")
Back to Journals » OncoTargets and Therapy » Volume 11
MicroRNA-425 promotes the development of lung adenocarcinoma via targeting A disintegrin and metalloproteinases 9 (ADAM9)
Authors Liu R, Wang F, Guo Y, Yang J, Chen S, Gao X, Wang X
Received 26 December 2017
Accepted for publication 4 May 2018
Published 16 July 2018 Volume 2018:11 Pages 4065—4073
DOI https://doi.org/10.2147/OTT.S160871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Ruibao Liu, Fang Wang, Yanchun Guo, Jianmei Yang, Shaoping Chen, Xin Gao, Xunguo Wang
Department of Oncology, Dongying People’s Hospital, Dongying, Shandong 257091, People’s Republic of China
Purpose: We aimed to investigate the roles of microRNA-425 (miR-425) in lung adenocarcinoma, as well as its possible regulatory mechanism.
Materials and methods: The miR-425 expression in lung adenocarcinoma tissues and cells was determined. The regulatory relationship between miR-425 and IL-6/STAT3 signaling was investigated. In addition, miR-425 was downexpressed in H1299 cells, and its effects on cell proliferation and apoptosis were determined. Furthermore, the target relationship between miR-425 and A disintegrin and metalloproteinases 9 (ADAM9) in lung adenocarcinoma cells was explored.
Results: The miR-425 was significantly downregulated in lung adenocarcinoma tissues and cells and was markedly inhibited by IL-6/STAT3 signaling. In addition, miR-425 expression was successfully overexpressed by transfection with pre-miR-425. Overexpression of miR-425 decreased the proliferation and colony formation of H1299 cells and promoted cell apoptosis markedly. Moreover, ADAM9 was revealed as a target of miR-425, and ADAM9 expression was negatively regulated by miR-425.
Conclusion: Our findings indicate that downregulation of miR-425 caused by IL-6/STAT3 signaling leads to loss of ADAM9 targeting, results in enhanced ADAM9 expression, and contributes to the development of lung adenocarcinoma. Thus, increasing miR-425 may be a promising therapeutic strategy for this disease.
Keywords: lung adenocarcinoma, microRNA-425, IL-6/STAT3 signaling, proliferation, apoptosis, A disintegrin and metalloproteinases 9
Introduction
Lung cancer is a common malignancy with high mortality,1 and the predominant form is non-small-cell lung cancer (NSCLC).2 Moreover, the 5-year overall survival rate of NSCLC is only 11%.3 Lung adenocarcinoma is the most common subtype of NSCLC, resulting in about 500,000 deaths per year worldwide.4 Therefore, it is imperative to elucidate the molecular mechanisms underlying lung adenocarcinoma.
MicroRNAs (miRNAs) and endogenous small noncoding RNAs (19–21 nucleotides) have emerged to mediate a variety of biological and pathological processes via regulating their target genes.5,6 In lung adenocarcinoma development, the key roles of crucial miRNAs are increasingly being disclosed. For instance, miR-200 is shown to suppress cell invasion and metastasis in lung adenocarcinoma;7 miR-483-5p promotes the invasive and metastatic properties of lung adenocarcinoma, and its upregulation results in the progression of this malignancy;8 miR-186 can play a tumor-suppressive role in lung adenocarcinoma, and its downregulation correlates with poor survival;9 and miR-27a and miR-224 can play crucial roles in regulating the chemoresistance of lung adenocarcinoma cells.10,11 Recently, aberrant expression of miR-425 was found to be involved in various cancers. It has been reported that high levels of miR-425 expression facilitate the tumorigenesis of esophageal squamous cell carcinoma;12 miR-425 can function as a tumor suppressor in inhibiting the metastasis of melanoma,13 and miR-425 can impact the oncogenic function in hepatocellular carcinoma.14 Despite these facts, limited data are available on the functions and possible mechanism of miR-425 in the pathogenesis of lung adenocarcinoma.
A disintegrin and metalloproteinases 9 (ADAM9) encode a member of the ADAM family.15 Members of this family are membrane-anchored proteins structurally related to snake venom disintegrins, and they have been implicated in a variety of biological processes involving cell–cell and cell–matrix interactions, including fertilization, muscle development, and neurogenesis.16 It has been confirmed to be closely related with lung cancer. Lin et al17 found that this gene could promote metastases of lung cancer by a plasminogen activator-based pathway. Besides, Chang et al18 also found that ADAM9 was a target for non-small cell lung cancer treatment.
In this study, miR-425 expression in lung adenocarcinoma tissues and cells was determined. The regulatory relationship between miR-425 and IL-6/STAT3 signaling was investigated. In addition, miR-425 was downexpressed in H1299 cells to further detect the roles of miR-425 in regulating cell proliferation and apoptosis. Furthermore, the target relationship between miR-425 and ADAM9 in lung adenocarcinoma cells was explored. The objective of this study was to explore the role and possible regulatory mechanism of miR-425 in lung adenocarcinoma development, and thus to provide a new insight into the development of effective molecular therapies for this disease.
Materials and methods
Tissue sample collection
From March 2015 to October 2016, 34 patients with lung adenocarcinoma who visited our hospital were recruited. Primary lung adenocarcinoma tissues and matching normal lung tissues were collected, quick-frozen in liquid nitrogen, and preserved at −80°C. The histological confirmation for these samples was performed by 2 pathologists. This study was approved by the ethical committee of our hospital, and written informed consent from all patients was obtained.
Cell culture and treatment
Normal lung epithelial cell line BEAS-2B and human lung adenocarcinoma cell lines H1299, H23, SPC-A1, and A549 were obtained from ATCC (Manassas, VA, USA). These cells were then maintained in Dulbecco’s Modified Eagle’s Medium (Sigma, St Louis, MO, USA) mixed with 10% fetal bovine serum (Welgene Biotech Company., Taipei, Taiwan) in a 37°C, 5% CO2 incubator.
To investigate the regulatory relationship between miR-425 expression and IL-6/STAT3 signaling, H1299 cells were stimulated with IL-1 (10 ng/mL, Sigma), IL-6 (30 ng/mL, Sigma), IFN-γ (10 ng/mL, Sigma), and TNF-α (10 ng/mL, Sigma) for 72 h, respectively. In addition, H1299 cells were treated with IL-6 (30 ng/mL) for different time durations (0, 12, 24, 48, and 72 h, respectively). Moreover, H1299 cells were treated with different doses of IL-6 (0, 1, 5, 10, 20, and 30 ng/mL, respectively) for 72 h. Besides, H1299 cells were stimulated with IL-6 (30 ng/mL) alone or combined with IL-6 and STAT3 inhibitor (S3I-201, 10 g/mL, Sigma) for 72 h.
Plasmid transfection
To explore the function of miR-425 in lung adenocarcinoma, H1299 cells were transfected with plasmids encoding pre-miR-425 or pre-miR-control (pre-miR-ctrl) that were purchased from GenePharma (Shanghai, People’s Republic of China) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). At 72 h posttransfection, cells were harvested for detecting transfection efficiency using quantitative reverse-transcription PCR (qRT-PCR).
Knockdown of ADAM9
To explore the function of ADAM9 in lung adenocarcinoma, H1299 cells were transfected with ADAM9 siRNA (5′-CCAGAGAAGUUCCUAUAUA-3′ and 5′-UAUAUAGGAACUUCUCUGG-3′) or ADAM9 siRNA-control (5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-AAACGUGACACACGUUCGGAGAA-3′) using Lipofectamine 2000 reagent (Invitrogen). At 72 h posttransfection, cells were harvested for detecting transfection efficiency using qRT-PCR.
Cell proliferation analysis
For MTT assay, H1299 cells were seeded onto 96-well plates with a density of 5×104 cells/well. After different time points (1, 2, 3, 4, 5, and 6 days) of transfection, the plates were added with 20 μL of MTT solution (0.5 mg/mL; Roche, Indianapolis, IN, USA), and incubation was continued for another 4 h at 37°C. To further dissolve the precipitated formazan, 100 μL of DMSO was used and the cells incubated for 15 min. The optical density value at 450 nm was then read with a Multiskan EX (Thermo, Vantaa, Finland) reader for calculating cell viability.
For colony formation assay, H1299 cells (100 cells/dish) were placed in 60 mm culture dishes at 72 h after transfection and then maintained in complete medium for 14 days. This was followed by fixation with methanol, staining of colonies with 0.1% crystal violet, and then counting of colonies containing more than 30 cells.
Detection of cell apoptosis
Cell apoptosis was assayed using the FITC/Annexin V Apoptosis Detection Kit (Invitrogen) by flow cytometry. Briefly, H1299 cells in different transfected groups were resuspended in annexin V-binding buffer. Next, H1299 cells were double stained with 10 μL Annexin V/FITC and 5 μL propidium iodide (PI) (10 mg/L) for 15 min away from light. By means of a FACS Calibur flow cytometer (Becton–Dickinson, San Jose, CA, USA), the apoptotic cells were then detected and analyzed.
Dual-luciferase reporter assay
The 3′-untranslated regions (UTRs) of AMAD19 containing miR-425 binding sites were amplified by Sangon Biotech (Shanghai, People’s Republic of China) and cloned downstream of the luciferase reporter in psiCHECK-2 vector (Promega, Madison, WI, USA). Using a site-directed mutagenesis kit (Stratagene, San Diego, CA, USA), the psiCHECK-2-ADAM9-mutated reporter construct with point mutations in the seed sequence was synthesized. Dual-luciferase reporter assay was performed using luciferase vectors constructed with ADAM9-3′-UTR alone or in the presence of pre-miR-425 or pre-miR-ctrl. At 48 h after treatment, luciferase activities of different treated groups were determined by the Dual-Glo luciferase assay kit (Promega). Firefly luciferase (FLuc) activity was used as reference for normalization. The recombinant vectors (psiCHECK-2-ADAM19 and psiCHECK-2-mut-ADAM19) were normalized to an empty psiCHECK-2 vector.
Real-time qRT-PCR
Using Trizol reagent (Invitrogen), total RNA was extracted. The quality was then determined by SMA 400 UV-VIS spectrophotometer (Merinton, Shanghai, People’s Republic of China). Following the manufacturer’s protocols of the PrimeScriptTM RT Master Mix Kit (RR036A; Takara, Kyoto, Japan), reverse transcription into cDNA was performed. For detecting the expression of miR-425, real-time qPCR was carried out using TaqMan microRNA assays (Applied Biosystems, Foster City, CA, USA). For analysis of ADMA9 expression, Power SYBR green PCR master mix (Applied Biosystems) was used. All qPCRs were performed on an ABI 7900HT series PCR machine (Applied Biosystems). Primers (forward and reverse) used for amplification in our study were as follows: miR-425, 5′-TCTACCGGTGTGCCCCTGACCCCCAGACA-3′ and 5′-TCTGAATTCAGCAGGGAAACCCAGGGGCA-3′; U6, 5′-CTCGCTTCGGCAGCACA-3′ and 5′-AACGCTTCACGAATTTGCGT-3′; ADAM9, 5′-GCTAGTTGGACTGGAGATTTGG-3′ and 5′-TTATTACCACAGGAGGGAGCAC-3′; STAT3, 5′-ACCAGCAGTATAGCCGCTT-3′, 5′-GCCACAATCCGGGCAATCT-3′; IL-6R, 5′-TG GTGGATGTTCCCCCCGAG-3′, 5′-TCCTGGGAATACTGGCACGG-3′; and β-actin, 5′-GCACTCTTCCAGCCTTCCTTCC-3′ and 5′-TCACCTTCACCGTTCCAGTTTTT-3′. The relative expression level of miR-425 was normalized to U6, and ADAM9, STAT3, and IL-6-R were normalized to β-actin, and calculated using the 2−ΔΔCt method.19
The plasmid was transfected to conduct luciferase assay, proliferation assay, cell colony counting and apoptosis assay were all according to the reagent instruction.
Statistical analysis
All experiments were repeated 3 times. The data are shown as the mean ± SD. Statistical analyses between groups were performed using 2-independent Student’s t-test and among groups using one-way analysis of variance and subsequent post hoc Turkey test. In addition, linear correlation analysis was performed to measure the correlation between miR-425 expression and AMAD9 levels in human lung adenocarcinoma tissues. All statistical analyses were conducted in SPSS 19.0 statistical software (IBM Corporation, Armonk, NY, USA), and statistical significance was set at P<0.05.
Results
miR-425 was downregulated in lung adenocarcinoma tissues and cells
To investigate whether miR-425 played a key role in lung adenocarcinoma, the miR-425 expression in human normal lung adenocarcinoma tissues and cells was determined. It was shown that miR-425 expression was significantly downregulated in human lung adenocarcinoma tissues relative to normal tissues (P<0.001, Figure 1A). Consistent results were obtained, demonstrating that miR-425 expression in lung adenocarcinoma cells (H1299, H23, SPC-A1, and A549) was also markedly lower than that in BEAS-2B cells (P<0.05, Figure 1B).
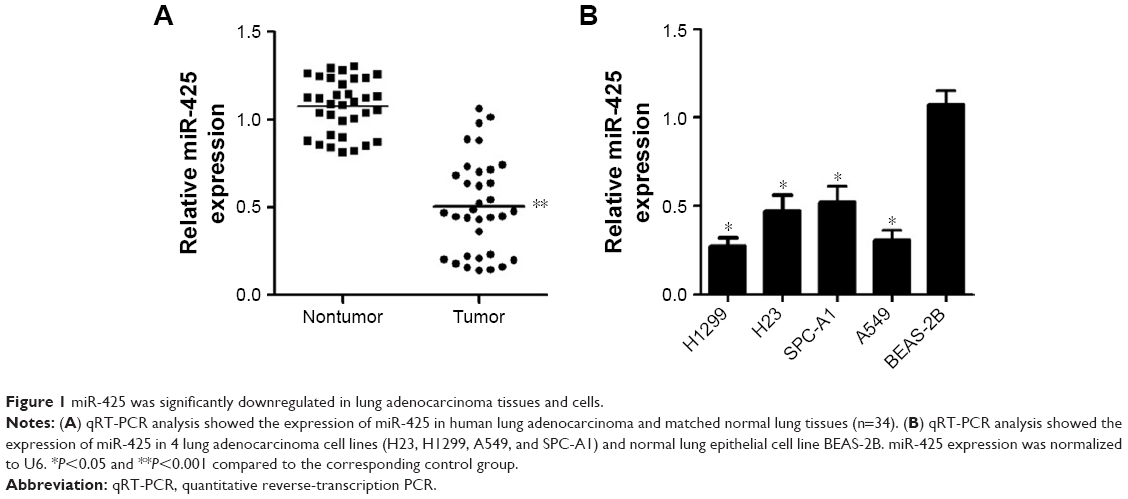 | Figure 1 miR-425 was significantly downregulated in lung adenocarcinoma tissues and cells. |
miR-425 expression was inhibited by IL-6/STAT3 signaling
The regulatory relationship between miR-425 expression and IL-6/STAT3 signaling in H1299 cells was investigated. We found that miR-425 expression was significantly decreased only after IL-6 treatment (P<0.05, Figure 2A). In addition, the results showed that IL-6 treatment significantly decreased miR-425 expression in a time- and dose-dependent manner (Figure 2B and C). Furthermore, H1299 cells were stimulated with IL-6 alone or combination with IL-6 and STAT3 inhibitor S3I-201 for 72 h. The results showed the S3I-201 treatment could significantly reverse the effects of IL-6 on the decreased miR-425 expression (P<0.01, Figure 2D). In addition, the expressions of STAT3 and IL-6R were significantly higher after IL-6 treatment (P<0.05, Figure 2E). miR-425 expression significantly negatively regulated the expression of ADAM9 (Figure 2F). Taken together, miR-425 expression was markedly inhibited by IL-6/STAT3 signaling and also negatively regulated the expression of ADAM9.
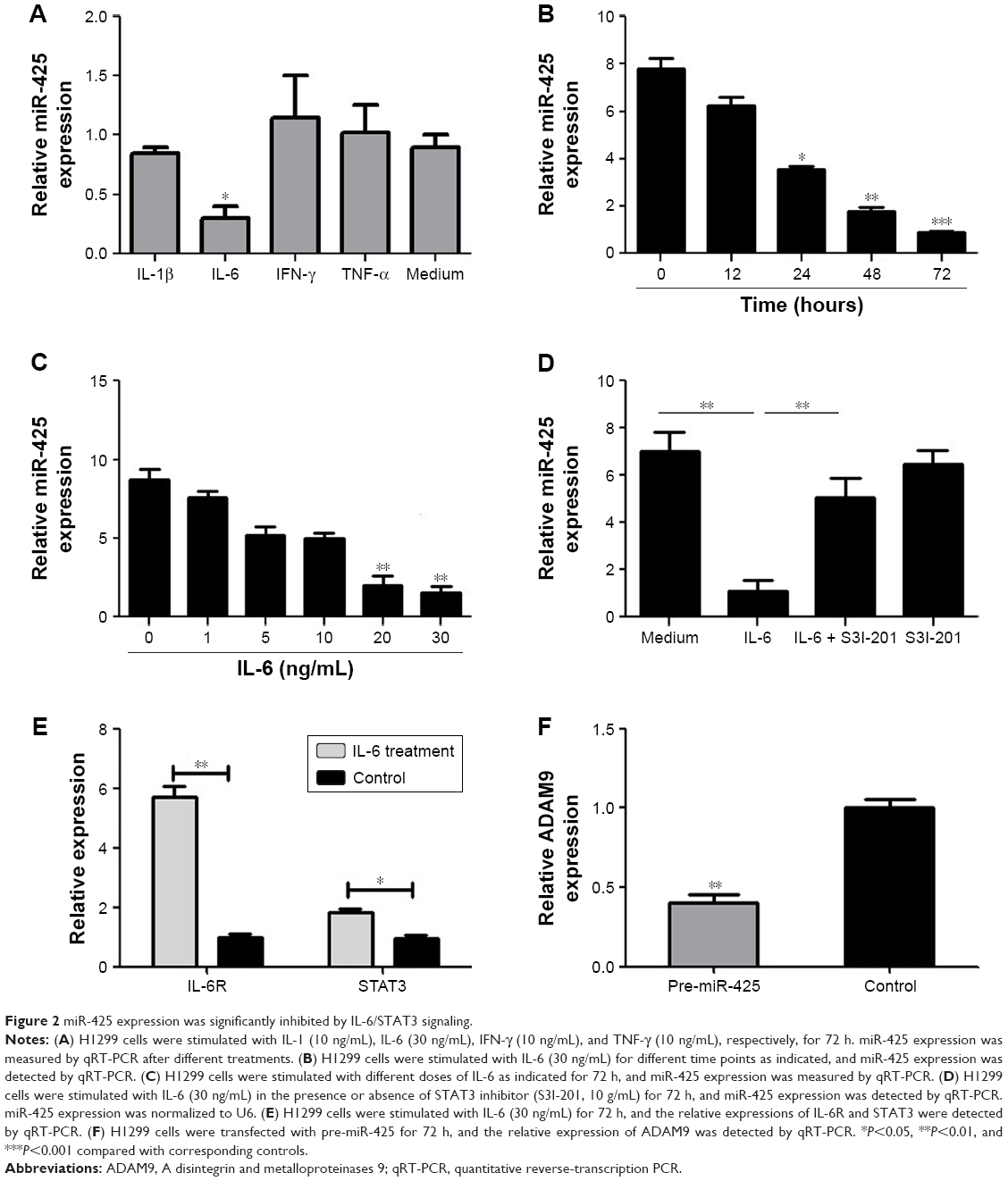 | Figure 2 miR-425 expression was significantly inhibited by IL-6/STAT3 signaling. |
Overexpression of miR-425 inhibited proliferation and induced apoptosis of H1299 cells
To further explore the role of miR-425 in lung adenocarcinoma, H1299 cells were transfected with pre-miR-425 to overexpress miR-425 expression. The high transfection efficiency was confirmed by qRT-PCR (Figure 3A). Moreover, MTT assay showed that, in comparison with the pre-miR-ctrl transfected group, the viability of pre-miR-425 transfected H1299 cells was significantly decreased with the increase of transfection time during an experimental period of 6 days after cell transfection (P<0.05, Figure 3B). By knockdown of ADAM9, the expression of ADAM9 was significantly decreased (P<0.05, Figure 3C). The viability of siRNA-transfected H1299 cells was significantly decreased with the increase of transfection time during an experimental period of 6 days after cell transfection (P<0.05, Figure 3D). Moreover, the colony formation ability of pre-miR-425-transfected H1299 cells was markedly lower than that of pre-miR-ctrl-transfected cells, and significant differences existed between the 2 groups (P<0.05, Figure 3E), further confirming that miR-425 overexpression could inhibit H1299 cell proliferation. Furthermore, we also detected the effects of miR-425 on cell apoptosis. The results showed that the percentages of apoptotic cells were obviously increased in the pre-miR-425-transfected group in comparison with the pre-miR-ctrl-transfected group (P<0.001, Figure 3F), indicating that overexpression of miR-425 significantly induced cell apoptosis.
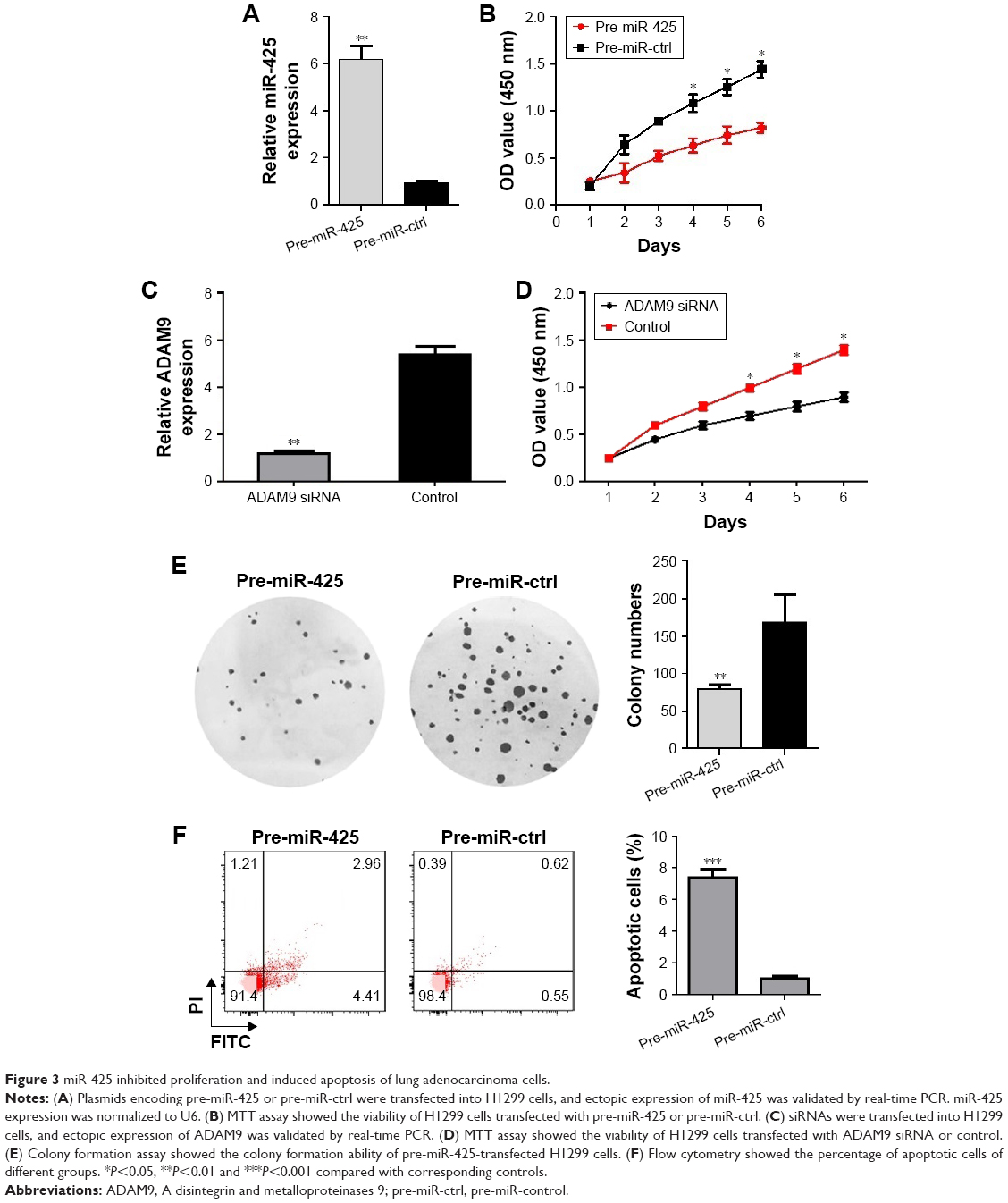 | Figure 3 miR-425 inhibited proliferation and induced apoptosis of lung adenocarcinoma cells. |
ADAM 9 was verified as a target of miR-425
To explore the mechanism of miR-425 in lung adenocarcinoma development, the potential targets of miR-425 were predicted using TargetScanHuman (Whitehead Institute for Biomedical Research, Cambridge, MA, USA). As shown in Figure 4A, a sequence alignment of miR-425 with reverse complementary ADAM9 was identified. To further verify the predicted target relationship between miR-425 and ADAM9, dual-luciferase reporter assay was performed. The expected results were obtained: that pre-miR-425 transfection could significantly inhibit the luciferase activity of ADAM9 (P<0.05, Figure 4B), but not mutated ADAM9. Moreover, the mRNA expression of ADAM9 was markedly downregulated in the pre-miR-425-transfected group compared to the pre-miR-ctrl-transfected group (P<0.01, Figure 4C). Importantly, compared with normal tissues, ADAM9 expression was significantly upregulated in human lung adenocarcinoma tissues (P<0.001, Figure 4D). Furthermore, linear correlation analysis confirmed the negative correlation between miR-425 expression and AMAD9 expression in human lung adenocarcinoma tissues (R = −0.7301, P<0.0001, Figure 4E). These data indicated that ADAM9 was a target of miR-425 and that ADAM9 expression was negatively regulated by miR-425.
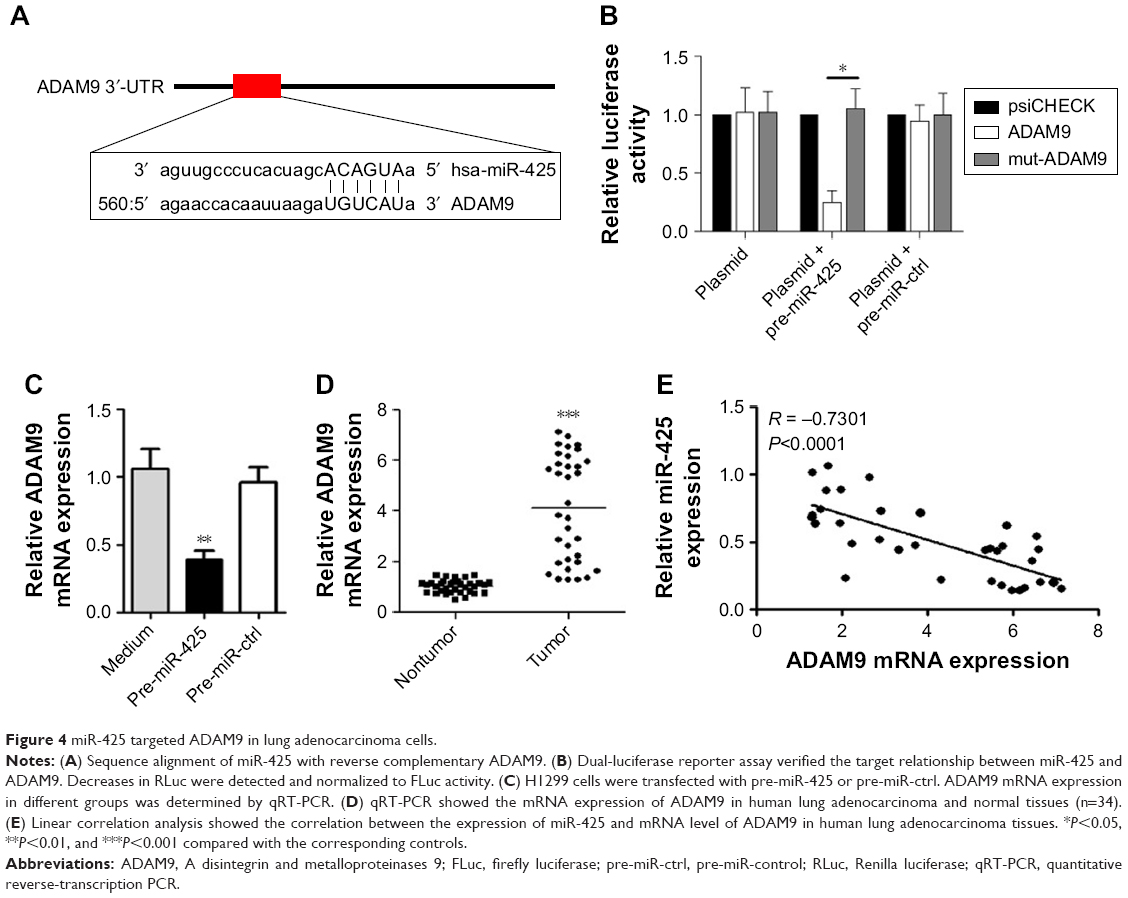 | Figure 4 miR-425 targeted ADAM9 in lung adenocarcinoma cells. |
Discussion
The emergence of miRNAs provided a new insight into their use as potential biomarkers and therapeutic targets for multiple cancers.20–23 Identification of crucial miRNAs in cancer development has become a research hotspot. In this study, miR-425 was found to be downregulated in lung adenocarcinoma tissues and cells, and was markedly inhibited by IL-6/STAT3 signaling. In addition, overexpression of miR-425 markedly decreased the proliferation and colony formation of H1299 cells, and markedly promoted H1299 cell apoptosis. Moreover, ADAM9 was a target of miR-425, and its expression was negatively regulated by miR-425. These data imply the important function of miR-425 in lung adenocarcinoma and merit further discussion.
In melanoma, miR-425 is confirmed to inhibit tumor metastasis.13 Consistent with this finding, miR-425 expression was also found to be downregulated in lung adenocarcinoma cells, and overexpression of miR-425 significantly decreased lung adenocarcinoma cell proliferation and induced apoptosis. Thus, we speculate that miR-425 also functions as a tumor suppressor in lung adenocarcinoma. In addition, the role of IL-6/STAT3 signaling in cancer development has attracted widespread attention.24,25 Tumor-promoting cytokines released by immune/inflammatory cells, including IL-6, act in a paracrine manner to activate transcription factors, such as STAT3, to achieve their effects in many types of cancer cells.26 Yeh et al27 revealed that STAT3 activation induced by IL-6 contributed to lung adenocarcinoma development. Furthermore, activation of IL-6R/JAK1/STAT3 pathway is a key mechanism to mediate the tumor-suppressive function of miR-26a on growth and metastasis of human hepatocellular carcinoma.28 In our study, miR-425 was decreased after IL-6 stimulation, which could be reversed by treatment with the STAT3 inhibitor S3I-201. It can therefore be speculated that miR-425 may be a downstream target of IL-6/STAT3 signaling, and miR-425 downregulation caused by IL-6/STAT3 signaling activation may contribute to the development of lung adenocarcinoma.
To further elucidate the possible mechanism of miR-425 in lung adenocarcinoma, the potential targets were explored, in which ADAM9 was identified. ADAM9 belongs to a family of transmembrane disintegrin-containing metalloproteinases (ADAMs), which are widely implicated in cell adhesion, cell–cell signaling, and cell migration,29,30 and can play diverse roles in pathological process of many diseases, including human cancers.31,32 It is reported that ADAM9 is highly expressed in gastric cancer tissues and may regulate the malignant behaviors of gastric cancer cells.33,34 Moreover, overexpression of ADAM9 is also found to promote the progression of colon cancer.35 In NSCLC, upregulation of ADAM9 is shown to correlate with brain metastasis.36 Sher et al37 also confirmed that ADAM9 regulation could contribute to brain metastasis in lung adenocarcinoma cells. Notably, Lin et al38 demonstrated the correlation between the enhanced expression of ADAM9 and the poor prognosis in patients with lung cancer. Given the crucial role of ADAM9, we speculate that miR-425 may inhibit proliferation and induce apoptosis of H1299 cells via targeting ADAM9.
Conclusion
Our findings reveal that downregulation of miR-425 caused by IL-6/STAT3 signaling leads to loss of ADAM9 targeting, results in enhanced ADAM9 expression, and contributes to the development of lung adenocarcinoma. However, there are some limitations in this study. In a future research, an apoptosis assay will be added to confirm cell death, and more miRNAs and genes will be included.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Sève P, Reiman T, Dumontet C. The role of betaIII tubulin in predicting chemoresistance in non-small cell lung cancer. Lung Cancer. 2010;67(2):136–143. | ||
Thomson CS, Forman D. Cancer survival in England and the influence of early diagnosis: what can we learn from recent EUROCARE results? Br J Cancer. 2009;101(Suppl 2):S102–S109. | ||
Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. | ||
Farazi TA, Hoell JI, Morozov P, et al. MicroRNAs in human cancer. In: Schmitz U, Wolkenhauer O, Vera J, editors. MicroRNA Cancer Regulation. Berlin: Springer; 2013:1–20. | ||
Tüfekci KU, Meuwissen RL, Genç S. The role of microRNAs in biological processes. Methods Mol Biol. 2014;1107:15–31. | ||
Roybal JD, Zang Y, Ahn YH, et al. miR-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011;9(1):25–35. | ||
Song Q, Xu Y, Yang C, et al. miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res. 2014;74(11):3031–3042. | ||
Cai J, Wu J, Zhang H, et al. miR-186 downregulation correlates with poor survival in lung adenocarcinoma, where it interferes with cell-cycle regulation. Cancer Res. 2013;73(2):756–766. | ||
Wang H, Zhu L, Wang R, Yang YC, Wang ZX. MiR-224 promotes the chemoresistance of human lung adenocarcinoma cells to cisplatin via regulating G 1/S transition and apoptosis by targeting p21 WAF1/CIP1. Br J Cancer. 2014;111(2):339–354. | ||
Li J, Wang Y, Song Y, Fu Z, Yu W. miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer. 2014;13:193. | ||
Liu L, Zhao Z, Zhou W, Fan X, Zhan Q, Song Y. Enhanced expression of miR-425 promotes esophageal squamous cell carcinoma tumorigenesis by targeting SMAD2. J Genet Genomics. 2015;42(11):601–611. | ||
Liu P, Hu Y, Ma L, Du M, Xia L, Hu Z. miR-425 inhibits melanoma metastasis through repression of PI3K-Akt pathway by targeting IGF-1. Biomed Pharmacother. 2015;75:51–55. | ||
He B, Li T, Guan L, et al. CTNNA3is a tumor suppressor in hepatocellular carcinomas and is inhibited by miR-425. Oncotarget. 2016;7(7):8078–8089. | ||
Wolfsberg TG, Primakoff P, Myles DG, White JM. ADAM, a novel family of membrane proteins containing A Disintegrin And Metalloprotease domain: multipotential functions in cell-cell and cell-matrix interactions. J Cell Biol. 1995;131(2):275–278. | ||
Scanlan MJ, Gordon CM, Williamson B, et al. Identification of cancer/testis genes by database mining and mRNA expression analysis. International Journal of Cancer. 2002;98(4):485–492. | ||
Lin CY, Chen HJ, Huang CC, et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74(18):5229–5243. | ||
Chang L, Gong F, Cai H, Li Z, Cui Y. Combined RNAi targeting human Stat3 and ADAM9 as gene therapy for non-small cell lung cancer. Oncol Lett. 2016;11(2):1242–1250. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using RT-qPCR and the 2−ΔΔ | ||
Acunzo M, Romano G, Wernicke D, Croce CM. MicroRNA and cancer – a brief overview. Adv Biol Regul. 2015;57:1–9. | ||
Cheng G. Circulating miRNAs: roles in cancer diagnosis, prognosis and therapy. Adv Drug Deliv Rev. 2015;81:75–93. | ||
Florczuk M, Szpechcinski A, Chorostowska-Wynimko J. miRNAs as biomarkers and therapeutic targets in non-small cell lung cancer: current perspectives. Target Oncol. 2017;12(2):1–22. | ||
Cho WC. MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol. 2010;42(8):1273–1281. | ||
Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41(16):2502–2512. | ||
Chang Q, Bournazou E, Sansone P, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15(7):848–862. | ||
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. | ||
Yeh HH, Lai WW, Chen HH, Liu HS, Su WC. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene. 2006;25(31):4300–4309. | ||
Yang X, Liang L, Zhang XF, et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58(1):158–170. | ||
Wildeboer D, Naus S, Amy sang QX, Bartsch JW, Pagenstecher A. Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol. 2006;65(5):516–527. | ||
Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20(2):126–137. | ||
Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007;98(5):621–628. | ||
Turner SL, Blair-Zajdel ME, Bunning RA. ADAMs and ADAMTSs in cancer. Br J Biomed Sci. 2009;66(2):117. | ||
Carl-McGrath S, Lendeckel U, Ebert M, Roessner A, Röcken C. The disintegrin-metalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int J Oncol. 2005;26(1):17–24. | ||
Kim JM, Jeung HC, Rha SY, et al. The effect of disintegrin-metalloproteinase ADAM9 in gastric cancer progression. Mol Cancer Ther. 2014;13(12):3074–3085. | ||
Li J, Ji Z, Qiao C, Qi Y, Shi W. Overexpression of ADAM9 promotes colon cancer cells invasion. J Invest Surg. 2013;26(3):127–133. | ||
Shintani Y, Higashiyama S, Ohta M, et al. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res. 2004;64(12):4190–4196. | ||
Sher YP, Wang LJ, Chuang LL, et al. ADAM9 up-regulates N-cadherin via miR-218 suppression in lung adenocarcinoma cells. PLoS One. 2014;9(4):e94065. | ||
Lin CY, Chen HJ, Huang CC, et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74(18):5229–5243. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.