Back to Journals » OncoTargets and Therapy » Volume 11
microRNA-26a induces a mitochondrial apoptosis mediated by p53 through targeting to inhibit Mcl1 in human hepatocellular carcinoma
Authors Gao X, Zhu Y ![]() , Li JH
, Li JH ![]() , Wang X, Zhang X, Yi C, Yang X
, Wang X, Zhang X, Yi C, Yang X ![]()
Received 27 December 2017
Accepted for publication 1 March 2018
Published 19 April 2018 Volume 2018:11 Pages 2227—2239
DOI https://doi.org/10.2147/OTT.S160895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Xiao-Mei Gao,* Ying Zhu,* Jian-Hua Li,* Xiang-Yu Wang, Xiao-Fei Zhang, Chen-He Yi, Xin Yang
Department of General Surgery, Huashan Hospital & Cancer Metastasis Institute, Fudan University, Shanghai, China
*These authors contributed equally to this work
Aim: We have previously found that microRNA-26a (miR-26a) is a potential tumor suppressor in hepatocellular carcinoma (HCC). In this study, we further explored the roles of miR-26a in HCC apoptosis.
Methods: miR-26a expression levels were detected in HCC tissues by real-time PCR. Statistical analysis was performed to explore the correlation between miR-26a expression and apoptotic cells and the antiapoptotic protein levels. In vitro assays were performed to investigate the roles of miR-26a in HCC apoptosis. The immunohistochemical staining analysis, Western blot, and luciferase reporter assay were performed to evaluate the relationship between miR-26a and its potential upstream regulating and downstream target genes. The potential mechanism of the combination treatment of interferon-α1b (IFN-α1b) and 5-fluorouracil (5-FU) was explored by in vitro and in vivo assays.
Results: miR-26a levels were significantly associated with the number of apoptotic cells and inversely correlated with the protein levels of Bcl-2, Bcl-xL, and Mcl1 in HCC tissues. Furthermore, miR-26a was proved to induce the mitochondrial apoptosis in vitro by directly targeting to inhibit Mcl1 in HCC cells. Moreover, p53 was demonstrated to mediate miR-26a-induced apoptosis, by activating its promoter in HCC. Meanwhile, the combination treatment of IFN-α1b and 5-FU could induce the expression of p53, which then upregulated miR-26a and downregulated Mcl1 levels, and finally promoted the apoptosis of HCC cells through a mitochondrial pathway.
Conclusion: These findings highlight the important and related molecular mechanism of miR-26a in the regulation of apoptosis and implicate the potential application of combination of IFN-α1b and 5-FU in HCC treatment.
Keywords: miR-26a, p53, Mcl1, mitochondrial apoptosis, hepatocellular carcinoma
Introduction
MicroRNAs (miRNAs) have been shown to play crucial roles in the tumorigenicity,1 proliferation,2 apoptosis,3 angiogenesis, invasion, and metastasis4–6 of hepatocellular carcinoma (HCC). In addition, many studies have also demonstrated that some miRNAs including miR-125,7 miR-181b,8 and miR-3659 take part in the apoptosis regulation and may be used as therapeutic targets for the other types of cancer.
Emerging data show that miRNA-26a (miR-26a) is downregulated and could serve as a potential tumor suppressor in several distinct cancer types including the carcinomas of thyroid, nasopharyngeal, breast, as well as liver;10–17 and restoration of miR-26a activity in HCC has been proposed as a novel treatment strategy.16 Recently, we have reported that miR-26a suppresses tumor growth and metastasis of HCC through IL-6–Stat3 signaling and is a novel prognostic marker and therapeutic target for HCC.18
Interferon (IFN) has various biological properties including immunomodulation and antitumor activity19,20 and is a useful adjuvant therapy to decrease the risk of tumor recurrence after HCC resection.21,22 The subgroup of patients with hepatitis B virus-related HCCs whose tumors had low expression of miR-26a had a better response to adjuvant IFN therapy versus patients whose tumors had high expression of miR-26a.17 In contrast, many studies suggest that 5-fluorouracil (5-FU) could significantly enhance the antitumor activity of IFN for HCC and combination of IFN-α and 5-FU could obviously increase the response for advanced HCC.23–26 Accordingly, understanding the related mechanism of this combination is important for future clinical applications.
In this study, we further investigated the functional mechanism of miR-26a in the regulation of HCC apoptosis and the potential application of the combination treatment of IFN-α1b and 5-FU in HCC.
Methods
Ethics approval and informed consent
This research was verified and ethically approved by the Ethics Committee of Fudan University (Shanghai, China). All patients provided written informed consent for the use of surgical specimens for pathological testing with operation consents. All animal care and experimental procedures were in strict accordance with recommendations of the Guide for the Care and Use of Laboratory Animals and were approved by the Committee on the Ethics of Animal Experiments of Fudan University (permit 2016-0-802-A186). The use of the cell lines was approved by the Ethics Committee of Fudan University.
Patients and specimens
A total of 58 patients who underwent curative resection for HCC at the Huashan Hospital & Cancer Metastasis Institute, Fudan University, from February 2014 to December 2015 were enrolled in this study. All patients did not receive any preoperative cancer treatment. Their clinicopathological characteristics are presented in Table S1. Clinical samples were collected from these patients.
Cell lines and culture conditions
HEK 293T and two high metastatic potential HCC cell lines including MHCC97-H and HCC-LM3 were used in this study as described in the Supplementary materials.
Vectors and cell transfections
The miRNA vectors were purchased from Genecopoeia (Guangzhou, China) including miR-26a expression vector (HmiR0044-pEZX-MR03), anti-miR-26a expression vector (HmiR-AN0354-pEZX-AM04), the control vector for miR-26a (CmiR0001-pEZX-MR03), and the negative control vector (CmiR-AN0001-AM01). The expression vectors for human Mcl1 and p53, wild-type and mutant 3′-untranslated region (UTR) segments of human Mcl1 mRNA, and the wild-type and mutant miR-26a-1 promoter were constructed as described in Table S2.
Transferase-mediated dUTP nick end-labeling (TUNEL) assay
We detected in situ apoptosis using the terminal deoxynucleotidyl TUNEL Kit (Keygen, Nanjing, China) according to the manufacturer’s instructions. This method can detect the fragmented DNA ends of apoptotic cells. For the quantification of apoptosis cells, five microscopic fields were randomly selected at 400× magnification and the average counts of TUNEL-positive cells were calculated.
Cell apoptosis analysis
HCC cells were seeded in six-well plates at 1×105 cells/well. Forty-eight hours after transfection or treated with 1,000 U/mL of IFN-α1b and 5 μg/mL of 5-FU, cells were stained with PE and Annexin IV (BD Pharmingen, San Jose, CA, USA). Apoptotic cells were analyzed by flow cytometry (Epics Altra; Beckman Coulter, Brea, CA, USA). All experiments were performed in triplicate.
Measurement of mitochondrial membrane potential
Cells were harvested by centrifugation at 300× g at 4°C for 5 minutes. Mitochondrial membrane potential (ΔΨm) was determined by staining the cells with JC-1 (Keygen) according to the manufacturer’s instructions. Mitochondrial ΔΨ was quantified by flow cytometry analysis (Epics Altra). Data were presented as the percentage of cells with the loss of ΔΨm. All experiments were performed in triplicate.
In vivo tumor apoptosis assays
The nude mice (BALB/c nu/nu, 4 weeks old) were randomly divided into the following two groups: control and IFN-α1b +5-FU group. HCC-LM3 cells (2×106 cells/mouse) were implanted subcutaneously into the flank of nude mice. When the tumor size reached to 5–10 mm in diameter (1 week later), the mice in the IFN-α1b +5-FU group were treated with 5-FU (Sigma-Aldrich Co., St Louis, MO, USA) (20 mg/kg/d, via intraperitoneal injection) and IFN-α1b (Kexin, Shenzhen, China) (3.0×105 U/d, via subcutaneous injection) for 6 weeks, while only phosphate-buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA, USA) was given to control. Six weeks later, the mice were sacrificed. Tumors were removed to evaluate their sizes and volumes (length/2× [the width]2), and then they were fixed in formalin and embedded in paraffin. Consecutive sections were made for every tumor block, and TUNEL assay was performed to assess the apoptosis of cancer cells.
Western blot
Cytosolic protein fractions were prepared using the cell mitochondria isolation kit (Beyotime, Shanghai, China). Total protein was extracted by lysing cells in RIPA buffer containing protease inhibitor. Western blot was performed as described in the Supplementary materials.
Immunohistochemical staining
The protein levels of Bcl-2, Bcl-xL, Mcl1, and p53 in tumor tissues were detected by immunohistochemistry. The details are presented in the Supplementary materials.
Luciferase reporter assays
The effect of miR-26a on the luciferase activity of Mcl1 containing a wild-type or mutant 3′-UTR was measured as described in the previous study.18 HEK 293T cells were seeded into 24-well plates 1 day before transfection, and 100 ng of wild-type or mutant miR-26a-1 promoter plasmids, 10 ng of Renilla plasmid, and 400 ng of p53 plasmids were co-transfected with Lipofectamine 3000. Then, the reporter activity was measured at 48 hours posttransfection using a Dual-Luciferase® Reporter Assay System (Promega Corporation, Fitchburg, WI, USA). Luciferase reporter assays were performed in duplicate and repeated in three independent experiments. Luciferase activity was detected using an Orion II Microplate Luminometer (Berthold Technologies, Bad Wildbad, Germany).
Statistical analysis
Statistical analysis was performed using SPSS 15.0 (SPSS Inc., Chicago, IL, USA). The differences between groups were analyzed using Student’s t-test (only two groups) or one-way analysis of variance (more than two groups were compared). The association of miR-26a expression with apoptotic cell number and the expression of apoptotic pathway-related protein in HCC patients was explored by Spearman’s correlation and χ2 tests. All statistical tests were two sided, and P-value <0.05 was considered statistically significant.
Results
The relationship between miR-26a expression and the apoptotic cell number in HCC tissues
miR-26a expression levels were detected by qRT-PCR and normalized against an endogenous control (U6 RNA) (P<0.01; Figure S1). The patients were further classified into low- and high-miR-26a groups according to the cutoff values of miR-26a, which were defined as its median level of the cohort. We next investigated whether there was any correlation between miR-26a expression levels and the number of apoptotic cells in HCC tissues. Apoptotic HCC cells were determined by TUNEL assays (Figure 1A). The TUNEL-positive cells were obviously much more in high-miR-26a HCC tissues than low-miR-26a HCC tissues (P<0.01; Figure 1B). Similarly, the percentage of TUNEL-positive cells was obviously higher in high-miR-26a HCC tissues than in low-miR-26a HCC tissues (P<0.01; Figure S2). Importantly, miR-26a mRNA levels were positively correlated with the number of TUNEL-positive cells in HCC tissues (r=0.5479, P<0.001; Figure 1C). This positive correlation between miR-26a levels and the number of apoptotic HCC cells (P=0.001) was further confirmed by χ2 tests (Table 1).
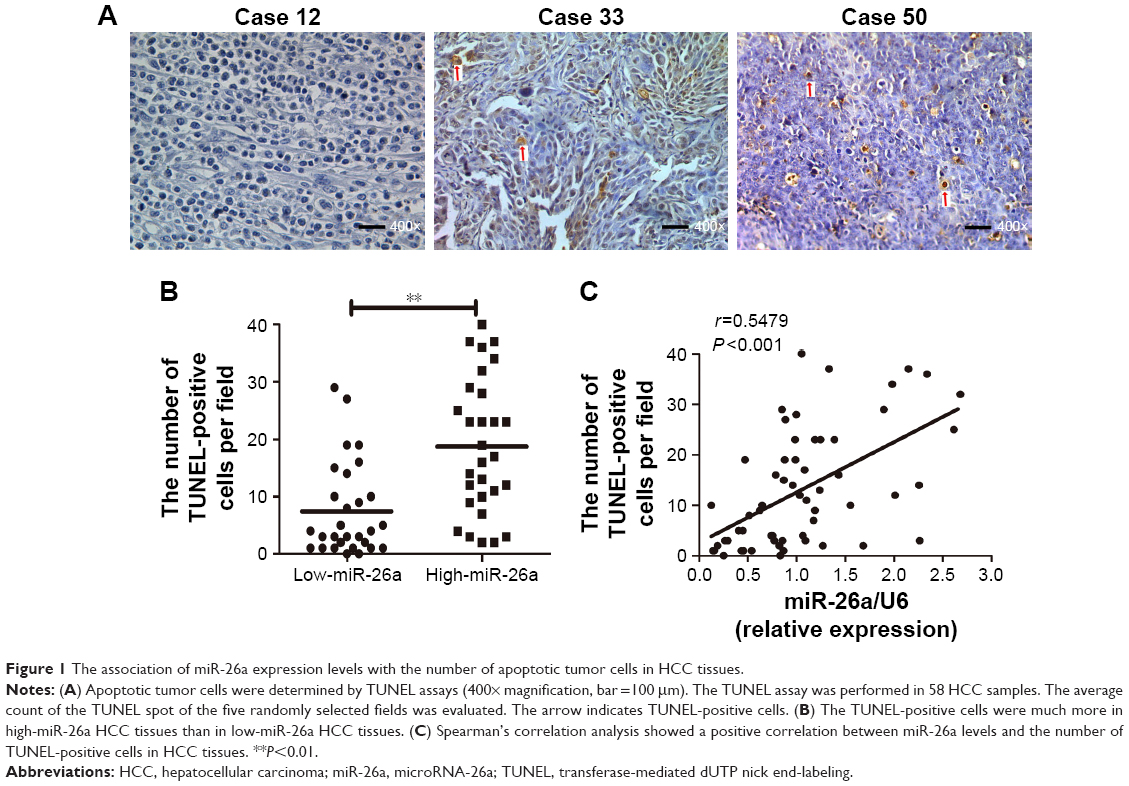 | Figure 1 The association of miR-26a expression levels with the number of apoptotic tumor cells in HCC tissues. |
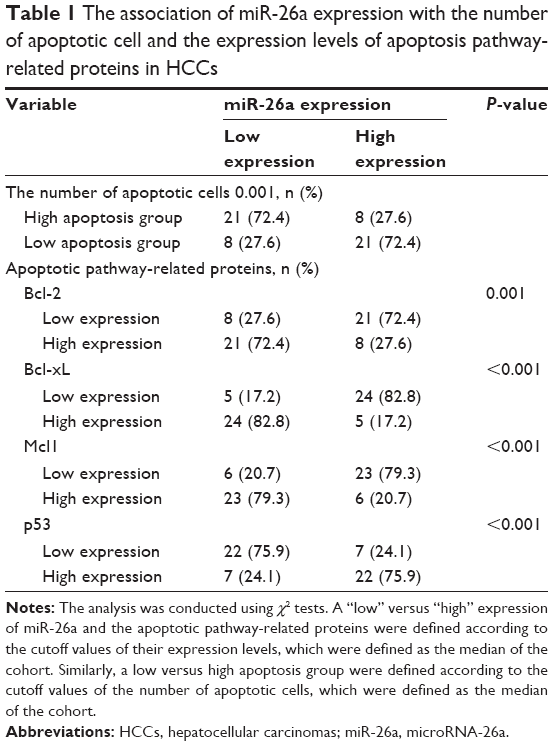 | Table 1 The association of miR-26a expression with the number of apoptotic cell and the expression levels of apoptosis pathway-related proteins in HCCs |
Upregulation of miR-26a induces the apoptosis of HCC cells through a mitochondrial pathway
To explore the functional roles of miR-26a in HCC apoptosis, we transfected the expressing vector of miR-26a into HCC-LM3 and MHCC97-H cells to upregulate the miR-26a expression. After transfection of miR-26a, both HCC-LM3 (26.3% versus 7.4%, P<0.01) and MHCC97-H (16.1% versus 6.1%, P<0.01) cells displayed higher apoptotic rates than control (Figure 2A). These suggest that miR-26a could induce HCC cells’ apoptosis.
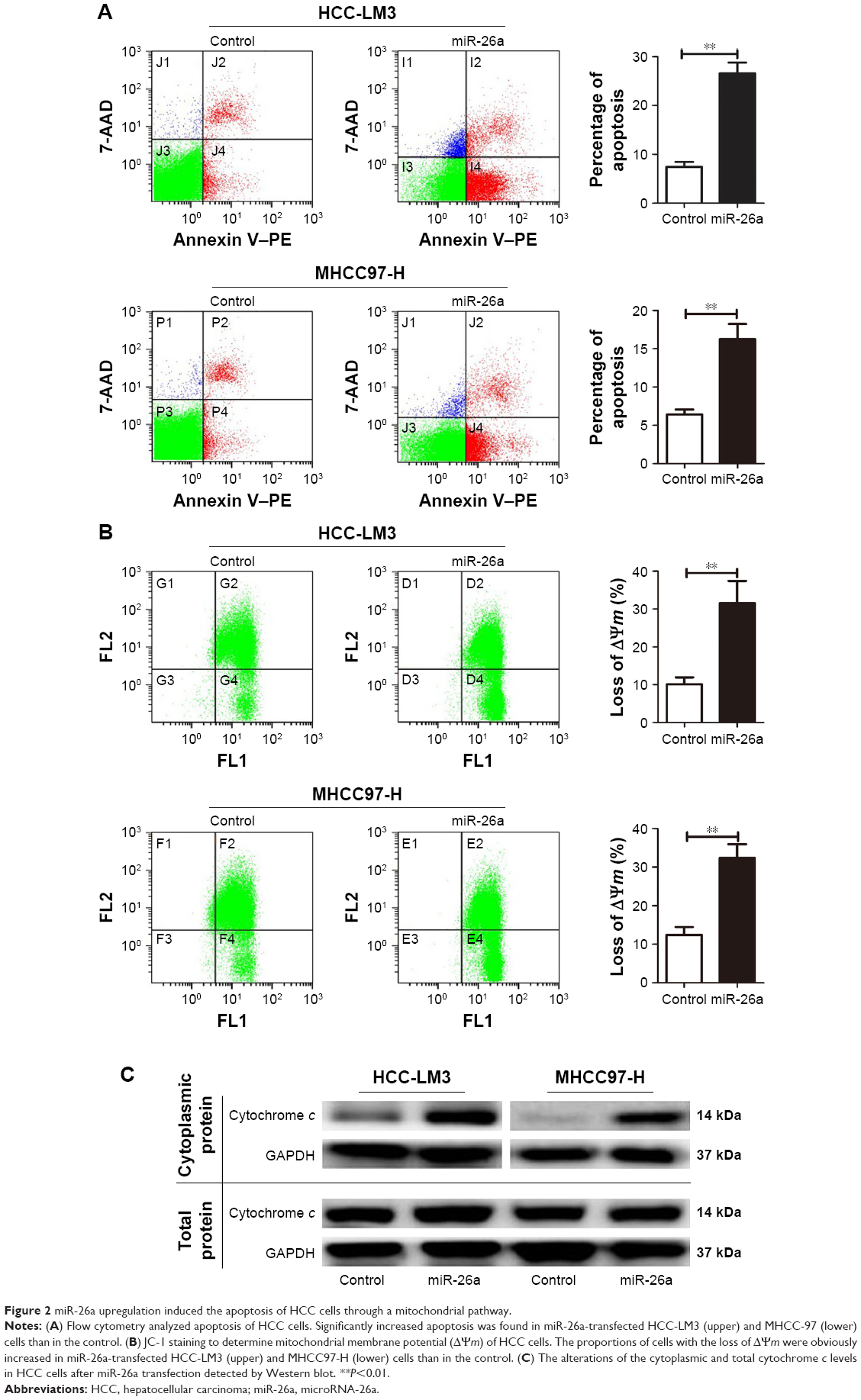 | Figure 2 miR-26a upregulation induced the apoptosis of HCC cells through a mitochondrial pathway. |
Since there have been some reports that miRNAs promote cell apoptosis through the mitochondrial pathway,1,7 we next examined the mitochondrial membrane potential (ΔΨm) by JC-1 assay and cytoplasmic cytochrome c release to evaluate whether upregulation of miR-26a triggered HCC cells’ apoptosis through a mitochondrial pathway. As shown in Figure 2B, the proportion of cells with the loss of ΔΨm were obviously increased in miR-26a-transfected HCC-LM3 (32.1% versus 10.5%, P<0.01) and MHCC97-H cells (33.6% versus 12.8%, P<0.01) than in the control. Moreover, miR-26a transfection induced a significant increase in the cytoplasmic cytochrome c levels, but not the total cytochrome c levels in HCC cells (Figures 2C and S3A), which suggests that the cytochrome c might be released from mitochondria in to cytoplasm. Taken together, these demonstrate that miR-26a may promote HCC apoptosis through a mitochondrial pathway.
Mcl1 is a direct target of miR-26a in promoting HCC apoptosis
Since miRNAs are mostly involved in a variety of biological processes through suppressing the expression of targeting mRNAs, and the proteins of Bcl-2 family are critical for the mitochondrial pathway-related apoptosis, we further evaluated the influence of miR-26a on the expression levels of the antiapoptotic proteins, including Bcl-2, Bcl-xL, and Mcl1, and determined whether miR-26a induces cell apoptosis by targeting these proteins. We found that the expression of Bcl-2, Bcl-xL, and Mcl1 was significantly decreased in high-miR-26a HCC tissues than in low-miR-26a tissues (P<0.01; Figure 3A); in contrast, miR-26a levels were closely associated with these antiapoptotic protein levels. To confirm this finding, we performed Spearman’s correlation analyses and found a statistically significant inverse correlation between miR-26a levels and the protein levels of Bcl-2 (r=−0.6259, P<0.001), Bcl-xL (r=−0.7452, P<0.001), and Mcl1 (r=−0.6940, P<0.001) in HCC tissues (Figure 3B). These correlations were further confirmed by χ2 tests (Table 1). After transfection of miR-26a, the expression levels of Bcl-2, Bcl-xL, and Mcl1 in HCC-LM3 and MHCC97-H cells were also obviously decreased compared with the control (Figures 3C and S3B). These results strongly suggest that miR-26a might induce HCC apoptosis by regulating these antiapoptotic proteins.
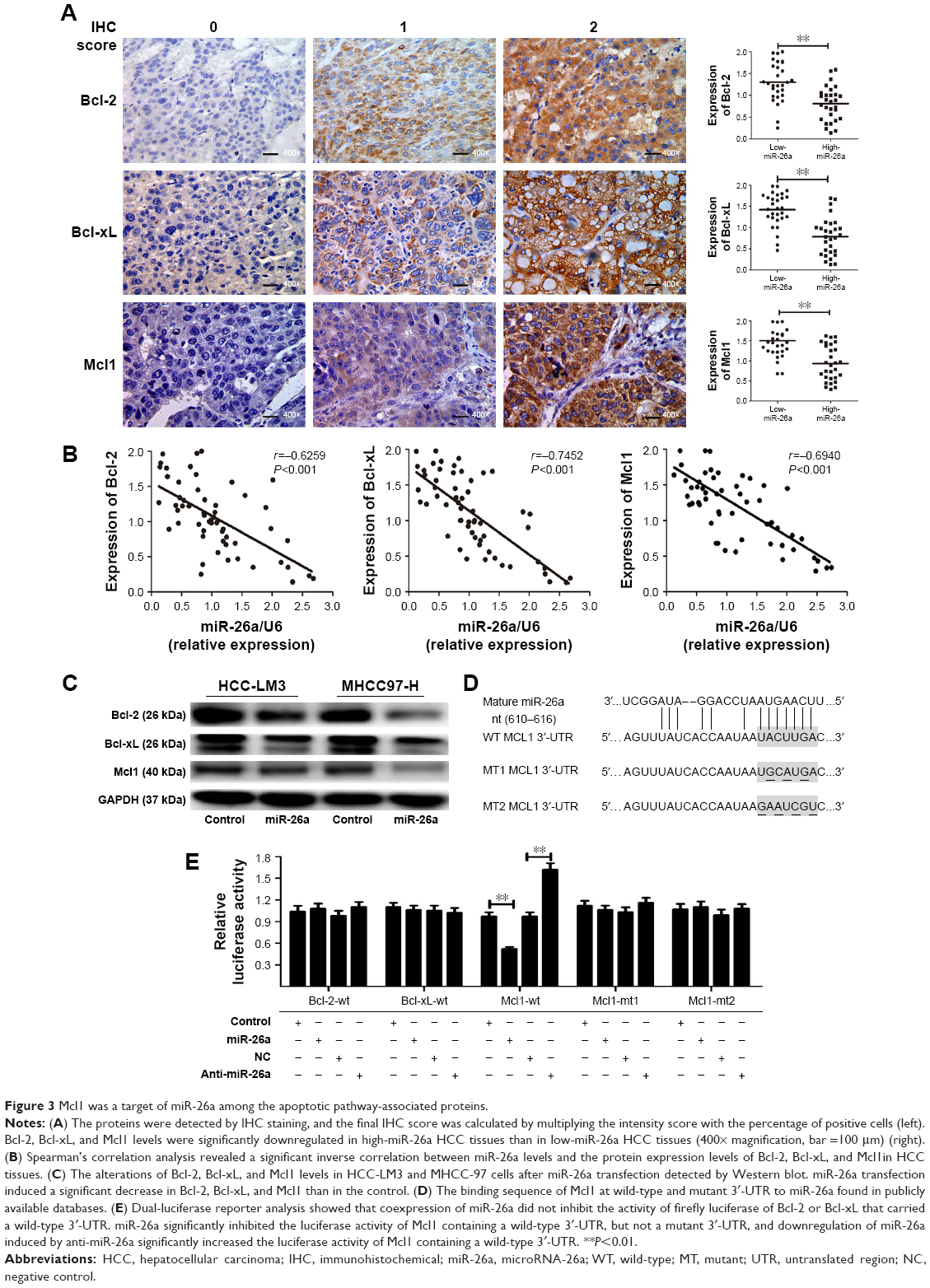 | Figure 3 Mcl1 was a target of miR-26a among the apoptotic pathway-associated proteins. |
To identify the potential target genes of miR-26a, Bcl-2, Bcl-xL, and Mcl1were selected for further validations. Among them, only the binding sequence of Mcl1 at wild-type 3′-UTR to miR-26a was found in publicly available databases including TargetScan, PicTar, and miRanda (Figure 3D), and Mcl1 might be the target gene of miR-26a. Dual-luciferase reporter analysis showed that coexpression of miR-26a significantly inhibited the luciferase activity of Mcl1 containing a wild-type 3′-UTR but did not suppress the activity of Mcl1 with a mutant 3′-UTR (P<0.01, Figure 3E). Also, downregulation of miR-26a induced by anti-miR-26a significantly increased the luciferase activity of Mcl1 containing a wild-type 3′-UTR (P<0.01, Figure 3E). However, miR-26a did not have a significant effect on the luciferase activity of Bcl-2 or Bcl-xL with a wild-type 3′-UTR. These indicate that miR-26a might inhibit Mcl1 expression through directly binding to its sequence at 3′-UTR in HCCs.
miR-26a increases cell apoptosis of HCC cells via targeting to inhibit Mcl1
To further confirm that Mcl1 was a functional target of miR-26a, we overexpressed Mcl1 in HCC-LM3 and MHCC97-H cells, which were miR-26a stable transfectants, and found that HCC-LM3 and MHCC97-H cells displayed obviously lower apoptotic rates than miR-26a transfectants (P<0.01) (Figures 4A and S3). Meanwhile, the proportion of cells with the loss of ΔΨm and the cytoplasmic cytochrome c levels significantly decreased in miR-26a-expressing HCC-LM3 and MHCC97-H cells after Mcl1 transfection than miR-26a transfectants (P<0.01, Figure 4B and C). These results strongly suggest that miR-26a might induce mitochondrial apoptosis of HCC by directly targeting Mcl1.
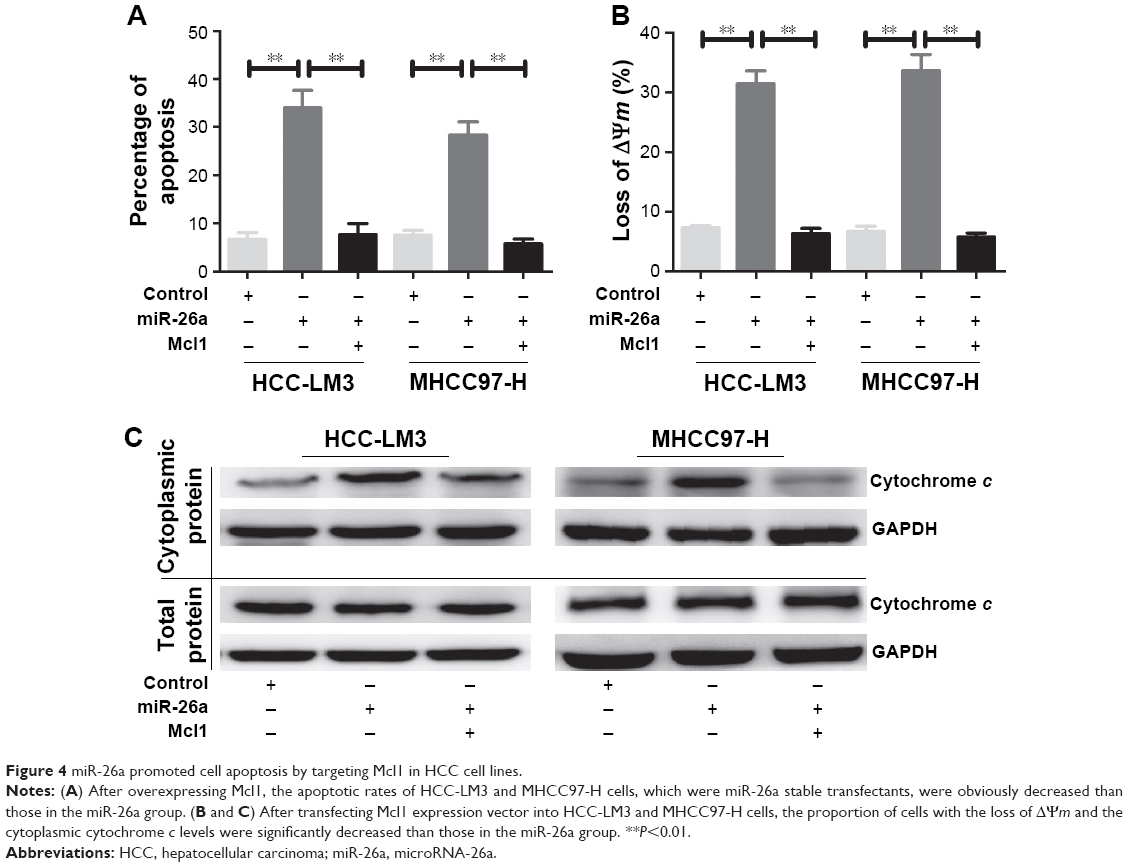 | Figure 4 miR-26a promoted cell apoptosis by targeting Mcl1 in HCC cell lines. |
p53 induced the mitochondrial apoptosis of HCC cells partly by activating miR-26a
We searched for candidate transcription factors of miR-26a promoter using the DECODE database and found that p53, a crucial apoptosis-inducing protein, could bind to the promoter region of miR-26a-1 rather than that of miR-26a-2 (Figure 5A).27 Moreover, p53 significantly activated the luciferase activity of miR-26a-1 containing a wild-type promoter sequence but did not increase the activity of miR-26a-1 with a mutant promoter region (P<0.01, Figure 5A). Moreover, overexpression of p53 (Figure S4) significantly upregulated the level of miR-26a in HCC-LM3 and MHCC97-H cells (P<0.01, Figure 5B), and a statistically significant positive correlation was revealed by Spearman’s correlation analysis between the mRNA levels of miR-26a and p53 level (r=0.7561; P<0.001, Figure 5C). And, this correlation was further confirmed by Fisher’s exact tests in HCC tissues (Table 1). These suggest that p53 plays an important role in activating the promoter activity and regulating the expression of miR-26a in HCC cells.
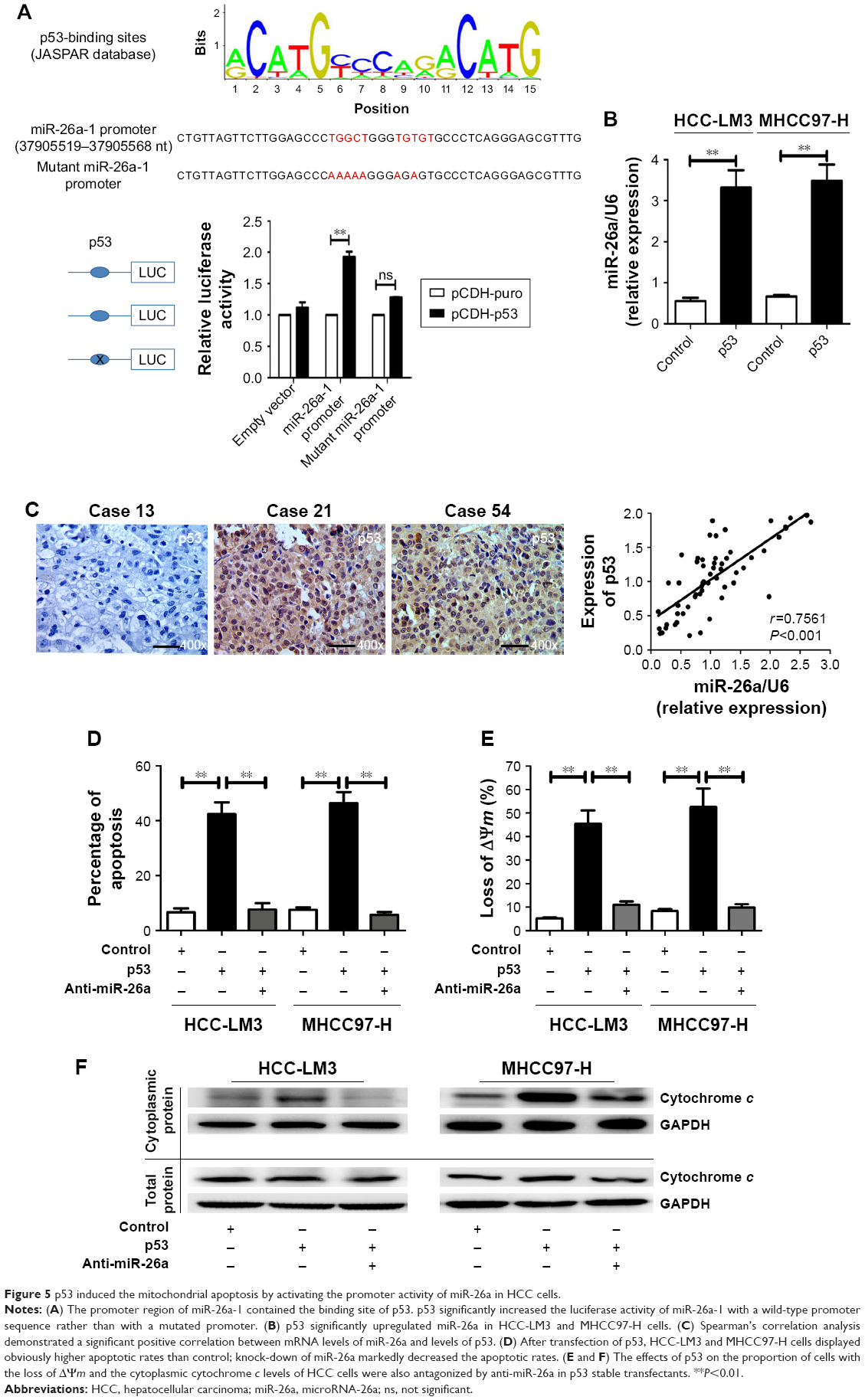 | Figure 5 p53 induced the mitochondrial apoptosis by activating the promoter activity of miR-26a in HCC cells. |
Next, we further explore whether p53 induces the mitochondrial apoptosis of HCC cells through activating miR-26a. As expected, transfection of p53 induced significantly higher apoptotic rates in HCC-LM3 and MHCC97-H cells than in the control (P<0.01, Figure 5D). However, when miR-26a was knocked down by anti-miR-26a, the apoptotic rate of the p53 stable transfecting HCC-LM3 and MHCC97-H cells (p53 transfectants) was markedly decreased (P<0.01, Figure 5D). Similarly, the effects of p53 on the proportion of cells with the loss of ΔΨm and the cytoplasmic cytochrome c levels of HCC cells were also antagonized by anti-miR-26a in p53 stable transfectants (P<0.01, Figures 5E and F and S3D). These provide further evidence to support that as an upstream functional mediator, p53 regulates the mitochondrial apoptosis of HCC cells at least in part by activating miR-26a.
Combination treatment of IFN-α1b and 5-FU induced cell apoptosis through upregulating miR-26a mediated by p53
The combination of IFN-α1b and 5-FU has been reported to be a useful treatment for patients with advanced HCCs.23–26 We next explored the HCC-LM3 and MHCC-97H cells with IFN-α1b in combination with 5-FU to explore whether their co-effects on cell apoptosis in HCC were induced by partly upregulating miR-26a mediated by p53 and provide clinical evidence for the combination of IFN-α1b and 5-FU in HCC.
After treated with the combination of IFN-α1b and 5-FU for 48 hours, an obviously higher ratio of apoptosis of both HCC-LM3 and MHCC-97H cells were detected compared with the control (P<0.01, Figure 6A). The proportion of HCC cells with the loss of ΔΨm and the levels of mitochondrial cytochrome c release in the IFN-α1b +5-FU group was also significantly higher than that in the control (Figures 6B and C and S3E). Similarly, in the nude mice models with subcutaneous implantation of HCC-LM3 cells, much smaller tumor volumes and obviously increased numbers of apoptotic HCC cells were found in the IFN-α1b +5-FU group than in the control (Figure 6D and E).
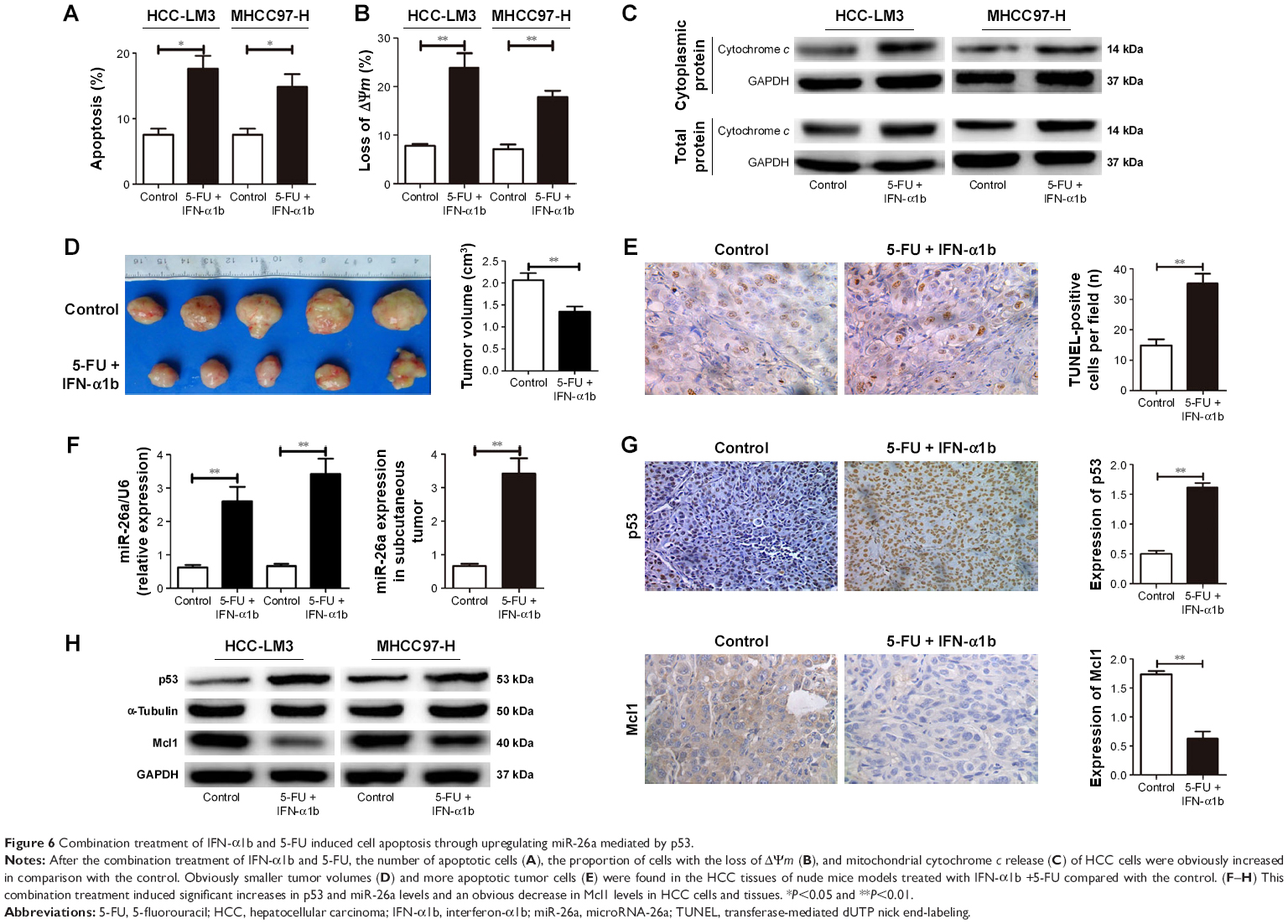 | Figure 6 Combination treatment of IFN-α1b and 5-FU induced cell apoptosis through upregulating miR-26a mediated by p53. |
Moreover, after the treatment with IFN-α1b +5-FU, significantly upregulated levels of p53 and miR-26a and obviously decreased Mcl1 level were detected in HCC cells of in vitro culture as well as tumor tissues of the subcutaneous implantation models (Figures 6F–H and S3E). Taking together, these results indicate that the combination treatment of IFN-α1b and 5-FU could promote HCC apoptosis, which, at least in part, is through inducing p53 expression, upregulating miR-26a levels, and thus downregulating Mcl1.
Discussion
Cancer cells evolve to evade apoptosis so that they can escape from the surveillance system, which is one of the important reasons of tumor progression.28,29 The induction and promotion of cancer cell apoptosis is one major strategy in cancer treatment. Therefore, exploring molecular targets involved in activating HCC cells’ apoptosis is helpful to develop new treatment strategies for HCC.
Many studies have demonstrated that miRNAs1,3,7–9 take part in the regulation of cancer cell apoptosis and may be used as therapeutic targets. miR-26a has been reported to regulate hepatocyte proliferation during liver regeneration,30 block G1/S transition,16,31 and suppress in vivo tumor growth.16,32 Restoration of miR-26a activity in HCC has been proposed as a novel treatment strategy.16 However, the molecular mechanisms by which miR-26a induces HCC apoptosis remain largely unclear.17 In this study, miR-26a was proved to involve in the mitochondrial apoptosis.
Many cancer therapies are prone to trigger the apoptosis through the intrinsic pathway, controlled by pro- and antiapoptotic Bcl-2 family proteins at the mitochondrion in tumor cells.29,33 Bcl-2, Bcl-xL, and Mcl1 maintain the integrity of the mitochondrial outer membrane and the endoplasmic reticulum/nuclear membrane.34 It has been reported that Bcl-2, Bcl-xL, and/or Mcl1 are upregulated, while Bax is downregulated in different types of cancers, which exerts an antiapoptotic function through the mitochondrial signaling pathway.35,36 During apoptosis, Bax and Bak form homo-oligomers within the mitochondrial membrane and probably breach its integrity, resulting in the release of cytochrome c.36 Caspase-9 is instead activated in the presence of ATP and cytochrome c by an allosteric change on a heptameric scaffold of apoptotic protease-activating factor 1 (Apaf1) proteins termed the apoptosome.37,38 Then, effector caspase-3 is activated by the initiator caspase-9.39 Ultimately, these effects promote mitochondrial apoptosis. In this article, we demonstrated that the upregulation of miR-26a decreased the expression levels of Bcl-2 and Bcl-xL, inhibited Mcl1 expression, and in turn promoted cytochrome c releasing from the mitochondria to the cytoplasm, which might increase the activities of mitochondrial apoptosis. These implicate the potential application of miR-26a in anticancer therapy.
Previous works have demonstrated that the p53-dependent augmentation of miR-16 and miR-26a expression levels is able to lead to the cell cycle arrest of G1/S and increase apoptosis of cancer cells.32,40 In the present study, based on the publicly available DECODE database, we found that the promoter region of miR-26a-1, but not miR-26a-2, contains the binding sequence to p53. And, p53 could activate the promoter activity to regulate the expression of miR-26a in HCC cells. More importantly, p53 is confirmed to induce the mitochondrial apoptosis of HCC cells at least in part by activating miR-26a. Therefore, these provide more clues to the functional mechanisms of p53 in HCC growth and progression.
IFN possesses a variety of biological properties including immunomodulation and antitumor activity as a useful adjuvant therapy to decrease the risk of tumor recurrence after curative resection of hepatitis B virus-related HCC.21 5-FU was conformed to induce cell apoptosis by p53-dependent way, and many studies suggest that 5-FU significantly enhanced the antitumor activity of IFN for HCC, and IFN-α1b in combination with 5-FU has been demonstrated to be an effective treatment for patients with advanced HCC.23–26 However, the detailed molecular mechanisms remain unknown. In the present study, using in vitro and in vivo studies, we have found that the combination treatments of IFN-α1b and 5-FU induced the expression of p53, which is able to upregulate miR-26a levels, and then downregulated Mcl1 to promote the apoptosis of HCC cells through a mitochondrial pathway. These are helpful for us to understand the underlying mechanism of the combination treatment of IFN-α1b and 5-FU and use more likely effective strategy for combating HCC.
Conclusion
Our results suggest that miR-26a promotes HCC apoptosis through a mitochondrial pathway by targeting the inhibition of Mcl1 and IFN-α1b in combination with 5-FU significantly suppressed tumor growth and promoted cells’ apoptosis through upregulating p53 and then inducing miR-26a expression. These provide more clues to understand the functional mechanisms of p53 and miR-26a in HCC growth and progression and to provide more molecular evidence for understanding of the underlying mechanism of the combination treatment of IFN-α1b and 5-FU, which is more likely an effective strategy for combating HCC.
Acknowledgments
This work was generously sponsored by Shanghai Sailing Program (15YF1401600) and China National Natural Science Foundation (81672365).
Disclosure
The authors report no conflicts of interest in this work.
References
Xiong Y, Fang JH, Yun JP, et al. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51(3):836–845. | ||
Fornari F, Gramantieri L, Ferracin M, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–5661. | ||
Su H, Yang JR, Xu T, et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69(3):1135–1142. | ||
Fang JH, Zhou HC, Zeng C, et al. MicroRNA-29b suppresses tumor angiogenesis, invasion, and metastasis by regulating matrix metalloproteinase 2 expression. Hepatology. 2011;54(5):1729–1740. | ||
Budhu A, Jia HL, Forgues M, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47(3):897–907. | ||
Aigner A. MicroRNAs (miRNAs) in cancer invasion and metastasis: therapeutic approaches based on metastasis-related miRNAs. J Mol Med (Berl). 2011;89(5):445–457. | ||
Zeng CW, Zhang XJ, Lin KY, et al. Camptothecin induces apoptosis in cancer cells via microRNA-125b-mediated mitochondrial pathways. Mol Pharmacol. 2012;81(4):578–586. | ||
Zhu W, Shan X, Wang T, Shu Y, Liu P. miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer. 2010;127(11):2520–2529. | ||
Nie J, Liu L, Zheng W, et al. microRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis. 2012;33(1):220–225. | ||
Lu J, He ML, Wang L, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71(1):225–233. | ||
Visone R, Pallante P, Vecchione A, et al. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene. 2007;26(54):7590–7595. | ||
Ciarapica R, Russo G, Verginelli F, et al. Deregulated expression of miR-26a and Ezh2 in rhabdomyosarcoma. Cell Cycle. 2009;8(1):172–175. | ||
Zhang B, Liu XX, He JR, et al. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32(1):2–9. | ||
Salvatori B, Iosue I, Mangiavacchi A, et al. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis. 2012;3:e413. | ||
Sander S, Bullinger L, Klapproth K, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–4212. | ||
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. | ||
Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361(15):1437–1447. | ||
Yang X, Liang L, Zhang XF, et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting IL-6-Stat3 pathway. Hepatology. 2013;58(1):158–170. | ||
Mazzaferro V, Romito R, Schiavo M, et al. Prevention of hepatocellular carcinoma recurrence with alpha-interferon after liver resection in HCV cirrhosis. Hepatology. 2006;44(6):1543–1554. | ||
Llovet JM, Sala M, Castells L, et al. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology. 2000;31(1):54–58. | ||
Someya T, Ikeda K, Saitoh S, et al. Interferon lowers tumor recurrence rate after surgical resection or ablation of hepatocellular carcinoma: a pilot study of patients with hepatitis B virus-related cirrhosis. J Gastroenterol. 2006;41(12):1206–1213. | ||
Sun HC, Tang ZY, Wang L, et al. Postoperative interferon alpha treatment postponed recurrence and improved overall survival in patients after curative resection of HBV-related hepatocellular carcinoma: a randomized clinical trial. J Cancer Res Clin Oncol. 2006;132(7):458–465. | ||
Sakon M, Nagano H, Dono K, et al. Combined intraarterial 5-fluorouracil and subcutaneous interferon-alpha therapy for advanced hepatocellular carcinoma with tumor thrombi in the major portal branches. Cancer. 2002;94(2):435–442. | ||
Ota H, Nagano H, Sakon M, et al. Treatment of hepatocellular carcinoma with major portal vein thrombosis by combined therapy with subcutaneous interferon-alpha and intra-arterial 5-fluorouracil; role of type 1 interferon receptor expression. Br J Cancer. 2005;93(5):557–564. | ||
Nagano H, Wada H, Kobayashi S, et al. Long-term outcome of combined interferon-alpha and 5-fluorouracil treatment for advanced hepatocellular carcinoma with major portal vein thrombosis. Oncology. 2011;80(1–2):63–69. | ||
Obi S, Yoshida H, Toune R, et al. Combination therapy of intraarterial 5-fluorouracil and systemic interferon-alpha for advanced hepatocellular carcinoma with portal venous invasion. Cancer. 2006;106(9):1990–1997. | ||
Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7(12):979–987. | ||
Fulda S .Regulation of apoptosis pathways in cancer stem cells. Cancer Lett. 2013;338(1):168–173. | ||
Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. | ||
Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. | ||
Zhou J, Ju W, Wang D, et al. Down-regulation of microRNA-26a promotes mouse hepatocyte proliferation during liver regeneration. PLoS One. 2012;7(4):e33577. | ||
Zhu Y, Lu Y, Zhang Q, et al. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res. 2012;40(10):4615–4625. | ||
Chen L, Zheng J, Zhang Y, et al. Tumor-specific expression of microRNA-26a suppresses human hepatocellular carcinoma growth via cyclin-dependent and -independent pathways. Mol Ther. 2011;19(8):1521–1528. | ||
Karst AM, Li G. BH3-only proteins in tumorigenesis and malignant melanoma. Cell Mol Life Sci. 2007;64(3):318–330. | ||
Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9(7):501–507. | ||
Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27(50):6398–6406. | ||
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. | ||
Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13(24):3179–3184. | ||
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9(2):423–432. | ||
Lezina L, Purmessur N, Antonov AV, et al. miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress. Cell Death Dis. 2013;4:e953. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.