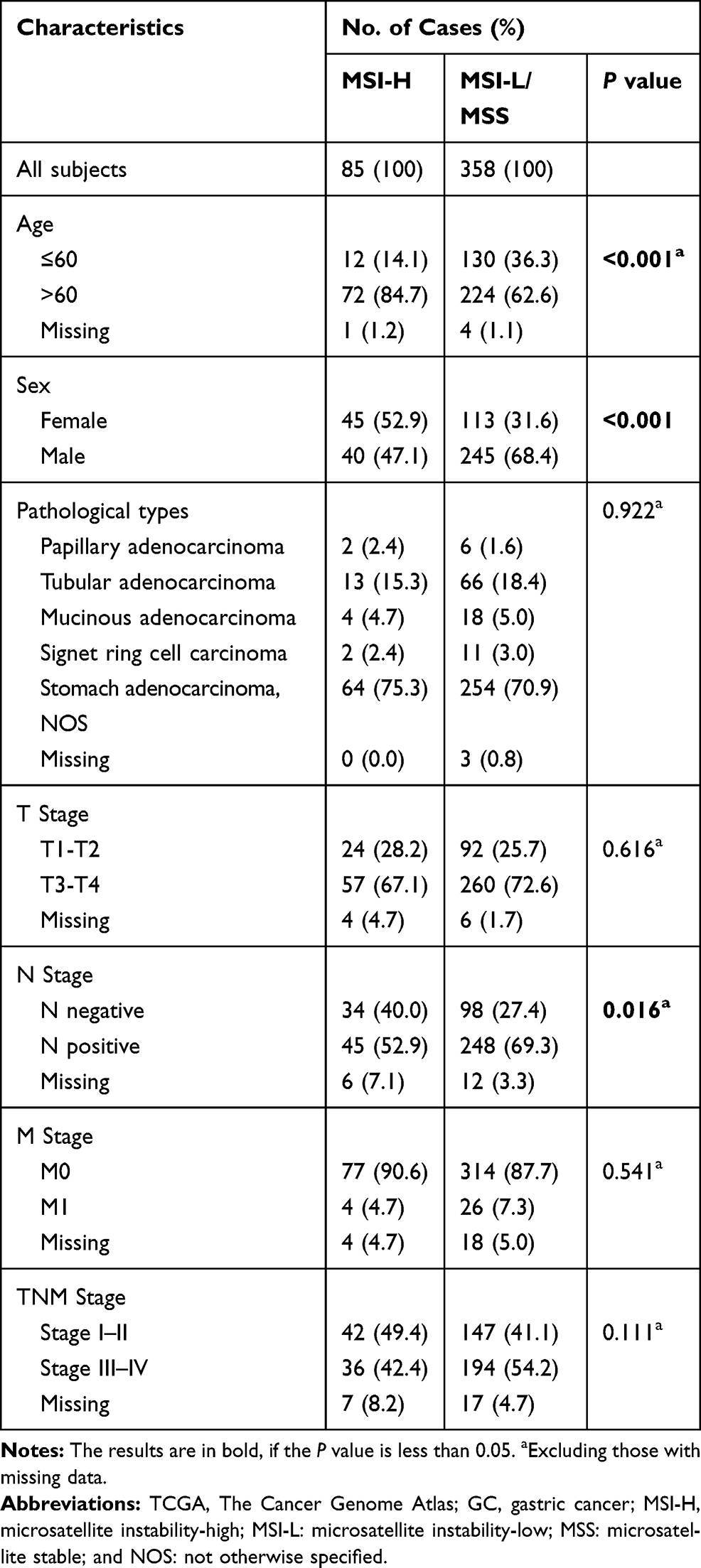 |
Table 2 Clinicopathologic Characteristics of TGCA GC Patients with MSI-H vs MSI-L/MSS |
 |
Figure 1 Mutated ACVR2A is strongly associated with MSI-H and high TMB. Notes: (A) The proportion of MSI-H in the ACVR2A mutation group was significantly higher than that in the ACVR2A wild-type group (p<0.001). (B) Our WES data also verified that there was a high proportion of MSI-H in the ACVR2A-mutated group (p<0.001). (C) In TCGA cohort, patients carried mutated ACVR2A had higher mutation frequency than patients carried wild-type ACVR2A. (D, E) Compared to the wild-type, mutated ACVR2A correlated with higher MSI scores and TMB derived from our data (p<0.001 and p=0.029, respectively).Abbreviations: ACVR2A, activin A receptor type 2A; MSI-H, microsatellite instability-high; TMB, tumour mutation burden; and WES, whole-exome sequencing. |
We investigated the correlation of ACVR2A mutation and clinical variables using data downloaded from TCGA and found that mutated ACVR2A was associated with age (p=0.001), sex (p=0.024), and mutation frequency (p<0.001). GCs with mutated ACVR2A had lower N stage, although the difference was not statistically significant (p=0.052) (Table 3). However, we did not observe an association in the FUSCC cohort owing to the small sample size and the small number of patients with ACVR2A mutations (Supplemental Table 2).
 |
Table 3 Clinicopathological Features in ACVR2A-Mutated and Wild-Type Groups of TCGA GC Patients |
The above results indicated that mutated ACVR2A may be an important reason for decreased LN metastasis in MSI-H GCs.
High ACVR2A Expression is Associated with Poor Prognosis, but Patients with ACVR2A Mutations Had Slightly Better Disease-Free Survival Than Patients with Wild-Type ACVR2A
To investigate the clinical significance of ACVR2A in GCs, we analysed the effect of ACVR2A expression or mutation on survival. Higher ACVR2A expression is associated with worse overall survival (OS) and progression-free survival (PFS) of GC patients (Figure 2A and B). Mutated ACVR2A did not affect overall survival, but patients with mutated ACVR2A had slightly better disease-free survival than patients with wild-type ACVR2A (p=0.187) (Figure 2C).
 |
Figure 2 High ACVR2A expression is associated with poor prognosis, while patients with mutated ACVR2A had slightly better PFS. Notes: (A) Survival analysis derived from the GEO dataset revealed worse OS (n=593) and PFS (n=359) in patients with high ACVR2A expression compared to those with low ACVR2A expression. (B) Survival analysis derived from the dataset from TCGA also revealed relatively poor OS (n=371) and PFS (n=215) in high ACVR2A expression patients. (C) Kaplan-Meier survival curves obtained from cBioPortal based on TCGA sequence data showed no difference in OS between the ACVR2A-mutated group and the wild-type group, but patients with mutated ACVR2A had slightly better PFS than patients with wild-type ACVR2A (p=0.187).Abbreviations: ACVR2A, activin A receptor type 2A; PFS, progression-free survival; OS, overall survival; GEO, Gene Expression Omnibus; and TCGA, The Cancer Genome Atlas. |
Two Polyadenine (A8) Tracts in Exon 3 and Exon 10 of the ACVR2A Coding Region are Frequent Sites of Mutation
To clarify mutations of ACVR2A in different populations, we detected gene mutations in 157 Chinese GC patients by WES and downloaded mutational and clinical data from the TCGA database. Our sequencing data showed that the mutation rate of ACVR2A in the Chinese population is 6.37% (10/157); four out of ten patients had nonsynonymous mutations in the coding region that led to ACVR2A protein change, and the other six patients had either silent mutations or mutations in the noncoding region that would not change ACVR2A protein. Some patients had two or more mutations in the ACVR2A gene, and most of the nonsynonymous mutations in the coding region were frameshift truncating mutations caused by nucleotide deletion (Figure 3A, Supplemental Table 3). In a TCGA Provisional dataset, ACVR2A mutations occurred in 14.94% (59/395) of patients, and 3.04% (12/395) of patients had more than one mutation in the ACVR2A gene. Similarly, the most frequent mutation type was frameshift truncation, and c.1309-1310delAA/1310delA and c.285delA were hotspot mutations (Figure 3B). According to the sequence of the ACVR2A coding region (Figure 3C), we discovered that the hotspot mutation sites are located in two polyadenine (A8) tracts, which are known as microsatellite loci.12 The top two most frequent mutations in ACVR2A in GC were c.1309-1310delAA/1310delA (p. K437Efs*19/Rfs*5, exon 10) and c.285delA (p. D96Tfs*54, exon 3), which were changed from A8 tracts to A7/A6 tracts.
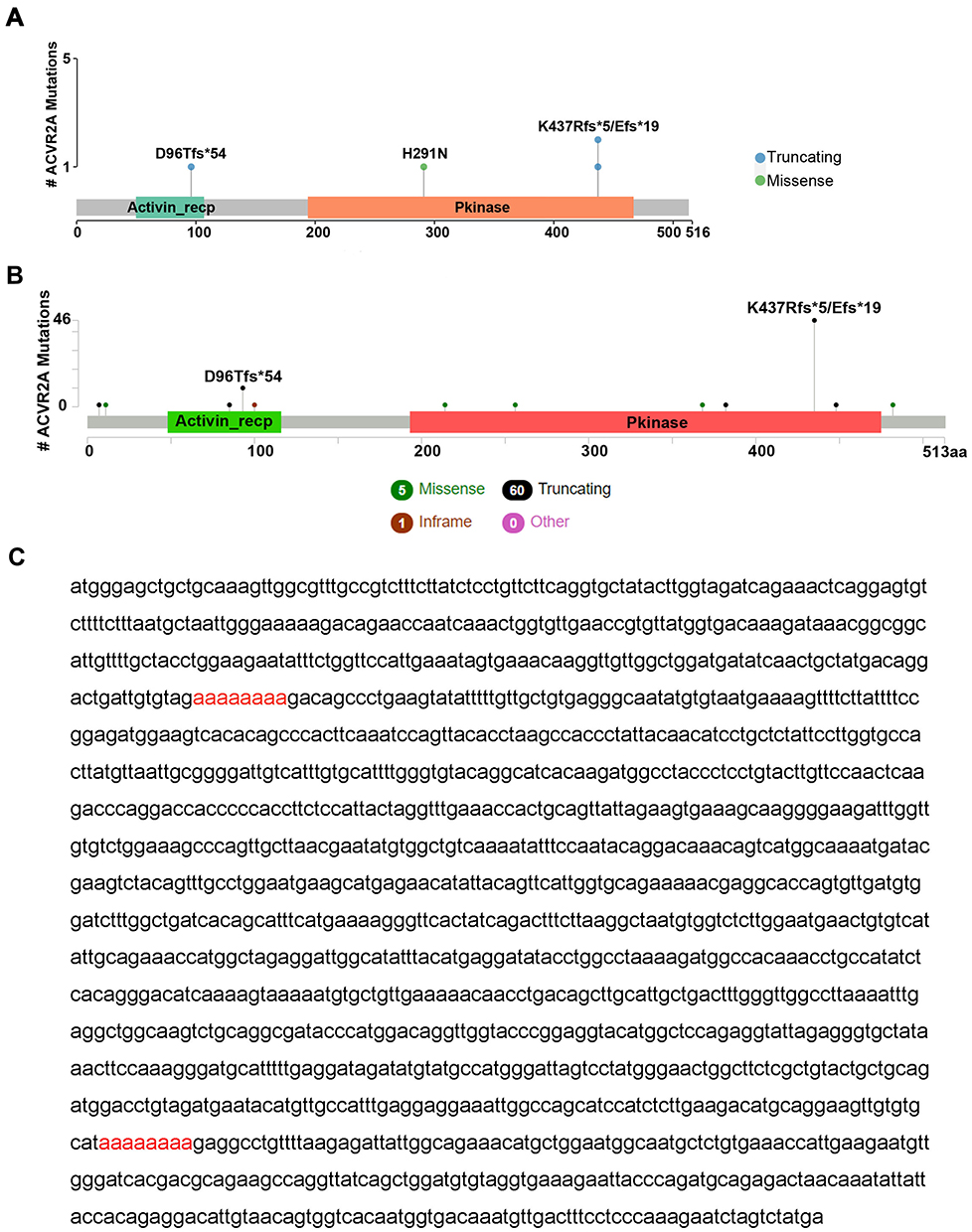 |
Figure 3 Two polyadenine (A8) tracts in the ACVR2A coding region are hotspot mutation sites. Notes: (A) In our sequenced samples, four patients had nonsilent mutations in the ACVR2A coding region, and three mutations were located in the two A8 tracts. (B) Fifty-nine out of 395 samples had nonsilent ACVR2A mutations in the provisional dataset from TCGA. Similarly, most mutations occurred in the two A8 tracts. (C) The coding sequence of ACVR2A contains two polyadenine (A8) tracts marked in red.Abbreviations: ACVR2A, activin A receptor type 2A; and TCGA, The Cancer Genome Atlas. |
Mutations That Occurred in Exon 10 Were Resistant to Nonsense-Mediated Decay and Could Produce Truncated ACVR2A Protein
We conducted in vitro experiments to verify the role of ACVR2A in the migration and proliferation of GC cells. First, we sequenced exon 3 and exon 10 of the ACVR2A gene in one human normal immortalized gastric mucosal cell line (GES-1) and four human GC cell lines (MGC803, HGC27, AGS, and MKN28), and we found no mutation in the two A8 tracts of ACVR2A in any of the cell lines (Supplemental Figure 1). Then, we detected the expression of ACVR2A in the five cell lines by PCR and Western blotting (Figure 4A and B). The MKN28 cell line, which showed relatively low expression of ACVR2A, was chosen for subsequent experiments. Stable cells overexpressing wild-type ACVR2A or one of three ACVR2A mutants (c. del1309-1310AA, c. del1310A, and c. del285A) were constructed by infecting MKN28 cells with the corresponding lentivirus. ACVR2A mRNA was significantly overexpressed in stable cells compared to the negative control (Figure 4C). We discovered that ACVR2A protein was overexpressed in cells transfected with wild-type ACVR2A; truncated ACVR2A protein was overexpressed in cells transfected with two mutants at exon 10, and two protein bands in each sample were detected. However, no protein band was found in the mutant of c.285delA, which is in exon 3 (Figure 4D). The protein patterns are shown in Figure 4E. Previous studies discovered that frameshift mutations occurring prior to the last exon of a gene lead to degradation of gene products through the process of nonsense-mediated decay (NMD),20,21 but nonsense transcripts with a coding microsatellite in the last exon or near the last exon-exon junction have intrinsic resistance to nonsense-mediated decay, leading to a source of truncated proteins in MSI-H tumours.22 ACVR2A is comprised of 11 exons; hence, our results coincide with previous findings. For the two protein bands in each sample, the upper band may be a glycosylated form of the ACVR2A protein, as the ACVR2A protein contains many glycosylation sites.
 |
Figure 4 Mutants that occurred in exon 10 were resistant to nonsense-mediated decay, leading to the production of truncated ACVR2A protein. Notes: (A, B) The expression of ACVR2A in the MKN28 cell line is lower than that in GES-1 and other gastric cancer cell lines. (C) ACVR2A mRNA was overexpressed in four cell lines infected with a lentivirus carrying wild-type ACVR2A or mutated ACVR2A. (D) Truncated ACVR2A proteins were detected in c.1310delA/1309-1310delAA mutants but not the c.285delA mutant. (E) Putative protein patterns of wild-type ACVR2A and three ACVR2A mutants are shown.Abbreviation: ACVR2A, activin A receptor type 2A. |
Wild-Type ACVR2A Probably Enhances the Migration of Gastric Cancer Cells by Activating Smads-Snail/Slug Signalling and Promoting EMT, but Truncated ACVR2A Lost This Function
To assess the potential effect of ACVR2A and its mutants on cell migration, we conducted a transwell chamber assay, which indicated that wild-type ACVR2A promoted the migration of gastric cancer cells, while none of the mutants could (Figure 5A). Meanwhile, we assessed the effect of ACVR2A and three mutants on gastric cancer cell proliferation and found that both wild-type ACVR2A and its mutants had no effect on cell proliferation, colony formation, or cell cycle (Figure 5B–D). To further explore the mechanism, we detected downstream Smad2/3 protein and its phosphorylation status; it is in the activin signalling pathway. We also detected important proteins involved in the EMT process (E-cadherin, Vimentin, Snail, and Slug), as activin signalling is similar to TGF-β signalling, which may induce EMT. The results showed that overexpression of wild-type ACVR2A but not ACVR2A mutants could upregulate p-Smad2/3, Snail, Slug, and Vimentin and downregulate E-cadherin, which showed that wild-type ACVR2A could activate the Smads-Snail/Slug pathway and promote EMT, while ACVR2A mutants lost this function (Figure 5E).
 |
Figure 5 Mutated ACVR2A lost the ability to promote gastric cancer cell migration, which is mediated by wild-type ACVR2A through the Snail/Slug-EMT pathway. Notes: (A) Overexpression of wild-type ACVR2A but not ACVR2A mutants promoted GC cell migration (crystal violet staining, 100X). (B–D) Overexpression of wild-type ACVR2A and mutated ACVR2A did not affect colony formation, cell proliferation, or the cell cycle. (E) Overexpression of wild-type ACVR2A but not ACVR2A mutants increased phosphorylation of downstream Smad2/3 and upregulated Snail and Slug, ultimately promoting EMT. *P<0.05.Abbreviations: ACVR2A, activin A receptor type 2A; GC, gastric cancer; and EMT, epithelial–mesenchymal transition. |
Discussion
MSI-H GCs have unique features, such as lower LN metastasis, resistance to fluorouracil-based chemotherapy, and the patients have better survival than those with MSI-L/MSS GCs.10,11,23 Our study also confirmed that MSI-H GCs have lower LN metastasis than MSI-L/MSS GCs. This phenomenon was partially attributed to mutated ACVR2A, as GC patients with ACVR2A mutations had a lower tendency toward LN metastasis, although the p value was slightly over 0.05. In addition, in vitro experiments verified that wild-type ACVR2A promoted the migration of gastric cancer cells, while mutated ACVR2A lost this function.
ACVR2A is located on chromosome 2 and comprises 11 exons; it contains two eight polyadenine DNA microsatellite sites in exon 3 and exon 10, which makes it highly predisposed to mutation in MSI-H GCs. The ACVR2A protein is a transmembrane receptor that is divided into an extracellular receptor region, a transmembrane region, and an intracellular kinase region, and the binding of activin to ACVR2A leads to the phosphorylation of downstream Smad and non-Smad signal transduction proteins.24 The truncating mutations occurring in ACVR2A exon 3 and exon 10 polyadenine tracts result in early termination of its messenger RNA translation, leading to protein degradation or generation of truncated ACVR2A proteins; further, none of the three mutants could activate downstream signalling pathways.
Previous studies have found that ACVR2A promotes the migration of colorectal cancer cells through the activin/Smad signalling pathway, leading researchers to believe that an ACVR2A truncation product produce by a mutation in exon 10 significantly reduced the expression of the ACVR2A protein, which weakens the function of ACVR2A in promoting cell migration.12,16 Our study also revealed that in gastric cancer, truncated ACVR2A lost its ability to promote cell migration. However, we found that frameshift mutants in exon 10 were resistant to nonsense-mediated decay and could produce truncated proteins. We thought the reason why previous studies hardly detected ACVR2A protein in most MSI-H colon cancers was that the anti-ACVR2A antibody used in that study targeted the C-terminus of the protein, which could not detect truncated ACVR2A protein.
Our mechanistic study showed that overexpression of wild-type ACVR2A could activate Smad-Snail/Slug signalling and promote EMT. The activin signalling participates in a variety of biological processes and activates signalling pathways in addition to the Smad protein-mediated pathway, including the MEKK1-JNK, p38-MAPK and ERK1/2 pathways.14,25,26 Further exploration is needed to confirm whether ACVR2A could enhance cell migration through non-Smad signalling pathways.
Our study demonstrated that LN metastasis was significantly decreased in MSI-H GC patients, and ACVR2A was the most vulnerable gene affected by mismatch repair deficiency in MSI-H GCs. As MSI-H emerged as a specific GC subtype that can significantly benefit from immune checkpoint inhibitor therapy, our research showed that ACVR2A can act as a solid diagnostic biomarker and provided insights into the mechanism underlying the unique clinical characteristics.
Conclusions
In summary, we demonstrated that the decreased LN metastasis of MSI-H GCs is partially due to the truncating mutation of ACVR2A, as wild-type ACVR2A could promote GC cell migration by activating the Smad-Snail/Slug-EMT pathway, but truncated ACVR2A lost this function. In addition, the rates of MSI-H and TMB in ACVR2A-mutated GCs were significantly higher than they were in MSI-L/MSS GCs, making the ACVR2A mutation a potential biomarker for identifying MSI-H and choosing patients suitable for immunotherapy in GC.
Abbreviations
ACVR2A, activin A receptor type 2A; LN, lymph node; TCGA, The Cancer Genome Atlas; MSI, microsatellite instability; MSI-H, microsatellite instability high; MSI-L, microsatellite instability low; MSS, microsatellite stable; GC, gastric cancer; TMB, tumour mutation burden; TGF-β, transforming growth factor-beta; EMT, epithelial mesenchymal transition; WES, whole-exome sequencing; DFS, disease-free survival; OS, overall survival; CDS, coding sequence; NMD, nonsense-mediated decay; 3ʹUTR, 3ʹ untranslated region; SNP, single nucleotide polymorphism; and FUSCC, Fudan University Shanghai Cancer Center.
Data Sharing Statement
All the authors confirmed that TCGA GC samples mutation data (n=395) were downloaded from the cBioPortal database (https://www.cbioportal.org/), and gene expression data, clinical information and MSI status information (n=443) of TCGA samples were downloaded from the FireBrowse database (https://www.firebrowse.org/). The survival analysis of 593 GC patients from the Gene Expression Omnibus (GEO) dataset (GSE14210, GSE15459, GSE22377, GSE 29272 and GSE51105; n=593) and 375 GC patients from the TCGA dataset were obtained from the KMPlot database (https://kmplot.com). Any readers are welcome to contact the corresponding authors to get the above detailed data analysis process. As for the FUSCC Chinese patients’ data, if anyone is interested in our whole exome data, he/she is welcome to contact us through the corresponding author’s email after acquired FUSCC Ethics Committee consent.
Ethics and Consent Statement
Our study was approved by the Institutional Medical Ethics Committee of FUSCC on 7 May 2018, and the protocol number was 050432-4-1805c. All samples were collected with written informed consent from patients, and our study protocol was conducted in accordance with the Declaration of Helsinki.
Consent for Publication
The consent for publication is exempt, because no details, images, or videos relating to individual participants are included in this manuscript.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81572331). Authors wish to thank Tissue Bank of FUSCC for their assistance in providing us with the GC samples.
Author Contributions
WG designed and guided the experiment. LZ, YY, ZG, ZW carried out the in vitro experiments. JZ and YY analysed the FUSCC GCs data. LZ, XQ performed the TCGA and GEO GC datasets analysis. LZ, JZ, XQ, and WG interpreted the results, prepared the figures and tables, wrote and revised the manuscript. All authors drafted and revised the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
Our study was funded by the National Natural Science Foundation of China (No. 81572331).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
3. Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature. 1980;283(5743):139–146. doi:10.1038/283139a0
4. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70(14):5649–5669. doi:10.1158/0008-5472.CAN-10-1040
5. Suzuki M, Tarin D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: clinical implications. Mol Oncol. 2007;1(2):172–180. doi:10.1016/j.molonc.2007.03.005
6. de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol. 2010;28(20):3380–3387. doi:10.1200/JCO.2009.27.0652
7. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
8. Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol. 2015;89(6):899–921. doi:10.1007/s00204-015-1474-0
9. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. doi:10.1038/nature13480
10. Smyth EC, Wotherspoon A, Peckitt C, et al. Mismatch repair deficiency, microsatellite instability, and survival: an exploratory analysis of the medical research council adjuvant gastric infusional chemotherapy (MAGIC) trial. JAMA Oncol. 2017;3(9):1197–1203. doi:10.1001/jamaoncol.2016.6762
11. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22(11):1342–1350. doi:10.1038/nm.4191
12. Jung B, Doctolero RT, Tajima A, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126(3):654–659. doi:10.1053/j.gastro.2004.01.008
13. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–172. doi:10.1038/cr.2009.5
14. Cocolakis E, Lemay S, Ali S, Lebrun JJ. The p38 MAPK pathway is required for cell growth inhibition of human breast cancer cells in response to activin. J Biol Chem. 2001;276(21):18430–18436. doi:10.1074/jbc.M010768200
15. Zhang Z, Zheng J, Zhao Y, et al. Overexpression of activin A inhibits growth, induces apoptosis, and suppresses tumorigenicity in an androgen-sensitive human prostate cancer cell line LNCaP. Int J Oncol. 1997;11(4):727–736. doi:10.3892/ijo.11.4.727
16. Jung BH, Beck SE, Cabral J, et al. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology. 2007;132(2):633–644. doi:10.1053/j.gastro.2006.11.018
17. Rossi MR, Ionov Y, Bakin AV, Cowell JK. Truncating mutations in the ACVR2 gene attenuates activin signaling in prostate cancer cells. Cancer Genet Cytogenet. 2005;163(2):123–129. doi:10.1016/j.cancergencyto.2005.05.007
18. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30(7):1015–1016. doi:10.1093/bioinformatics/btt755
19. Guo WJ, Zeng MS, Yadav A, et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. 2007;67(11):5083–5089. doi:10.1158/0008-5472.CAN-06-4368
20. Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5(2):89–99. doi:10.1038/nrm1310
21. Ionov Y, Matsui S, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proc Natl Acad Sci U S A. 2004;101(5):1273–1278. doi:10.1073/pnas.0307276101
22. Williams DS, Bird MJ, Jorissen RN, et al. Nonsense mediated decay resistant mutations are a source of expressed mutant proteins in colon cancer cell lines with microsatellite instability. PLoS One. 2010;5(12):e16012. doi:10.1371/journal.pone.0016012
23. Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219–3226. doi:10.1200/JCO.2009.27.1825
24. Namwanje M, Brown CW. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol. 2016;8:7. doi:10.1101/cshperspect.a021881
25. Zhang L, Deng M, Parthasarathy R, et al. MEKK1 transduces activin signals in keratinocytes to induce actin stress fiber formation and migration. Mol Cell Biol. 2005;25(1):60–65. doi:10.1128/MCB.25.1.60-65.2005
26. Dean M, Davis DA, Burdette JE. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017;391:114–124. doi:10.1016/j.canlet.2017.01.011
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.