")
Back to Journals » OncoTargets and Therapy » Volume 13
MicroRNA-16 Inhibits Glioblastoma Growth in Orthotopic Model by Targeting Cyclin D1 and WIP1
Authors Wang H, Pan J, Yu L, Meng L, Liu Y, Chen X
Received 18 February 2020
Accepted for publication 11 September 2020
Published 23 October 2020 Volume 2020:13 Pages 10807—10816
DOI https://doi.org/10.2147/OTT.S250369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Heng Wang,1,* Jun Pan,2,* Lisheng Yu,3 Linghu Meng,4 Yue Liu,4 Xin Chen4
1Department of Gastrointestinal Surgery/Pediatric Surgery, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei 442000, People’s Republic of China; 2Department of Traditional Chinese Medicine, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei 442000, People’s Republic of China; 3Department of Neurosurgery, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325000, People’s Republic of China; 4Department of Neurosurgery, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei 442000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xin Chen
Department of Neurosurgery, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei 442000, People’s Republic of China
Tel +86-13872821952
Email [email protected]
Introduction: To examine the molecular mechanism by which miRNA-16 (miR-16) suppresses glioblastoma in vitro and in vivo.
Methods: Gene expression of miR-16 in normal brain tissues and human glioma cell lines was examined. To characterize the functional role of miR-16 in vitro, miR-16 was ectopically expressed in U87 cells by lentiviral transduction. Expression of miR-16 downstream targets cyclin D1 and Bcl-2 in U87 was studied using Western blotting. Cell proliferation and clonogenic property were examined using CCK-8 and clone formation assay, respectively. Migration and invasiveness of U87 was studied using wound-healing assay and transwell assay, respectively. In vivo tumorigenic properties of the miR-16-transduced U87 cells were examined in an orthotopic xenograft model. Immunohistochemistry was performed to examine cyclin D1, WIP1 and CD31 expressions.
Results: Expression of miR-16 was reduced in glioblastoma cell lines compared to normal human brain tissues. Ectopic miR-16 expression reduced cyclin D1 and Bcl-2 in U87 cells. miR-16 also induced apoptosis, reduced cell proliferation and clone formation. Furthermore, miR-16 suppressed U87 migration in wound-healing assay and invasion across transwell membrane in vitro. In an orthotopic tumor model, overexpression of miR-16 inhibited tumor growth in vivo was accompanied with reduction in cyclin D1 and WIP1 expression in the xenografts. CD31 expression in miR-16-overexpressed xenografts was also decreased. The determined microvessel density of the miR-16 overexpression group was significantly lower than those groups treated with vehicle and empty vector.
Discussion: MicroRNA-16 exhibits inhibitory effects of glioblastoma. MicroRNA-16 and its downstream targets could be potential therapeutic targets for treatment of glioblastoma.
Keywords: microRNA-16, glioblastoma, cyclin D1, WIP1, angiogenesis, apoptosis, invasion
Introduction
Glioblastoma is one of the most common primary malignant brain tumors in adults. The overall age-adjusted incident rate ranges from 4.67–5.73 per 100,000 persons.1 Surgical resection remains the main therapeutic option for patients with glioblastoma, although the procedure is challenging. In many of the cases, the tumor margins are hardly defined because of the invasive nature of the tumors. Other therapies such as radiation, chemotherapy, and immunotherapy are also used to treat patients.2,3 Nevertheless, the prognosis of glioblastoma is still poor, and the recurrence rate is high.4 The median survival time of patients was approximately 14 months.5 Given the limited success of the standard treatment in prolonging survival in patients with glioblastoma, new therapeutic strategies are needed.
MicroRNAs (miRNAs) are small (~22 nt) single-stranded RNAs that play an important role in post-transcriptional gene regulation. MicroRNAs usually bind to the 3ʹUTR (untranslated region) of the target mRNAs, leading to translational repression. It is predicted that more than one-third of human genes are regulated by miRNAs.6 The expressions of miRNAs at normal levels are essential to maintain normal cellular functions. Many of the studies have implicated the relationship between deregulated miRNAs and cancers.7 In more than 50% of chronic lymphocytic leukemia cases, miR-15 and miR-16 are deleted or downregulated.8 miR-21 was the first oncogenic miRNA identified that can target multiple tumor suppressor genes, such as PTEN.9
Reports have shown that miR-16 is frequently deleted or downregulated in cancer, including chronic lymphocytic leukemia,8 prostate cancer,10 and lung cancer.11 Therefore, miR-16 is considered a key tumor-suppressive miRNA. In glioblastoma, the role of miR-16 remains largely unknown. To better understand the functions of miR-16 in human glioblastoma, the present study investigated the expression of miR-16 and its target genes in glioblastoma cell lines. The effects of miR-16 on cell cycle, apoptosis, cell migration, and invasiveness were also evaluated in vitro and in vivo.
Materials and Methods
Cell Lines, Human Tissue Samples and Reagents
Human glioblastoma cell lines A172, LN229, U87, U251, U343 and U373 were obtained from the cell bank of the Chinese Science Academy, and weres maintained in DMEM supplemented with 10% FBS. Normal human brain tissue was collected from patients who received third ventricular ostomy in our hospital. The use of human tissue in the present study has been reviewed and approved by the Ethics Committee of Renmin Hospital, Hubei University of Medicine, with written informed consent obtained from the patients. Lentiviruses expressing GFP and GFP with miR-16 were constructed by Shanghai Genechem (Shanghai, China). Rabbit anti-human cyclin D1, Bcl-2, WIPI, CD31 and beta-actin polyclonal antibodies were purchased from Santa Cruz (Santa Cruz, USA).
Ectopic Expression of miR-16 in U87
U87 cells were divided into three treatment groups. Normal control (CON) received no treatment, while negative control (NC) and overexpression group (OE) were transduced with lentivirus expressing GFP and miR-16-GFP. Cells were seeded into six-well plates at a density of 5×104/well, and were transduced with respective lentiviruses at an MOI of two. The transduction efficacy was determined by counting the number of GFP-positive cells 72 h after the transduction. All transductions in the present study achieved an efficacy >80%.
Intracranial Orthotopic Glioma Model
Nude mice (n=18) were divided into CON (n=6), NC (n=6), and OE (n=6) groups. To establish the orthotopic model, nude mice were first anesthetized by intraperitoneal injection of chloral hydrate (10% v/v), fixed on a stereotactic instrument, and with their scalp sterilized. After the skull was exposed, a hole 2.5 mm right to rhabdoid suture and 1 mm front to coronal suture was made. Respective U87 cells (ie CON, NC, or OE) were slowly injected in a volume of 10 µL. The scalp was sutured and disinfected after surgery. Body weights of the mice were measured twice a week. All mice were sacrificed for collection of brains three weeks after U87 injection. Tumor volume was determined by caliper measurement using the formula V=(W(2)×L)/2, where V, W, and L were the volume, width, and length of the tumor, respectively. After measurement, brain tissues were formalin-fixed and paraffin-embedded for analysis. The use of animals, the study design and the experimental procedures involving animals have been reviewed and approved by the Ethics Committee of Hubei University of Medicine before the study. Procedures involving animals and their care were conducted in conformity with NIH guidelines (NIH Pub. No. 85–23, revised 1996) and was approved by Animal Care and Use Committee of Hubei University of Medicine. Our study covers the 3Rs (refinement, replacement, and reduction) and also outlines the procedures dealing with humane endpoints and pain management.
Histology and Immunohistochemistry
Tissue sections with a thickness of 4 µm were prepared for morphological examination using H&E staining and immunohistochemistry (IHC). To perform IHC, after antigen retrieval, tissue sections were incubated with primary antibodies against cyclin D1 (1:100), WIP1 (1:100) or CD31 (1:100) overnight at 4°C. Reactivity signals were developed using HRP-conjugate and DAB.
Microvessel Density (MVD) Determination
CD31 signals were employed to determine the microvessel density within the orthotopic xenograft using the Weidner method. In this method, any single endothelial cell or cell mass stained by antibodies, regardless of whether a lumen is formed, as long as there is a clear boundary between surrounding microvessels, tumor cells, and other connective tissues, can be considered as a countable microvessel. Any microvessels within the tumor sclerosis and soft tissues in tumor junction were not counted. Blood vessels with smooth muscle walls and lumen diameters >8 red blood cell diameters were also excluded. The number of microvessels were counted from 10 microscopic fields with 200× magnification.
DAPI Staining
U87 cells were seeded into 24-well plates, and were fixed with 4% paraformaldehyde when their confluency reached 80%. DAPI staining was done in the dark for 10 min. Apoptotic nucleus in each high-power field were counted using a fluorescence microscope.
Annexin V-APC Flow Cytometry
PBS-washed U87 cells were stained with 5 µL annexin V-APC (1×105–1×106 cells/staining) in the dark for 15 min. The percentage of annexin V-APC-positive cells were subsequently analyzed using flow cytometry.
Wound-Healing and Transwell Assays
U87 cells were grown into a confluency of 80% in 24-well plates. After wounds were made, the closure of the wounds was observed at 0, 6, 12, and 24 h. To perform transwell assay, diluted matrigel was added to the upper chamber of the transwell (Corning, USA), where 1×105 U87 cells were seeded without FBS. The lower chamber of the transwell was filled with DMEM medium supplemented with 10% FBS. After incubation for 24 h, the cells at the bottom of the membrane were methanol-fixed and were then stained with 0.1% crystal violet for 30 min. Stained cells were counted under 200× magnification. An average number was taken from five random microscopic fields.
Cell Proliferation and Clone Formation Assays
Proliferation of U87 cells was determined using CCK-8 assay, which was performed at 0, 24, 48, 72, and 96 h after the seeding of the cells into 96-well plates. To examine the clone formation, U87 cells were plated into six-well plates at a density of 300–400 cells/well for culture in 3 mL complete medium for 14 days. The resulting clones, which consisted of at least 50 cells within, were methanol-fixed and then stained with 0.1% crystal violet for 30 min. Stained clones were counted from five random microscopic fields.
Real-time PCR
Total RNA was extracted and transcribed. Gene amplification was performed using SYBR Premix Ex TaqTM (Takara, Japan). The PCR cycling consisted of an initial activation step at 95°C for 30 seconds, and 40 cycles of 95°C for five seconds and 60°C for 30 seconds. Housekeeping gene U6 was included as the internal control. Gene expression was calculated using 2−∆∆Ct approach. The oligonucleotide for miR-16 amplification was synthesized by Guangzhou RiboBio (Guangzhou, China).
Western Blotting
Whole-cell lysate of U87 cells was resolved on SDS-PAGE, transferred on PVDF, and finally probed with primary antibody against cyclin D1 (1:500 dilution) and Bcl-2 (1:500 dilution). After incubation with HRP-conjugate, signal was developed using ECL reagent. Beta-actin was included as the internal reference.
Statistical Analysis
All data were analyzed using statistical software SPSS version 20.0. One way ANOVA and t-test were used to compare data between groups. Significant difference was indicated by P-value <0.05.
Results
Expression of miR-16 in Normal Human Brain Tissues and Glioblastoma Cell Lines
Real-time PCR was employed to examine miR-16 expression in normal human brain tissue and multiple glioblastoma cell lines (Figure 1). Results clearly showed that miR-16 was substantially suppressed in six glioblastoma cell lines when compared to normal brain tissues. The differences between normal brain tissues and the cell lines were statistically significant (P<0.05). Among the cell lines, U87 cells expressed the lowest miR-16 level and therefore was selected for the functional characterization study of miR-16.
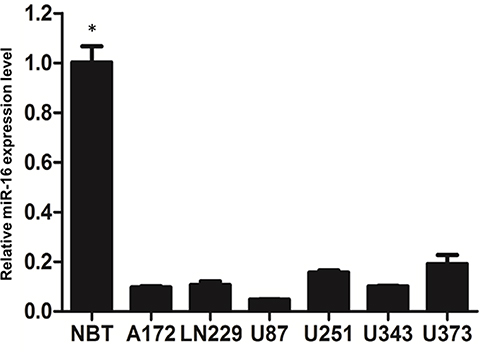 |
Figure 1 Expression of miR-16 in normal brain tissues and glioblastoma cell lines was assessed using real-time PCR. Data are represented as expression levels relative to that of normal brain tissues (NBT). *P<0.05 comparing to different glioblastoma cell lines. Shown are the representative data set of three independent experiments. |
Ectopic Expression of miR-16 Modulated Cyclin D1 and Bcl-2 in U87
To characterize the functions of miR-16 in U87, the cells were transduced with lentiviruses expressing either GFP plasmid alone or GFP plasmid containing miR-16. Seventy-two hours after the transduction, expression of GFP was observed under fluorescence microscope, showing the transduction efficiencies of NC and OE groups were greater than 90% (Figure 2A). The expression of miR-16 in the OE group was validated using real-time PCR (Figure 2B). miR-16 was expressed in the OE group but not in CON or NC groups. The upregulation of miR-16 in OE group when compared to CON and NC groups was significant (P<0.05).
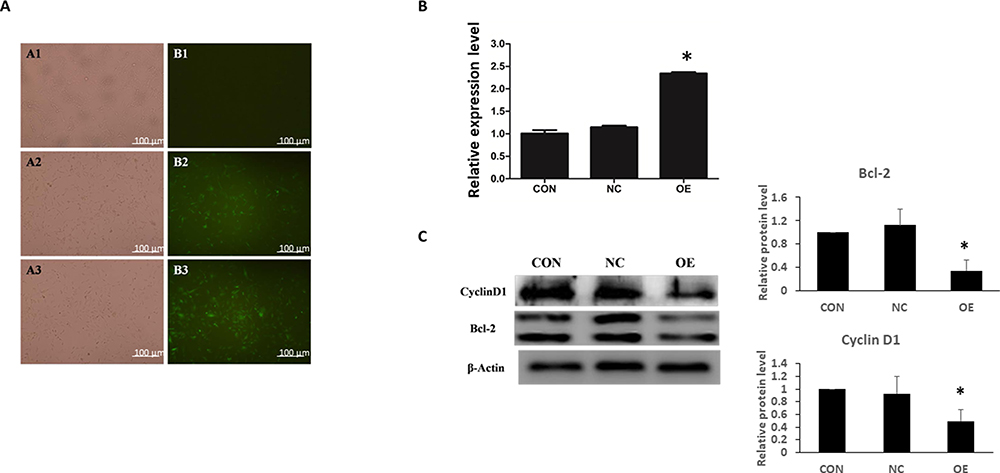 |
Figure 2 Ectopic expression of miR-16 in U87 cells by lentiviral transduction. U87 cells were treated with transduction reagent alone (CON), lentivirus with GFP plasmid (NC), and lentivirus with GFP-miR16 (OE). (A) At 72 h after transduction, the expression of GFP was monitored under fluorescence microscope to determine the transduction efficiency. (B) Six days after the transduction the expression level of miR-16 in different groups was assessed using real-time PCR. *P<0.05 compared to CON and NC groups. (C) Expression of cyclin D1 and Bcl-2 in U87 cells was examined using Western blotting. The expression was quantified and compared between groups. *P<0.05 compared to CON and NC groups. Shown are the representative data sets of three independent experiments. |
Molecular targets modulated by miR-16 were also examined (Figure 2C). Western blotting revealed that overexpression of miR-16 suppressed Bcl-2 and cyclin D1 levels in U87 cell line. When compared to CON and NC groups, the reduction in OE group was statistically significant (P<0.05).
miR-16 Induction of Apoptosis in U87
The induction of apoptosis in U87 by miR-16 was studied using DAPI and annexin V-APC staining (Figure 3). In the CON and NC groups, U87 cells demonstrated intact nuclear morphology with bright blue fluorescence signal but in the OE group, U87 exhibited typical apoptotic morphological changes (Figure 3A). The number of abnormal nuclei was quantified. The proportion of atypical nuclei was significantly higher in the OE group than in the CON and NC groups (P<0.05). The induction of apoptosis in U87 cells by miR-16 was further validated using annexin V-APC staining (Figure 3B). Results indicated that the percentage of annexin V-APC-positive cells was significantly increased in the OE group when compared to the CON and NC groups (P<0.05). miR-16 overexpression (ie OE) resulted in 8% apoptotic cells, but treatment with mock transfection (ie CON) and empty vector (ie NC) could lead to about 2% and 5% apoptotic cells, respectively.
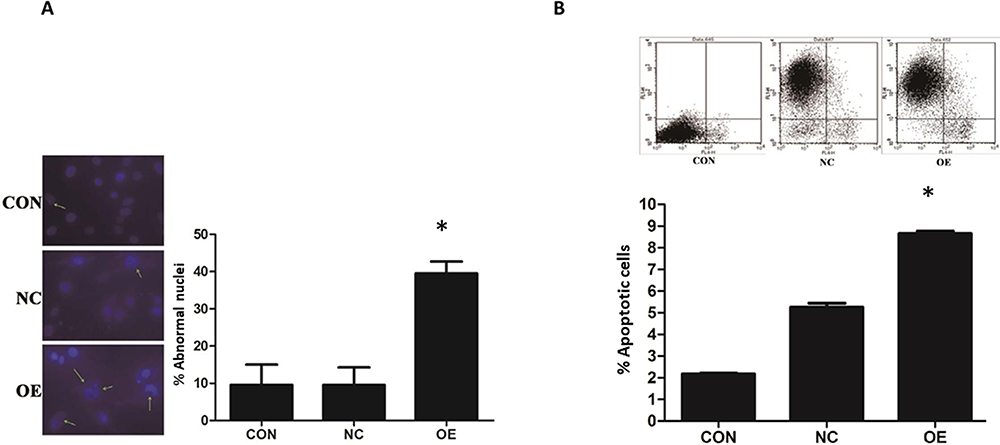 |
Figure 3 MiR-16 induction of apoptosis in U87 cells. (A) DAPI staining was utilized to observe the heterotypic cell nucleus and morphological changes of apoptosis. The atypical nuclei are arrowed. The percentage of abnormal nuclei in each group was quantified and compared. *P<0.05 comparing to CON and NC groups. (B) Annexin V-APC staining was used to validate the induction of apoptosis in U87 by miR-16. The positively stained cells were quantified using flow cytometry to calculate the percentage of apoptotic cells. *P<0.05 compared to CON and NC groups. Shown are the representative data sets of three independent experiments. |
Suppressive Effect of miR-16 on U87 Migration and Invasion
Whether miR-16 would affect the migration and invasion of U87 cells were examined using wound-healing and transwell assays, respectively (Figure 4). In wound-healing assay, the closure of the wound in different groups was monitored at 6, 12, and 24 h after the start of the assay. Results showed that in the CON and NC groups, the wounds started to close at 12 hand were completed closed at 24 h (ie 100% closure) (Figure 4A). In the OE group, the healing was suppressed, resulting in a closure of about 70%. The reduction in wound closure in OE, when compared with the CON and NC groups, was statistically significant (P<0.05). In the transwell assay, obvious transmigration across the chamber membrane was noted in the CON and NC groups, however, the process was significantly reduced in the OE group (Figure 4B). The number of migrated cells in CON, NC, and OE groups was 120.44±16.62, 113.67±24.99, and 37.6±5.18, respectively. The decrease in migrated cells in the OE group when compared to the CON and NC groups was statistically significant (P<0.05).
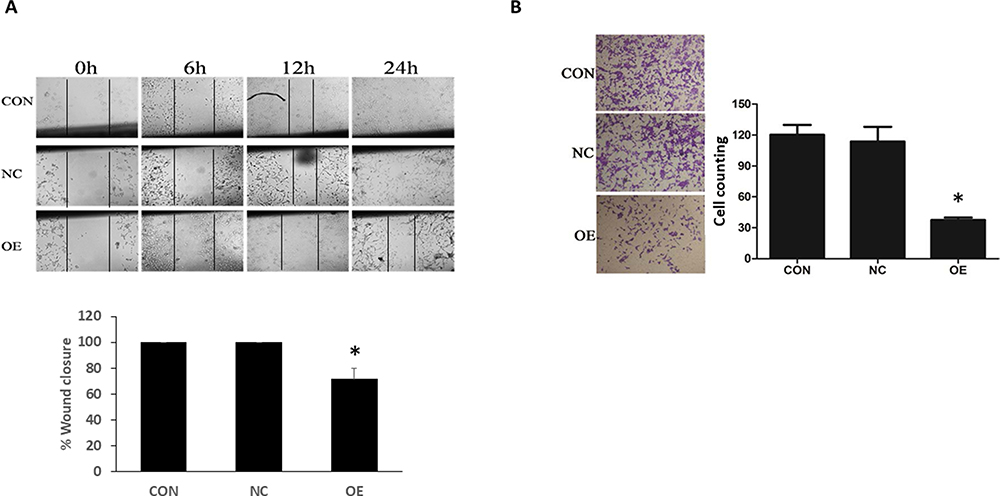 |
Figure 4 Inhibition of U87 migration and invasion by miR-16. (A) The migration ability of U87 cells was examined using wound-healing assay at different time points: 0, 6, 12, and 24 h from the start of experiment. The healed wound at 24 h was quantified as percentage of wound closure. *P<0.05 compared to CON and NC groups. (B) Transwell assay was done to examine the invasion ability of U87 cells. The transmigrated cells were counted and compared between groups. *P<0.05 compared to CON and NC groups. Shown are the representative data sets of three independent experiments. |
Inhibitory Effect of miR-16 on U87 Cell Proliferation and Clone Formation
The effect of miR-16 on clone formation and cell proliferation was also investigated (Figure 5). It was illustrated that in the CON and NC groups, there were more than 100 clones formed but in the OE group, the number of clones formed was below 50 (Figure 5A and B). Whether miR-16 would affect the proliferation of U87 cells was studied using CCK-8 assay (Figure 5C). U87 cells of the CON and NC groups proliferated more than five folds over four days, however, those of the OE group could proliferate only by two folds. The reduction in proliferation rate in the OE group was statistically significant (P<0.05).
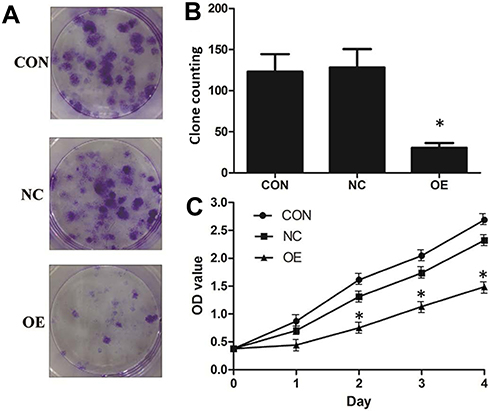 |
Figure 5 Suppression of cell proliferation and clonogenicity of U87 cells by miR-16. (A) Clone formation was allowed for 14 days after lentiviral transduction. Formed clones were stained by crystal violet. (B) Formed clones were counted and compared between groups. *P<0.05 compared to CON and NC groups. (C) Proliferation of U87 after lentiviral transduction was monitored over four days using CCK-8 assay. *P<0.05 compared to CON and NC groups. Shown are the representative data sets of three independent experiments. |
miR-16 Suppressed in vivo Orthotopic Xenograft Growth
The effect of miR-16 on in vivo tumor growth was examined in an intracranial orthotopic glioma xenograft model, in which the formed tumor would protrude on the surface of the brain, shifting significantly the midline of the brain (Figure 6A). In the present study, H&E staining showed that tumors were formed in the basal ganglia directly under the puncture hole. Most of the gliomas in the OE group were small tumors restricted to the injection site, while those in the NC and CON groups were large tumors compressing brain ventricles (Figure 6B). In some NC and CON mice, the ventricles even disappeared with the midline significantly shifted. In gliomas, the nuclei were heteromorphic and strongly stained. More dividing nuclei were also seen. Notably, more new blood vessels were observed in the stroma of the tumors.
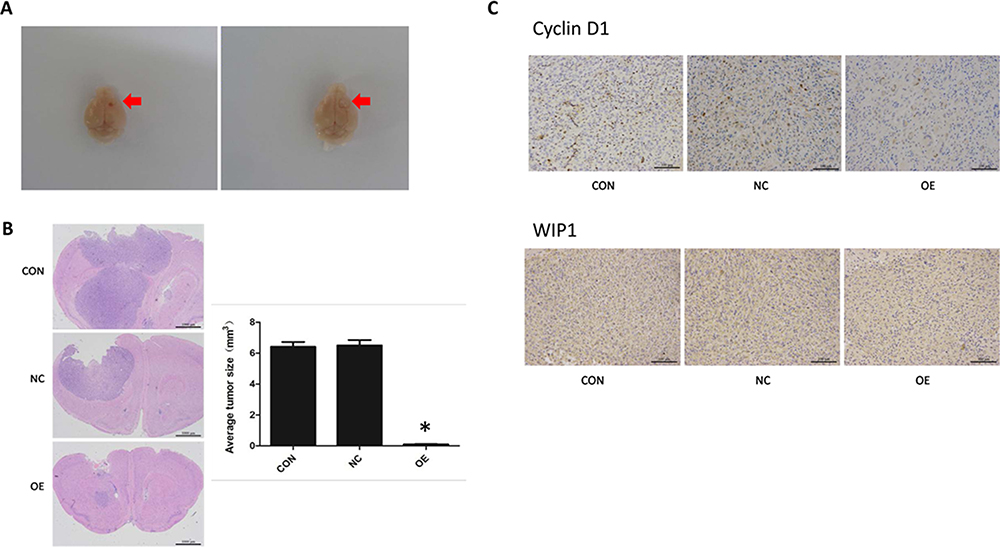 |
Figure 6 Inhibition of orthotopic tumor growth by miR-16. (A) Formation of orthotopic glioblastoma in nude mouse brains. The site of tumor formation is arrowed. (B) Histology of brain tumors was examined using H&E staining. The size of the tumors was measured and compared. *P<0.05 comparing to CON and NC groups. (C) Immunohistochemistry was done to assess cyclin D1 and WIP1 expression in orthotopic tumors. Magnification 200×. Shown are the representative data sets of three independent experiments. |
The volumes of orthotopic xenograft formed in different groups were measured. The volume of the OE group was substantially reduced (P<0.05) compared to the CON and NC groups (CON, 6.413±0.910 mm3; NC, 6.500±1.408 mm3; OE, 0.106±0.044 mm3).
Suppressive Effect of miR-16 on Cyclin D1 and WIP1 in Orthotopic Xenograft
Cyclin D1 and WIP1 expression in the xenograft was examined using IHC (Figure 6C). Reactivity towards cyclin D1 was mainly localized in the nucleus. Positive staining was seen in the CON and NC groups but in the OE group the positivity was significantly lower. Likewise, the expression of WIP1 was found restricted in the nucleus and was upregulated in the CON and NC groups.
Reduction in Tumor Angiogenesis by miR-16
CD31 is expressed in the endothelial cells of blood vessels in normal and tumor tissues. To examine the effect of miR-16 on new blood vessel formation in glioblastoma, CD31 expression in the orthotopic xenograft was examined using IHC (Figure 7). It was demonstrated that the reactivity towards CD31 was reduced in the OE group compared to CON and NC groups. The MVD of each group was determined based on the CD31 signal. The MVD of the OE group was obviously reduced (P<0.05) compared to the CON and NC groups (OE, 11.20±1.47; CON, 99.40±2.48; NC, 95.30±3.09).
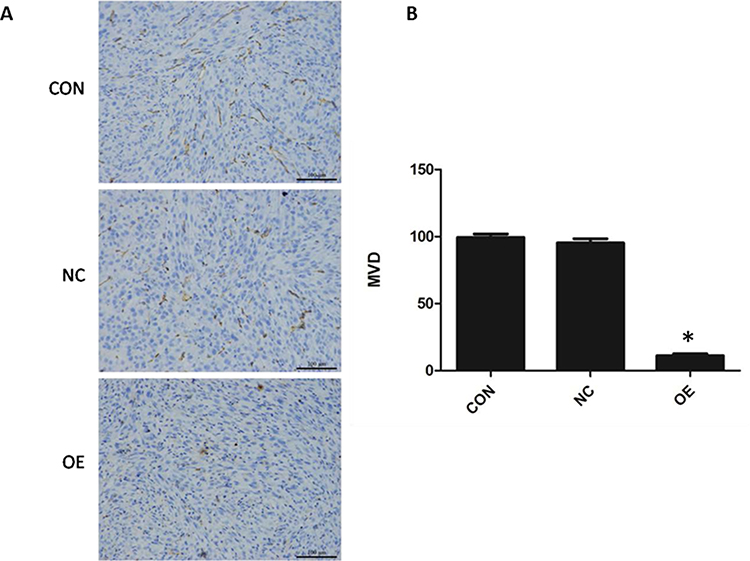 |
Figure 7 Suppression of tumor angiogenesis by miR-16. (A) Expression of endothelial marker CD31 was evaluated using immunohistochemistry. Magnification 200×. (B) Based on the signal density, the microvessel density (MVD) was calculated and compared between groups. *P<0.05 compared to CON and NC groups. Shown are the representative data sets of three independent experiments. |
Discussion
The present study characterized the functional role of miR-16 in glioblastoma in vitro and in vivo. Expression of miR-16 was shown downregulated in glioblastoma cells compared to normal brain tissue. Results from in vitro assays showed that miR-16 could promote apoptosis, reducing tumor cell invasiveness and suppressing cancer cell proliferation and clonogenicity. The orthotopic model suggested that miR-16 could suppress tumor growth and reduce blood vessel formation within tumors. All of these findings collectively highlighted the importance of miR-16 in suppressing glioblastoma. The potential targets of miR-16 were first identified by an in silico prediction using different databases including, TargetScan, miRbase, and miRDB. Among the identified, cyclin D1, Bcl-2, and WIP1 were confirmed as the downstream targets of miR-16 in this study. miR-16 and its downstream targets likely hold promise as therapeutic targets for treatment of glioblastoma and other cancers.
The aggressive growth of malignant tumors including glioblastoma is partially attributed to the dysregulation of cell cycle. In normal cells cell cycle is tightly regulated involving different mediators including cyclin D1. Cyclin D1 is encoded by the CCND1 gene in humans, and its main function is to promote the entry of G1 from G0 phase. Aberrant activation of cyclin D1/CDK4/CDK6 is reported to deregulate the normal cell cycle control and in turn leads to tumor formation.12 CCND1 gene amplification has been reported in 15–40% of breast cancer, lung cancer, melanoma, and oral squamous cell carcinoma.13 Notably, in patients with malignant tumors, overexpression of cyclin D1 is associated with shorter survival and increased risk of metastasis.14 Deficiency of cyclin D1 could prevent spontaneous cancer development in mice.15 Recent studies have identified miR-16 as one of the important modulators on cyclin D1/CDK4/CDK6. Goretti et al reported that ectopic expression of miR-16 precursor could significantly downregulate CCND1 in endothelial progenitor cells.16 In prostate and lung cancers the expression of miR-16 and cyclin D1 negatively correlated with each other.10 MiR-16 could reduce CDK6 expression in leukemia.17 Despite these important findings, whether and how miR-16 would modulate CCND1 in glioblastoma has remained unclear. The present study indicated ectopic miR-16 expression could suppress cyclin D1 expression in vitro and in vivo. Suppression of CCND1 by miR-16 inhibited orthotopic tumor growth in mice, probably due to the blockade of cell cycle at G0 phase.
In addition to cyclin D1, the present study suggested that miR-16 would affect WIP1 in glioblastoma. WIP1 (wild-type p53-induced phosphatase 1, or named as protein phosphatase 1D), encoded by PPMID,18 is a serine/threonine phosphatase that is implicated as a novel oncogene in multiple cancers including neuroblastoma, beast and ovarian cancers.19–21 WIP1 was reported to promote malignant progression, and its inactivation was shown to inhibit mammary tumorigenesis.22 Like cyclin D1, the role of WIP1 in glioblastoma has remained to be fully elucidated. In this study, overexpression of miR-16 reduced significantly WIP1 expression in orthotopic tumors. The reduction of WIP1 expression was accompanied by inhibited in vivo tumor growth. Taken together, dysregulated miR-16-WIP1 axis would contribute to the malignant growth of glioblastoma. Given cyclin D1 and WIP1 were both modulated by miR-16, a better understanding of the functional interaction between these two miR-16 targets in glioblastoma would be important.
Tumor tumorigenesis provides a supportive environment for the aggressive growth and malignant invasion of glioblastoma, especially if the size of tumor exceeds 2 mm.23 MicroRNAs can substantially affect tumor angiogenesis. Downregulation of microRNA processing enzymes like Dicer in endothelial cells significantly elevated angiogenic factors like VEGFR1, VEGFR2, Tie-2 and eNOS, in turn, promoting angiogenesis.24 MicroRNA-16 was reported to suppress pituitary adenoma growth, of which the mechanism was likely attributed to the inhibition of RARS by miR-16, elevating antiangiogenic p43.25 To establish the suppressive role of miR-16 in angiogenesis in glioblastoma, the present study examined CD31 level in orthotopic tumors overexpressing miR-16. CD31 is a commonly used biomarker for endothelial cells of blood vessels.26 Ectopic expression of miR-16 in the present orthotopic tumor model could reduce CD31 expression, implicating the suppressive role of miR-16 in tumor angiogenesis in glioblastoma. The reduction in tumor angiogenesis would eventually lead to significant suppression on in vivo tumor growth.
To summarize, the present study characterized the functional role of miR-16 in suppressing glioblastoma. In vitro studies suggested that miR-16 would inhibit cell proliferation and clone formation, suppressing invasion and promoting apoptosis. Ectopic miR-16 expression could also suppress in vivo tumor growth and tumor angiogenesis in orthotopic glioblastoma model. The antitumor effect of miR-16 was probably attributed to its suppression on cyclin D1, Bcl-2, WIP1 and CD31. Angiogenesis is shaped by the balance between pro-angiogenic (eg vascular endothelial growth factor, platelet derived growth factor, etc) and antiangiogenic (eg endostatin,thrombospondin-1, etc) factors.23 Whether miR-16 would affect these factors in glioblastoma have remained unclear. MicroRNA-16 was shown to suppress cyclin D1. It is imperative to examine if miR-16 would affect CDK4 and CDK6. This stage of research has focused on U87 cells and U87 orthotopic xenograft, which could hardly represent the heterogeneous nature of human glioblastoma. The results of the present work will be further validated in other glioblastoma cell lines. Furthermore, the findings of this work would be strengthened by a rescue experiment in which miR-16 and cyclin D1 were co-transfected. Nevertheless, the present study has provided insights into the role of miR-16 and other microRNAs in malignant tumors of the central nervous system. MicroRNA-16 and its regulated signaling axis could be therapeutic targets for treatment of glioblastoma.
Abbreviations
CON, control; IHC, immunohistochemistry; miR-16, miRNA-16; miRNAs, MicroRNAs; MVD, microvessel density NC, negative control OE, overexpression group; qRT-PCR, quantitative real-time PCR; UTR, untranslated region.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by Natural Science Foundation of Zhejiang Province (No. LY13H160033) and Wenzhou Municipal Science and Technology Bureau (2014Y0105).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology. 2014;16:896–913.
2. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411.
3. Hafazalla K, Sahgal A, Jaja B, Perry JR, Das S. Procarbazine, CCNU and vincristine (PCV) versus temozolomide chemotherapy for patients with low-grade glioma: a systematic review. Oncotarget. 2018;9:33623–33633.
4. Nieder C, Adam M, Molls M, Grosu AL. Therapeutic options for recurrent high-grade glioma in adult patients: recent advances. Crit Rev Oncol Hematol. 2006;60:181–193.
5. Wang J, Hu G, Quan X. Analysis of the factors affecting the prognosis of glioma patients. Open Med. 2019;14:331–335.
6. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139.
7. Iorio MV, Croce CM. Causes and consequences of microRNA dysregulation. Cancer Jl. 2012;18:215–222.
8. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529.
9. Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658.
10. Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277.
11. Bandi N, Zbinden S, Gugger M, et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009;6(9):5553–5559.
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
13. Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10:59–64.
14. Thomas GR, Nadiminti H, Regalado J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. Int J Exp Pathol. 2005;86:347–363.
15. Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118(4):477–491. doi:10.1016/j.cell.2004.07.025
16. Goretti E, Rolland-Turner M, Leonard F, Zhang L, Wagner DR, Devaux Y. MicroRNA-16 affects key functions of human endothelial progenitor cells. J Leukoc Biol. 2013;93:645–655.
17. Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171.
18. Li J, Yang Y, Peng Y, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31:133–134.
19. Saito-Ohara F, Imoto I, Inoue J, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–1883.
20. Hirasawa A, Saito-Ohara F, Inoue J, et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res. 2003;9:1995–2004.
21. Castellino RC, De Bortoli M, Lu X, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245–256.
22. Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–350.
23. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410.
24. Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. Dicer is required for embryonic angiogenesis during mouse development. J Biol Chem. 2005;280:9330–9335.
25. Bottoni A, Piccin D, Tagliati F, Luchin A, Zatelli MC, Degli Uberti EC. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J Cell Physiol. 2005;204:280–285.
26. Ilan N, Madri JA. PECAM-1: old friend new partners. Current Opinion Cell Biol. 2003;15:515–524. doi:10.1016/S0955-0674(03)00100-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.