Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
MicroRNA-126a-5p Exerts Neuroprotective Effects on Ischemic Stroke via Targeting NADPH Oxidase 2
Authors Tan Y, Zhou F, Yang D, Zhang X, Zeng M, Wan L
Received 23 November 2020
Accepted for publication 14 May 2021
Published 25 June 2021 Volume 2021:17 Pages 2089—2103
DOI https://doi.org/10.2147/NDT.S293611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Taro Kishi
Yu Tan,1,* Feng Zhou,2,* Dejiang Yang,1 Xiaowei Zhang,1 Meihong Zeng,1 Lei Wan1
1Department of Neurology, The Third Affiliated Hospital of Nanchang University, Nanchang City, Jiangxi Province, 330008, People’s Republic of China; 2Department of Neurology, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai City, Guangdong Province, 519000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yu Tan
Department of Neurology, The Third Affiliated Hospital of Nanchang University, No. 128 Xiangshan Road, Nanchang City, Jiangxi Province, 330008, People’s Republic of China
Email [email protected]
Background: Ischemic stroke is a destructive cerebrovascular disorder related to oxidative stress; NOX2 is a major source for ROS production; and miR-126a-5p is involved in several diseases, such as abdominal aortic aneurysm. We investigated the role of miR-126a-5p in regulating NOX2 in ischemic stroke.
Methods: MiR-126a-5p and NOX2 were examined in the brains of rats subjected to cerebral ischemia/reperfusion (I/R) by RT-PCR and Western blot. MiR-126a-5p agomir was delivered to examine the effects of miR-126a-5p on I/R injury. The neurological deficit, infarct volume, and brain water content were evaluated. NOX activity, ROS production, and MDA and SOD levels were detected to assess oxidative stress. H&E staining was used to examine cell state. Apoptosis was evaluated by TUNEL, caspase-3 activity, and cleaved-caspase-3 protein level. The relationship between miR-126a-5p and NOX2 was analyzed by bioinformatics and luciferase reporter assay. MiR-126a-5p mimic, miR-126a-5p inhibitor, or pcDNA-NOX2 were transfected in SH-SY5Y cells to further assess the effects of miR-126a-5p on OGD/R-induced cells injury.
Results: NOX2 was upregulated and miR-126a-5p was down-regulated in the brains of I/R rats. MiR-126a-5p agomir obviously reduced the neurological deficit, infarct volume, brain water content, oxidative stress, and apoptosis in I/R rats. MiR-126a-5p targeted NOX2 directly and regulated NOX2 negatively. Moreover, miR-126a-5p mimic elevated cell viability and inhibited oxidative stress and apoptosis in OGD/R-treated SH-SY5Y cells, while miR-126a-5p inhibitor had the opposite effects. NOX2 overexpression antagonized the protective effects of miR-126a-5p mimic on OGD/R-induced cell injury.
Conclusion: MiR-126a-5p is a novel potential target for ischemic stroke therapy due to its protection against cerebral I/R injury via directly targeting NOX2.
Keywords: oxidative stress, NOX2, miR-126a-5p, cerebral ischemia/reperfusion
Introduction
Stroke is one of the major causes of disability and leading causes of mortality worldwide.1 Although endovascular thrombectomy and thrombolysis mediated by tissue plasminogen activator (tPA) have significantly improved the survival of patients at the acute stage of ischemic stroke,2 therapeutic strategies for ameliorating neurological recovery after stroke remain inadequate.3 Oxidative stress is a phenomenon caused by the imbalance between the generation and accumulation of reactive oxygen species (ROS) in tissues and cells and the capability of biological circumstance to detoxify those responsive products.4 Oxidative stress has emerged as a vitally detrimental factor in ischemic tissues and compromises genome integrity, leading to DNA injuries, death of neuronal, vascular, and glial cells, and neurological recovery impairments following stroke.5 Therefore, suppression of ischemic oxidative stress can be a promising therapeutic approach in restorative administration for stroke.
MicroRNAs (miRNAs) are endogenous small non-coding RNAs with a length of approximately 22 nucleotides and play critical roles in post-transcriptional gene regulation via sponging target messenger RNAs at the complementary sites of the 3ʹ-untranslated region.6 Recently, accumulating shreds of evidence have revealed that miRNAs function as translational inhibitors to participate in ischemic stroke pathogenesis,7 such as inflammation, oxidative stress, excitotoxicity, neurogenesis, and apoptosis.8 For example, miR-124 exerts its effects on age-related ischemic encephalopathy, including ischemic stroke, through modulating neuroinflammation, neuronal excitability, oxidative stress, synaptic plasticity, apoptosis, and axonal growth.9 MiR-137 confers neuroprotective impacts on ischemic stroke by alleviating inflammatory, apoptotic, and oxidative pathways via Src-dependent MAPK pathway.10 However, due to the potent and wide tissue distribution of miRNAs, the description of cell-specific targets and their functions remain elusive, especially in the complex brain environment.
Recently, it has been studied and identified that NADPH oxidases (NOXs) function as essential ROS producers after ischemic brain injury.11 The isoforms, such as NOX1, NOX2, and NOX4, are elevated by various neurodegenerative factors, and down-regulation of NOXs by pharmacological or genetic approaches is neuroprotective against brain tissue damages due to stroke.12 Previous research has reported that miR-652 protects rats against oxidative stress injuries following cerebral ischemia/reperfusion through targeting NOX2 directly.13 In addition, in a mouse cholestasis model, miR-126a deletion promotes hepatic inflammation and aging,14 indicating it might be a drug target for therapy of hepatic failure. Moreover, miR-126a-5p reduces the level of a disintegrin and metallopeptidase (ADAM) with thrombospondin type 1 motif 4 (ADAMTS-4) and restricts abdominal aortic aneurysm development in mice.15 In neonatal pulmonary hypertension, miR-126a-5p is implicated in endothelial-to-mesenchymal transition induced by hypoxia.16 However, the function of miR-126a-5p and the interaction between miR-126a-5p and NOX2 in oxidative stress and functional restoration following ischemic stroke are unclear.
This study established a mouse model and a cell model to study the impacts of miR-126a-5p on brain injury and neurological outcomes after cerebral ischemia/reperfusion and the potential underlying molecular mechanisms. Moreover, we validated the interaction between NOX2 and miR-126a-5p and demonstrated that miR-126a-5p functions as a sponge of NOX2 to inhibit oxidative stress and improve neurological recovery.
Materials and Methods
Animal Experiment
Sprague-Dawley rats (male, 250–300g) were obtained from the Laboratory Animal Center of the University of Science and Technology of China. All rats were acclimatized to the environments with a humidity of 65% and a 12h/12h light-dark cycle at room temperature and assigned into 4 groups with 6 per group randomly: Sham group, I/R group, NC agomir group, and miR-126a-5p agomir group. A cerebral ischemia/reperfusion (I/R) injury model was established in all rats except those in the sham group. Before I/R operation, rats in the NC agomir group and miR-126a-5p agomir group were fixed to the stereotaxic apparatus. A brain infusion cannula was implanted into the left lateral ventricle (bregma: −0.22mm; lateral: 1mm; dorsoventral: 3mm). Then intracerebroventricular infusion of NC agomir (5 nmol) or miR-126a-5p agomir (5 nmol) was delivered to the brain using a micro-osmotic pump (Model 1004, Alzet, Cupertino, CA, USA) that was connected to the cannula, at a rate of 200 μL/min. Two days later, rats in I/R group, NC agomir group, and miR-126a-5p agomir group were subjected to I/R operation. In brief, under light anesthesia by intraperitoneally injecting sodium pentobarbital (40 mg/kg body weight), the left internal carotid artery was separated. Then a 4–0 surgical nylon monofilament with a round tip was inserted into the anterior cerebral artery to occlude the blood flow of the internal carotid artery, posterior cerebral artery, anterior cerebral artery, and middle cerebral artery. Two hours after occlusion, the filament was removed. The blood reperfusion was performed for 24 hours while maintaining rat body temperature at 37°C and free of ileus or peritonitis during the whole surgical operation. Rats in the sham group were exposed to the same process except for internal carotid artery occlusion with filament. After reperfusion, the neurological deficit score and infarct volume were evaluated prior to harvesting brain tissues for other analysis. Procedures operated in this research were completed in keeping with the Laboratory Animal Safety guidelines of Nanchang University and approved by the Ethics Committee of the Institutional Animal Care and Use Committee of Nanchang University.
Cell Culture
SH-SY5Y cells were purchased from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China) and validated by STR profiling prior to the experiments. Human embryonic kidney 293T cells were from the American Type Culture Collection (ATCC). Cells were maintained in DMEM with 10% FBS, 1% sodium pyruvate, 1% glutamate, 1% non-essential amino acids, 1% penicillin, and 1% streptomycin in an incubator with 5% CO2 at 37°C and plated in 12-well plates for assessment of miRNA functions, luciferase reporter analysis, apoptosis, Western blot, and real-time PCR.
Oxygen Glucose Deprivation/Reoxygenation (OGD/R)
To simulate cerebral I/R damage in vitro, the OGD/R model was constructed using SH-SY5Y cells. After removal of culture medium, cells were washed with glucose-free DMEM (Sigma Aldrich, St. Louis, MO, USA) twice and moved in a hypoxic incubator with N2/CO2 (95:5) for 10 min at 37°C to decrease oxygen content to < 1%. After that, cells were cultured in glucose-free DMEM for 2 h at 37°C for glucose deprivation. Subsequently, cells were cultured in a standard culture medium for reoxygenation in an incubator with 5% CO2 under normoxic conditions for 24 h. Cells without OGD/R procedure were used as the control.
Measurement of Neurological Deficits, Infarct Volume and Brain Water Content
Neurological performance was measured with a 5-point rating scale before euthanization: 0 for no observable deficit, 1 for failure to extend the left forepaw, 2 for decreased grip strength of left forepaw, 3 for circling to the left on pulling the tail, and 4 for spontaneous circling. The brain infarct was assessed with 2,3,5-triphenyl tetrazolium chloride (TTC) staining. In brief, after sectioned into 4 coronal sections, brain sections (0.2–0.3 cm) were immersed at 37°C in 2% TTC solution for 0.5 h. The infarct volume was analyzed based on the lesion thickness and areas of the sections using Image J software and presented as the percentage to the total hemisphere. For measurement of brain water contents, the infarct brain hemisphere was weighed before and after dried in a desiccating oven overnight at 105°C on an electronic scale as the wet weight and dry weight, respectively. The total brain water was measured using the formula: (wet weight - dry weight)/wet weight × 100%.
Bioinformatic Analysis
The possible target of NOX2 and association between miRNAs and NOX2 were predicted using microrna.org (http://www.microrna.org/microrna/home.do) and Target Scan (http://www.targetscan.org/vert_72/).
Evaluation of NOX and Caspase-3 Enzymatic Activities and Oxidative Stress Indicators
NOX enzyme activity was measured using the NADPH oxidase activity quantification kit (Shanghai Genmed Pharmaceutical Technology Co., Ltd., Shanghai, China). Briefly, oxidized cytochrome C was mixed with cell supernatant or tissue lysates, transferred into a quartz cuvette, and kept at 30°C for 3 min. The NOX substrate NADPH was then added to initiate the reaction. After reacting at 30°C for 15 min, the absorbance at 550 nm was determined on a spectrophotometer (Bio-Rad Laboratories, USA), and NOX activity was calculated as cytochrome c reduction per min.
Caspase-3 activity was determined using a caspase-3 activity assay kit (Beyotime Institute of Biotechnology) following the manufacturer’s instructions. Briefly, 90 μL caspase-3 substrate Ac-DEVD-pNA working solution was mixed with 10 μL cell lysate or tissue lysate and incubated at 37°C for 1 h. The absorbance at 405 nm was measured on a multifunction reader (Varioskan LUX, ThermoFisher, US). The caspase-3 activity was exhibited as U/g protein, and the amount of enzyme reacting with Ac-DEVD-pNA (1.0 nmol/per hour) was defined as 1 U.
Oxidative stress was assessed by the levels of reactive oxygen species (ROS), malondialdehyde (MDA), and superoxide dismutase (SOD). ROS, MDA and SOD levels in cells and brain tissues were measured as the optical density at 450 nm using corresponding commercial kits (Abcam, Cambridge, MA, USA) following the manufacturer’s protocols on a spectrophotometer (Bio-Rad Laboratories, USA).
Cell Apoptosis
Cellular apoptosis in brain tissues was measured using One Step TUNEL Apoptosis Kits (Beyotime, China). In brief, brain tissue sections were fixed in 4% paraformaldehyde at room temperature for 10 min, washed with PBS, and further fixed in paraformaldehyde plus acetic acid for 5 min at 4°C. After that, they were washed with PBS, equilibrated in equilibration buffer, and incubated with TUNEL reaction mixture containing terminal deoxynucleotidyl transferase (TdT) at 37°C for 1 h. The negative control was treated similarly and incubated with TUNEL reaction mixture without TdT. The TUNEL-positive cells were examined and photographed at ×400 magnification under an epifluorescence microscopy Nikon Eclipse 80i and presented as the percentage of the total cells.
The apoptosis of SH-SY5Y cells was detected by Hoechst 33258 staining using a commercial kit (Sigma‐Aldrich, St. Louis, MO, USA) following the guideline and flow cytometry. For Hoechst 33258 staining, SH-SY5Y cells were fixed in 4% paraformaldehyde for 15 min. After washed with PBS, cells were incubated with Hoechst 33258 staining solution at room temperature for 5 min and observed under a fluorescence microscope. The apoptotic cells were counted measured in a blinded manner and represented as the proportion to the total cells.
Cell Transfection
The empty vector (pcDNA-NC) and NOX2-overexpressing vector (pcDNA-NOX2) were from Sangon Biotech (Shanghai, China). The miR-126a-5p mimic and inhibitor for functional gain or reduction, respectively, were obtained from RiboBio (Guangzhou, China). For cell transfection, miR-126a-5p mimic, miR-126a-5p inhibitor, or pcDNA-NOX2 were mixed with transfection agent Lipofectamine 2000. After 20 min, the mixture was added into the cultured cells in 24-well plates to reach a concentration of 100 nmol/well. Following the transfection for 6 h, cells were cultured for another 48 h in normal media and collected for measurement of enzyme activity, protein, mRNA, and others.
Hematoxylin and Eosin (H&E) Staining
Brain tissues were collected immediately at 24h after cerebral I/R injury and were cut along the coronal plane at 4 mm behind chiasma. The forebrains were fixed with 4% paraformaldehyde and embedded in paraffin. Afterward, the coronal sections (6 μm thick) of the dorsal hippocampus (−3.8 mm from bregma) were subjected to H&E staining.
Luciferase Reporter Gene Experiment
The 3ʹ-UTR fragments of NOX2 containing the predicted wild-type or the mutated miR-126a-5p binding site were inserted into pmirGLO luciferase reporter vector (Abnova, Taipei, Taiwan) downstream of the firefly luciferase gene. The recombinant constructs and NC miRNA or miR-126a-5p mimics, were co-transfected into 293T cells. At 48 h of post-transfection, luciferase activity was measured with a luminometer according to the manufacturer’s instructions.
miR-126a-5p Agomir Delivery by Intracerebroventricular Infusion
To explore the role of miR-126a-5p in cerebral I/R injury, miR-126a-5p overexpression in vivo was managed in accordance with the instruction as previously stated.17,18 In brief, miR-126a-5p agomir was dissolved in PBS to 100 nmol/mL. Under anesthesia, rats were fixed to the stereotaxic apparatus, and a brain infusion cannula was implanted into the left lateral ventricle (bregma: −0.22 mm; lateral: 1 mm; dorsoventral: 3 mm) and connected to a micro-osmotic pump (Model 1004, Alzet, Cupertino, CA, USA). Then NC agomir (5 nmol) or miR-126a-5p agomir (5 nmol) was delivered to the brain via intracerebroventricular infusion using the micro-osmotic pump (Model 1004, Alzet, Cupertino, CA, USA) at 200 μL/min. After 2 days of intracerebroventricular infusion, rats were exposed to I/R. Under anesthesia, rats were sacrificed, and the brain tissues were collected for Western blot, qRT-PCR, TTC staining, and other analysis.
Cell Viability Assay
Cell viability was measured using 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay. In brief, cells were seeded into 96-well culture plates (1×104 cells/well). At the end of each treatment, 50 μL MTT (5 mg/mL) was added, and cells were incubated at 37°C for another 4 h. After removal of the supernatant, 150 μL DMSO was added into each well to dissolve the insoluble purple formazan product, and the absorbance (A) value was detected with a microplate reader (Epoch, BioTek, USA) at 570 nm for quantification. Cell viability was calculated using the formula Cell viability (%) = (A570nmsample-A570nmblank)/(A570nmcontrol-A570nmblank)×100 and presented as the proportion to the control group.
qRT-PCR
Total RNAs in brain tissues and cells were isolated using Trizol reagent (Solarbio, Tongzhou District, Beijing, China). RNA concentration and purity were measured spectrophotometrically. The relative mRNA and miRNA expression levels were measured using 100 ng RNA as the template and the TransScript® Green Two-Step qRT-PCR SuperMix (Transgen, Beijing, China) and the TransScript Green miRNA Two-Step qRT-PCR SuperMix (Transgen, Beijing, China), respectively, on a 7500 FAST Real-Time PCR System (Bio-Rad Co., USA). The primers for miR-126a-5p, NOX2, U6, and GAPDH were listed in Table 1. The relative expression was evaluated using the 2−ΔΔCt method, and data were presented as the ratio of miR-126a-5p to U6 or NOX2 mRNA to GAPDH mRNA.
 |
Table 1 Sequences of Primers Used in qRT-PCR |
Western Blot
The concentration of protein samples extracted from tissues and cells was determined using the BCA protein assay kit (Pierce Company). After mixed with the loading buffer, proteins were denaturized at 99°C for 5 min, separated on 12% SDS-PAGE, and transferred onto PVDF membranes. Then membranes were blocked for 2 hours at room temperature in 5% nonfat milk followed by incubation with primary anti-NOX2 (1:500; Abcam), anti-cleaved-caspase-3 (1:300; Abcam), and anti-GAPDH (1:1000; Santa Cruz) antibodies at 48°C for 16 h. Afterward, the membranes were incubated with secondary antibodies for 1 h at room temperature, and the blots were quantified with the ECL Plus reagent (Millipore, Burlington, MA) and exposed to X-film with GAPDH as the internal control.
Statistical Analysis
Data were analyzed using statistical software SPSS 20.0 and expressed as the mean ± standard deviation (SD) of three biological replicates. Difference between two groups was evaluated with Student’s t-test, while the difference among multiple groups was determined with one-way ANOVA followed by Tukey’s post hoc test. A p < 0.05 was considered statistically significant.
Results
Cerebral I/R Injury Resulted in miR-126a-5p Downregulation, NOX2 Upregulation, and Oxidative Stress in Rat Brain Tissues
As NOX expression variation played a critical role in response to cerebral I/R injury, we emphasized NOX2 in this study. The NOX2 and miR-126a-5p levels were measured in the brains of rats suffering from cerebral I/R. As shown in Figure 1A–C, I/R operation remarkably increased neurological function scores (P < 0.01, Figure 1A), infarct volume (P < 0.01, Figure 1B), and brain water content (P < 0.01, Figure 1C) after reperfusion for 24h, implying the presence of ischemic injury. Then we found that in I/R rats, SOD activity was decreased after 24 h reperfusion (P < 0.01), and MDA content was significantly increased accompanied with ROS outburst (P < 0.01, Figure 1D). Besides, the mRNA and protein levels of NOX2 were noticeably upregulated in I/R injury tissues (P < 0.01, Figure 1E and F). These results indicated that NOX2 alteration was associated with oxidative stress in rats exposed to I/R injury. Since miRNAs were involved in regulating genes, including NOX2, bioinformatic analysis was performed to forecast the possible targets of NOX2 and found that miR-29b-5p, miR-188-3p, miR-532-3p, miR-150-5p, miR-126a-5p, and miR-327 were the possible NOX2 targets (Figure 1G). Measurement of these miRNAs showed that miR-126a-5p expression was apparently reduced in brain tissues of rats suffered from I/R injury (P < 0.01, Figure 1G). These results implied that miR-126a-5p downregulation might result in NOX2 upregulation and led to ROS upregulation. Therefore, miR-126a-5p was further evaluated as a potential target of NOX2.
 |
Figure 1 Cerebral I/R injury resulted in elevated NOX2 and oxidative stress and reduced miR-126a-5p in rats. Animals (Sprague-Dawley rats, male) experienced Sham or I/R (2h/24h) operation. (A) Neurological scores. (B) Infarct volume measured with TTC staining. (C) Brain water content. (D) SOD activity, MDA content, and ROS production. (E) The mRNA expression of NOX2 measured using qRT-PCR. (F) NOX2 protein level assessed with Western blot. (G) MiRNAs expressions by qRT-PCR. Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
Injection of miR-126a-5p Agomir Reduced Brain Injury in Rats
To confirm the effects of miR-126a-5p on brain injury, miR-126a-5p agomir was administrated into rats. The administration efficiency of miR-126a-5p agomir was confirmed by qRT-PCR (P < 0.01, Figure 2A). The results showed that miR-126a-5p agomir remarkably reduced the elevated neurological scores, infarct volumes, and brain water content in I/R rats (P < 0.05, P < 0.01, Figure 2B–D), suggesting it alleviated the brain injury of I/R rats. TTC staining revealed that rats in the sham group had normal neuronal structures in the hippocampal CA1 region, as presented by three or four layers of pyramidal cells arranged in a clear and tight order and large and regular nuclei in neurons (Figure 2E). By contrast, in rats exposed to cerebral I/R, neurons of hippocampal CA1 were injured selectively and extensively, as supported by chromatin condensation, pyramidal neuronal shrinkage in nuclei, and reactive gliosis (Figure 2E). These neuronal damages suffering from cerebral I/R were decreased by miR-126a-5p agomir (P < 0.01, Figure 2E). These results indicated that miR-126a-5p overexpression inhibited brain injuries in I/R rats.
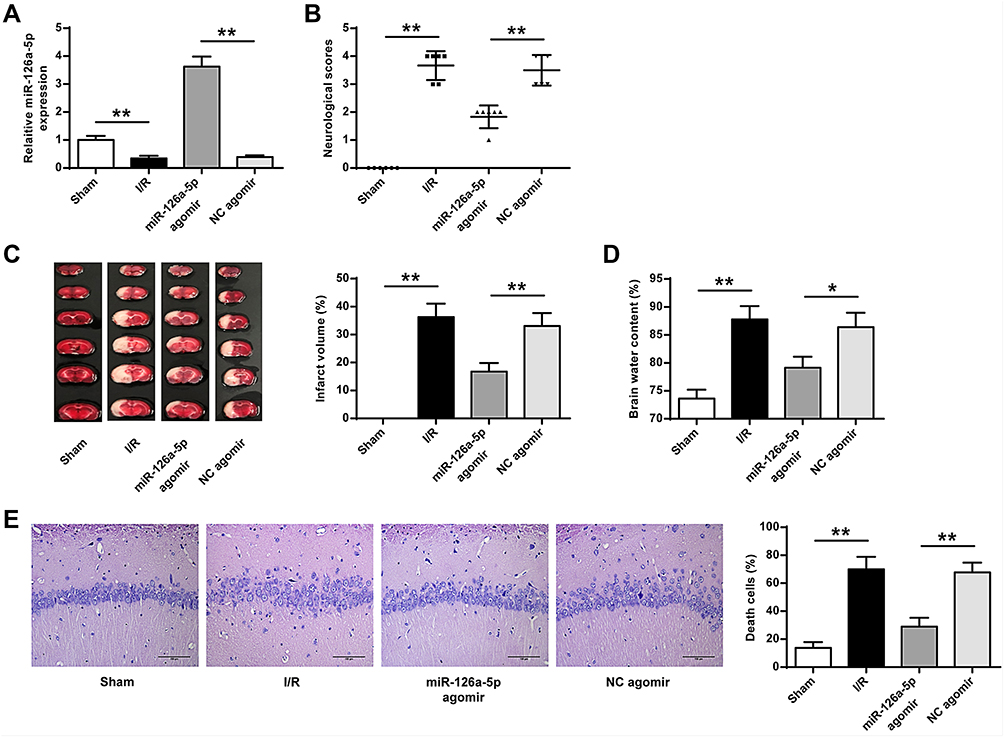 |
Figure 2 MiR-126a-5p agomir injection into rats alleviated brain injury. NC agomir or miR-126a-5p agomir was delivered into lateral ventricle. Rats were exposed to I/R (2h/24h) or Sham surgery after 2 days of the intracerebroventricular infusion. (A) Efficiency of miR-126a-5p agomir transfection was evaluated by qRT-PCR. (B) Neurological scores. (C) Infarct volume determined with TTC staining. (D) Brain water content. (E) Sections of hippocampal CA1 regions detected by H&E staining (×200). Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
Injecting miR-126a-5p Agomir into Rats Reduced Oxidative Stress and Apoptosis in Brain Tissues of I/R Rats
Considering the role of miR-126a-5p in modulating NOX2 expression, we investigated the impacts of miR-126a-5p agomir on oxidative stress and apoptosis in vivo. We first evaluated NOX2 expression and found that miR-126a-5p upregulation reduced NOX2 expression in the brain tissues of I/R rats, while miR-126a-5p negative control agomir showed no effect on NOX2 expression (P < 0.01, Figure 3A and B). Similarly, miR-126a-5p overexpression remarkably downregulated NOX activity, reduced MDA and ROS levels and increased SOD activity (P < 0.05, P < 0.01, Figure 3C and D). These results indicated that miR-126a-5p was a modulator in brain tissues against oxidative stress. Then we found that miR-126a-5p overexpression reduced caspase-3 expression in brain tissues after I/R treatment (P < 0.05, P < 0.01, Figure 3E). Consistently, caspase-3 enzyme activity was also decreased (P < 0.05, Figure 3F). In the TUNEL labelling experiment, we observed more TUNEL positive cells in rat brains suffered from I/R, indicating that cell apoptosis in brain tissues was effectively upregulated by I/R (P < 0.01, Figure 3G). Additionally, miR-126a-5p overexpression obviously decreased the number of TUNEL positive cells in I/R rats, but no obvious change was observed in the negative control agomir group (P < 0.05, Figure 3G). These results implied that miR-126a-5p expression exerted a protective role in oxidative stress and apoptosis in brain tissues.
 |
Figure 3 MiR-126a-5p agomir injection into rats inhibited oxidative stress and apoptosis in brain tissues. NC agomir or miR-126a-5p agomir was delivered into the lateral ventricle. Rats experienced I/R (2h/24h) or Sham operation after 2 days of the intracerebroventricular infusion. Brains were gained for the subsequent detections. (A and B) mRNA (A) and protein (B) expression of NOX2 detected by qRT-PCR and Western blot. (C) Total NOX enzyme activity. (D) SOD activity, MDA content, and ROS production. (E) Protein expression of caspase-3 detected by Western blot. (F) Caspase-3 enzyme activity. (G) Hippocampal positive cell numbers evaluated by TUNEL staining (×400). Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
NOX2 Served as a Direct Target of miR-126a-5p
We intended to verify the functional targets of miR-126a-5p to further confirm the association between miR-126a-5p and NOX2. Bioinformatics analysis revealed a seed region for miR-126a-5p at the 3′-UTR of NOX2 (Figure 4A) indicating that NOX2 might be a target of miR-126a-5p. To confirm the prediction, a dual-luciferase reporter assay was operated on 293T cells co-transfected with a reporter vector of 3ʹ-UTR fragments of NOX2 containing either the wild-type or the mutated putative miR-126a-5p binding site and miR-126a-5p mimics. The results revealed that miR-126a-5p overexpression decreased luciferase activity of the reporter vector containing the wild-type NOX2 3ʹ-UTR (P < 0.01, Figure 4B), but not mutated NOX2 3ʹ-UTR in miR-126a-5p overexpression cells (Figure 4B). These data implied that miR-126a-5p directly bound to 3ʹ-UTR of NOX2 at the predicted binding site. After that, we measured the impact of miR-126a-5p on NOX2 expression and found that miR-126a-5p overexpression noticeably inhibited mRNA (P < 0.01, Figure 4C) and protein (P < 0.01, Figure 4D) expressions of NOX2. These data indicated that NOX2 was a direct target gene of miR-126a-5p.
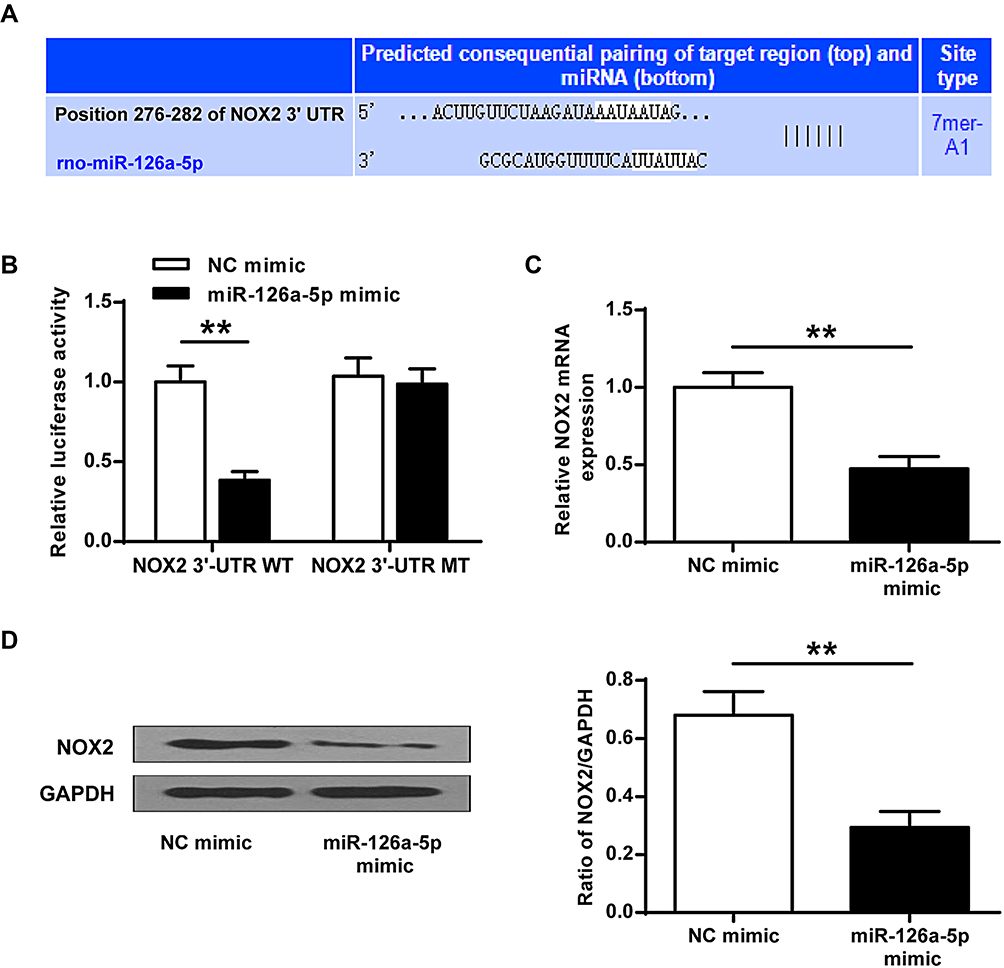 |
Figure 4 NOX2 functioned as a direct target gene of miR-126a-5p. (A) Sequence alignment between miR-126a-5p and NOX2 3ʹ-UTR. (B) The luciferase reporter vector of NOX2-WT or NOX2-MT was co-transfected with NC mimic or miR-126a-5p mimic in 293T cells. After 48h transfection, the relative luciferase activity was evaluated. (C and D) SH-SY5Y cells were treated with miR-126a-5p mimic or NC mimic for 48h. mRNA (C) and protein (D) levels of NOX2 regulated by miR-126a-5p in SH-SY5Y cells measured by qRT-PCR and Western blot. Data were presented as mean±SD. **P < 0.01. n=6. |
Oxidative Stress Was Suppressed by Exogenous miR-126a-5p Mimic in SH-SY5Y Cells
We next verified whether miR-126a-5p alteration was related to ROS production in OGD/R cell model in vitro. In alignment with animal results, miR-126a-5p level was evidently reduced in SH-SY5Y cells suffered from OGD/R in comparison with the control group (P < 0.01, Figure 5A). The miR-126a-5p mimic noticeably increased miR-126a-5p level in SH-SY5Y cells after OGD/R administration in comparison by controls or NC miRNA (P < 0.01, Figure 5A), indicating that these mimics worked successfully. Cell viability was obviously decreased in OGD/R group (P < 0.01) compared to the control group, while it was increased in miR-126a-5p mimic group (P < 0.05) compared to the NC mimic group (Figure 5B). Then we detected NOX2 mRNA and protein levels and found that NOX2 expression was effectively upregulated in the OGD/R group and downregulated in the miR-126a-5p mimic group compared with the corresponding grou (P < 0.01, Figure 5C and D). The total enzyme activity of NOX was noticeably upregulated in OGD/R-treated cells and downregulated in cells transfected with miR-126a-5p mimic (P < 0.05, P < 0.01, Figure 5E). By contrast, SOD activity was remarkably decreased in the OGD/R cells but increased in the miR-126a-5p mimic group (P < 0.05, P < 0.01, Figure 5F). Moreover, in agreement with the NOX activity, ROS production and MDA content were upregulation in the OGD/R cells and downregulation in the miR-126a-5p mimic group (P < 0.01, Figure 5F). Importantly, miR-126a-5p inhibitor had the opposite effects to miR-126a-5p mimic on cell viability, NOX2 level, NOX and SOD activities, ROS production, and MDA level (Supplementary Figure 1 A–F). These findings suggested that miR-126a-5p upregulation was related to NOX2 downregulation.
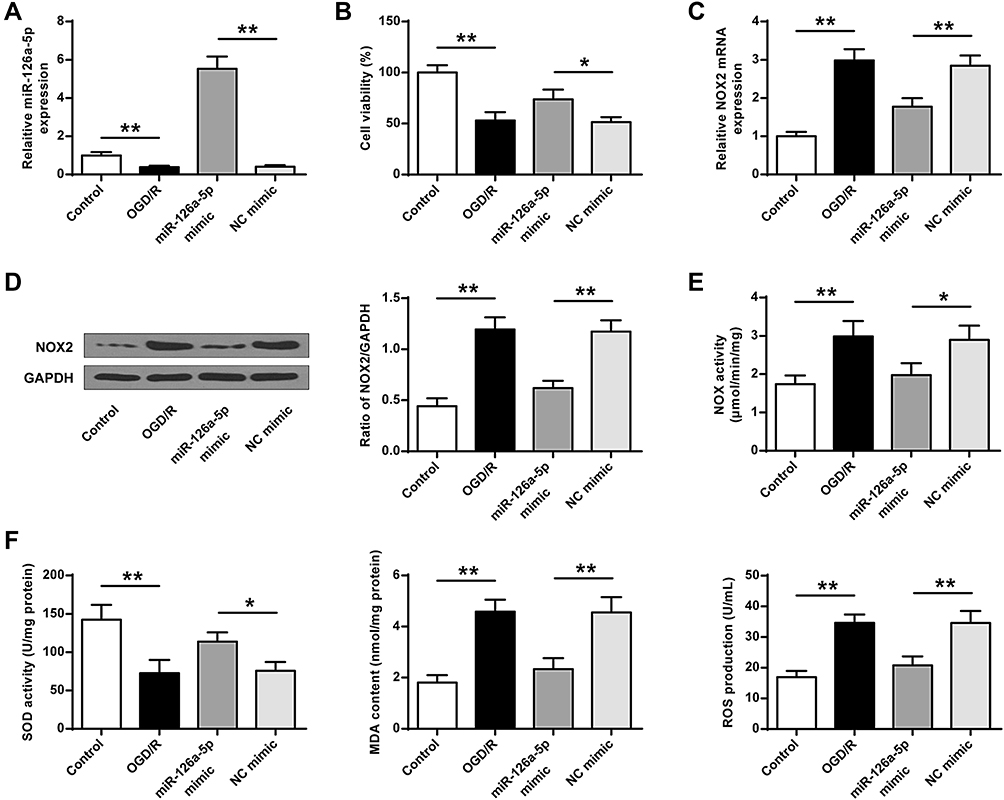 |
Figure 5 Exogenous miR-126a-5p mimic reduced oxidative stress. NC mimic or miR-126a-5p mimic was transfected into SH-SY5Y cells for 48h. Then cells were exposed to OGD/R (2h/24h). (A) Efficiency of miR-126a-5p mimic transfection was evaluated by qRT-PCR. (B) MTT assay used for cell viability evaluation. (C and D) mRNA (C) and protein (D) expressions of NOX2 detected by qRT-PCR and Western blot. (E) Total NOX enzyme activity. (F) SOD activity, MDA content, and ROS production. Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
Exogenous miR-126a-5p Mimic Decreased Apoptosis in SH-SY5Y Cells
We further analyzed the effect of miR-126a-5p mimic on cell apoptosis using OGD/R cell model. The protein level and activity of caspase-3 were upregulated in the OGD/R group compared with the sham control group and decreased in the miR-126a-5p mimic group compared with the NC mimic group (P < 0.05, P < 0.01, Figure 6A and B). Hoechst staining demonstrated more apoptotic cells after OGD/R administration, and miR-126a-5p overexpression noticeably protected OGD/R-treated cells from apoptosis compared with the negative control (P < 0.01, Figure 6C). Similarly, miR-126a-5p overexpression markedly decreased cell death in OGD/R-treated cells (P < 0.05, P < 0.01, Figure 6D) and miR-126a-5p inhibition further elevated the increased protein level and activity of caspase-3 induced by OGD/R. Besides, OGD/R-induced cell apoptosis was aggravated after incubation with miR-126a-5p inhibitor (Supplementary Figure 2A–D). These results proved that miR-126a-5p overexpression played a protective role against apoptosis or death of OGD/R-treated SH-SY5Y cells.
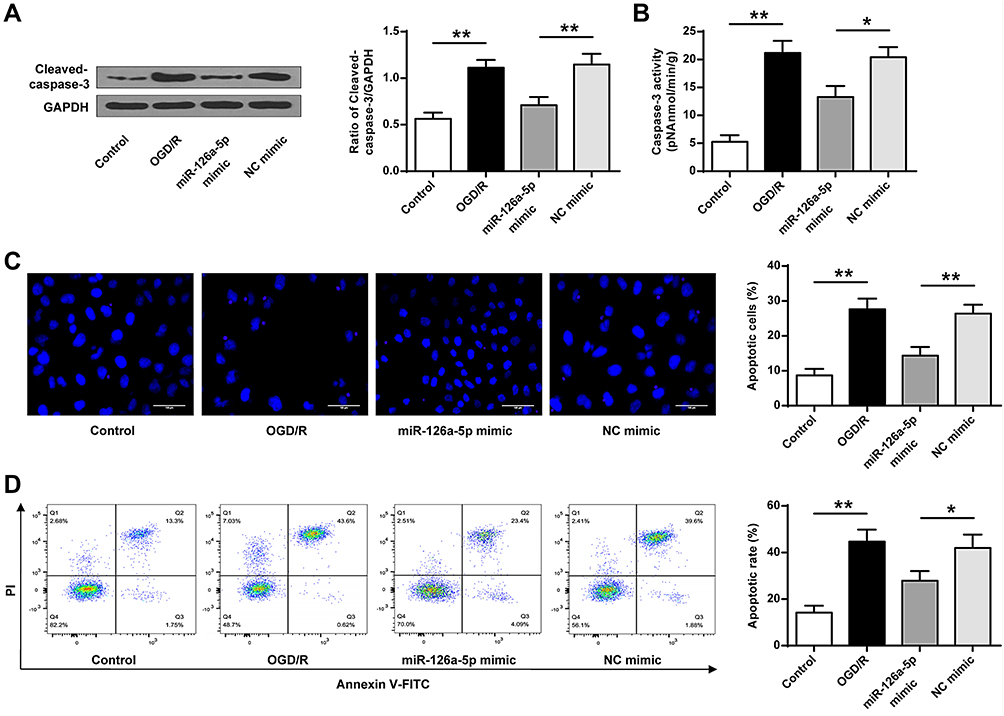 |
Figure 6 Exogenous miR-126a-5p mimic led to decreased apoptosis. NC mimic or miR-126a-5p mimic was transfected into SH-SY5Y cells for 48h. Then cells experienced OGD/R (2h/24h). (A) Protein expression of caspase-3 detected by Western blot. (B) Enzyme activity of caspase-3. (C) Cell apoptosis calculation by Hoechst 33258 staining (×200). (D) Representative image of apoptosis evaluation with flow cytometry. Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
Exogenous miR-126a-5p Mimic Suppressed Oxidative Stress and Apoptosis by Targeting NOX2 in SH-SY5Y Cells
At last, we explored the neuroprotective effects of NOX2 modulated by miR-126a-5p in ischemic brain injury. MiR-126a-5p mimic and pcDNA-NOX2 were co-transfected into OGD/R-treated SH-SY5Y cells. The efficiency of pcDNA-NOX2 transfection was evaluated by Western blot (P < 0.01, Figure 7A). MTT assay illustrated an upregulated proliferation of miR-126a-5p mimic-transfected cells. However, NOX2 overexpression apparently inhibited this increase (P < 0.05, P < 0.01, Figure 7B). Moreover, NOX2 activity was upregulated in cells co-transfected with miR-126a-5p mimic and pcDNA-NOX2 compared with cells co-transfected with miR-126a-5p mimic and pcDNA-NC (P < 0.05, P < 0.01, Figure 7C). Similarly, MDA content and ROS production were increased while SOD activity was remarkably downregulated in cells co-transfected with miR-126a-5p mimic and pcDNA-NOX2 compared to cells co-transfected with miR-126a-5p mimic and pcDNA-NC (P < 0.05, P < 0.01, Figure 7D). Additionally, the protein expression and enzyme activity of caspase-3 were apparently upregulated in miR-126a-5p mimic+pcDNA-NOX2 group in comparison with miR-126a-5p mimic+pcDNA-NC group (P < 0.05, P < 0.01, Figure 7E and F). In line with that, the apoptotic rate was obviously increased in miR-126a-5p mimic+pcDNA-NOX2 group (P < 0.05, P < 0.01, Figure 7G). These results implied that miR-126a-5p mimic inhibited oxidative stress and apoptosis through directly targeting NOX2 in SH-SY5Y cells.
 |
Figure 7 Exogenous miR-126a-5p mimic suppressed oxidative stress and apoptosis by targeting NOX2 in SH-SY5Y cells. (A) The pcDNA-NOX2 or pcDNA-NC was transfected into SH-SY5Y cells for 48h. Efficiency of pcDNA-NOX2 transfection was evaluated using Western blot. (B–G) miR-126a-5p mimic, pcDNA-NOX2 or pcDNA-NC was transfected into SH-SY5Y cells for 48h, alone or in combination. Then cells were exposed to OGD/R (2h/24h). (B) Cell viability determined by MTT assay. (C) Total NOX enzyme activity. (D) SOD activity, MDA content, and ROS production. (E) Expression level of caspase-3 protein by Western blot. (F) Enzyme activity of caspase-3. (G) Cell apoptosis measurement of flow cytometry and the representative images. Data were presented as mean±SD. *P < 0.05, **P < 0.01. n=6. |
Discussion
Oxidative stress is an essential pathological mechanism in response to cerebral ischemia stroke and contributes to the impaired neurological outcomes following stroke through damaging genome integrity, resulting in DNA lesions and neural cell death.5 It has been reported that miRNAs play crucial roles in various pathogenic processes after ischemic stroke, including inflammation, oxidative stress, excitotoxicity, neurogenesis, and apoptosis.8,19 miRNAs are emerging diagnostic biomarkers and function as innovative therapeutic targets for the damages in the central nervous system by modulating their target genes negatively.20 In the present work, cerebral I/R injury upregulated NOX2 and oxidative stress and decreased miR-126a-5p in rat’s brain tissues. MiR-126a-5p overexpression reduced brain injury, oxidative stress, and apoptosis, which eventually resulted in neurological protection in rats against ischemic stroke. Our findings also indicated that exogenous miR-126a-5p suppressed OGD/R-induced oxidative stress and apoptosis through targeting NOX2 in SH-SY5Y cells. To our best knowledge, our study is the first to examine the functions of miR-126a-5p in I/R animal model and OGD/R cell model of ischemic stroke. Our work identifies a previously uncharacterized interaction of miR-126a-5p and NOX2 that mediates stroke-induced oxidative stress in vivo and in vitro. These findings indicate that miR-126a-5p can serve as an innovative potential therapeutic target for ischemic stroke. A previous study has shown that miR-126a-5p reduces ADAMTS-4 expression and restricts the abdominal aortic aneurysm formation in mice.15 Another research has shown that miR-126a-5p participates in endothelial-to-mesenchymal transition stimulated by hypoxia in neonatal pulmonary hypertension.16 Function enrichment analysis indicates that stroke‑prone spontaneously hypertensive (SHRSP)‑specific D‑box binding PAR bZIP transcription factor (Dbp) in rats is associated with circadian rhythm, which is predicted to be regulated by rno‑miR‑126a‑5p.21 It has been shown that 13 miRNAs, including miR-126a-5p, are reduced in retinas in rd10 animal model of retinitis pigmentosa (RP) when mice are treated systemically with pentazocine (PTZ), but are elevated in retinas in rd10/sigma 1 receptor (Sig1R)−/- mice.22 Other than these observations, we detected substantial decline of miR-126a-5p in the ischemic hemisphere of I/R. miRNAs participate actively in a variety of pathophysiological cascades in ischemic stroke, including oxidative stress and apoptosis.20 For instance, an antagomir to miRNA-106b-5p inhibits oxidative stress and apoptosis to ameliorate cerebral injury induced by ischemia and reperfusion in rats.17 MiRNA-410 exerts a neuroprotective role against oxidative stress-induced apoptosis through suppression of TIMP2-dependent MAPK pathway following ischemic stroke in mice.23 MiRNA-424 suppresses oxidative stress to protect against focal cerebral injury induced by ischemia/reperfusion in mice.24 Long non-coding RNA (lncRNA) ZFAS1 modulates miR-582-3p to protect against neuronal injury and regulate oxidative stress, inflammation, apoptosis, and NO level after cerebral I/R damage.25 MiRNA-195 diminishes ischemic stroke induced-neuronal apoptosis in rats by suppressing the activation of JNK signaling mediated by KLF5.26 Similar to the above-mentioned studies, our study systematically demonstrated that miR-126a-5p overexpression induced potent anti-apoptosis and anti-oxidative stress effects after reperfusion of the ischemic brain, eventually enhanced neurological recovery, reduced infarct volumes, and decreased brain water content following ischemic stroke. Cell experiments revealed further that exogenous miR-126a-5p mimic inhibited oxidative stress and diminished apoptosis in SH-SY5Y cells. The present work focused on exploring miR-126a-5p regulation on oxidative stress, apoptosis, and neurological recovery following ischemic stroke. Thus, it is likely that the decreased brain miR-126a-5p could influence the environmental neural cells in cerebral ischemia in vivo. To our best knowledge, this study is the first to validate that miR-126a-5p acts as a neuroprotective factor in ischemic stroke, illustrating a novel function of miR-126a-5p in the regulation of oxidative stress and apoptosis. A better understanding of miR-126a-5p role in oxidative stress and apoptosis may accelerate our recognition of novel targets for oxidative stress and/or apoptosis associated-diseases in the central nervous system.
To date, NOX2 and NOX4 are the major NOX isoforms contributing to ROS generation and cerebral ischemia pathology.12 Studies have demonstrated that NOX2 is involved in post-ischemic oxidative stress, apoptosis, and neurological recovery and that NOX2 downregulating exerts a neuroprotective effect on brain injury mediated by inflammatory cytokines.27 It is identified that several miRNAs potentially target NOX2 and NOX4 genes, such as miRNA-126a, miRNA-132, miRNA-29a, and miRNA-29c, which are considerably reduced following transient middle cerebral artery occlusion (tMCAO) in cerebral area.28 Currently, various miRNAs are identified to regulate NOX. MiR-204-3p diminishes oxidative stress and memory deficits via targeting NOX4 in APP/PS1 mice, and miR-204-3p elevation and/or NOX4 suppression may be a prospective therapeutic approach for Alzheimer’s Disease (AD).29 MiR-141 relieves LPS-stimulated inflammatory damages via elevating NOX2 and further restraining p38 MAPK and NF-κB signaling in WI-38 fibroblasts.30 Neural progenitor cell (NPC)-exosomes (EXs) regulates miR-210 downstream NOX2 and VEGFR2 signaling to relieve endothelial oxidative stress and dysfunction.31 We examined the target genes of miR-126a-5p and identified that miR-126a-5p directly sponges 3ʹ-UTR of NOX2 at the predicted binding site, as revealed by dual-luciferase reporter assay. As additional proof, our mechanistic experiment displayed that miR-126a-5p overexpression decreased NOX2 expression at both mRNA and protein levels in SH-SY5Y cells treated with OGD/R, suggesting that the downregulated NOX2 signaling pathway by miR-126a-5p overexpression might alleviate stroke-induced oxidative stress in the brain and function as a restorative response against ischemic brain injury.
Consistent with our findings, other miRNAs have been reported to modulate ischemia and/or reperfusion injury by targeting NOX2. For instance, miR-124-5p/NOX2 axis regulates the inflammatory microenvironment, ROS production, and neurons apoptosis, ultimately altering cerebral I/R injury.32 MiR-652 can protect animals from oxidative stress damage following cerebral ischemia/reperfusion by targeting NOX2 directly.13 Angiotensin-converting enzyme 2 (ACE2)-primed endothelial progenitor cells (EPCs)-exosomes (EXs) have protective impacts on hypoxia/reoxygenation injury in aging endothelial cells (ECs) via their carried miR-18a and successive inhibition of NOX2/ROS signaling.33 miR‑532‑3p downregulation leads to oxidative stress injury following cerebral ischemia/reperfusion by targeting NOX2 directly.34 In the present study, we also observed that the neuroprotective functions mediated by miR-126a-5p in ischemic brain injury were noticeably antagonized by NOX2 overexpression, suggesting NOX2 overexpression and its upstream regulator miR-126a-5p could synergistically modulate oxidative stress and apoptosis. Our results indicated that upregulated miR-126a-5p could suppress oxidative stress and apoptosis through targeting NOX2 directly. Thus, we proposed that miR-126a-5p triggered anti-apoptotic and anti-oxidative stress effects in neural cells might contribute to neuronal protection during ischemic stroke through modulating NOX2. Nevertheless, our existing evidence cannot rule out the probability that other anti-oxidative stress mechanisms may be also implicated in the miR-126a-5p-mediated suppression of oxidative stress, which calls for further comprehensive investigations.
Conclusion
In conclusion, the present study demonstrates that miR-126a-5p overexpression inhibits cerebral oxidative stress following experimental ischemic stroke. It appears that miR-126a-5p can be an encouraging pharmacological target to ameliorate stroke-induced neurological recovery via inhibition of cerebral oxidative stress through regulating NOX2. Thus, miR-126a-5p upregulation may shed light on the development of novel therapeutics for ischemic stroke.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author: Yu Tan, Department of Neurology, the Third Affiliated Hospital of Nanchang University, No.128 Xiangshan Road, Nanchang City, Jiangxi Province, 330008, P. R. China. E-mail: [email protected].
The data are not publicly available due to their containing information that could compromise the privacy of research participants.
Ethical Approval
The methods applied in this study were approved by the Animal Care Committee of the Third Affiliated Hospital of Nanchang University. Procedures operated in this research were completed in keeping with the standards set out in the Announcement of Helsinki and laboratory guidelines of research in China.
Author Contributions
Yu Tan, Feng Zhou: study concepts, literature research, clinical studies, data analysis, experimental studies, manuscript writing and review; Dejiang Yang: study design, literature research, experimental studies and manuscript editing; Xiaowei Zhang: definition of intellectual content, clinical studies, data acquisition and statistical analysis; Meihong Zeng: data acquisition, manuscript preparation and data analysis; Lei Wan: data acquisition and statistical analysis.
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
All other authors have no conflicts of interest. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
References
1. Johnson CO, Nguyen M, Roth GA; Collaborators GBDS. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):439–458. doi:10.1016/S1474-4422(19)30034-1
2. Phipps MS, Cronin CA. Management of acute ischemic stroke. BMJ. 2020;368:l6983. doi:10.1136/bmj.l6983
3. Raffin E, Hummel FC. Restoring motor functions after stroke: multiple approaches and opportunities. Neuroscientist. 2018;24(4):400–416. doi:10.1177/1073858417737486
4. Pizzino G, Irrera N, Cucinotta M, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. doi:10.1155/2017/8416763
5. Li P, Stetler RA, Leak RK, et al. Oxidative stress and DNA damage after cerebral ischemia: potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology. 2018;134(Pt B):208–217. doi:10.1016/j.neuropharm.2017.11.011
6. Bulygin KV, Beeraka NM, Saitgareeva AR, et al. Can miRNAs be considered as diagnostic and therapeutic molecules in ischemic stroke pathogenesis? Current status. Int J Mol Sci. 2020;21(18):6728. doi:10.3390/ijms21186728
7. Koutsis G, Siasos G, Spengos K. The emerging role of microRNA in stroke. Curr Top Med Chem. 2013;13(13):1573–1588. doi:10.2174/15680266113139990106
8. Li G, Morris-Blanco KC, Lopez MS, et al. Impact of microRNAs on ischemic stroke: from pre- to post-disease. Prog Neurobiol. 2018;163–164:59–78.
9. Liu X, Feng Z, Du L, et al. The potential role of MicroRNA-124 in cerebral ischemia injury. Int J Mol Sci. 2019;21(1):120. doi:10.3390/ijms21010120
10. Tian R, Wu B, Fu C, Guo K. miR-137 prevents inflammatory response, oxidative stress, neuronal injury and cognitive impairment via blockade of Src-mediated MAPK signaling pathway in ischemic stroke. Aging (Albany NY). 2020;12(11):10873–10895.
11. Zhang L, Wu J, Duan X, et al. NADPH oxidase: a potential target for treatment of stroke. Oxid Med Cell Longev. 2016;2016:5026984. doi:10.1155/2016/5026984
12. Ma MW, Wang J, Zhang Q, et al. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener. 2017;12(1):7.
13. Zuo ML, Wang AP, Song GL, Yang ZB. miR-652 protects rats from cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Biomed Pharmacother. 2020;124:109860. doi:10.1016/j.biopha.2020.109860
14. Yan Y, Qin D, Hu B, et al. Deletion of miR-126a promotes hepatic aging and inflammation in a mouse model of cholestasis. Mol Ther Nucleic Acids. 2019;16:494–504. doi:10.1016/j.omtn.2019.04.002
15. Li L, Ma W, Pan S, et al. MiR-126a-5p limits the formation of abdominal aortic aneurysm in mice and decreases ADAMTS-4 expression. J Cell Mol Med. 2020;24(14):7896–7906. doi:10.1111/jcmm.15422
16. Xu YP, He Q, Shen Z, et al. MiR-126a-5p is involved in the hypoxia-induced endothelial-to-mesenchymal transition of neonatal pulmonary hypertension. Hypertens Res. 2017;40(6):552–561. doi:10.1038/hr.2017.2
17. Li P, Shen M, Gao F, et al. An antagomir to MicroRNA-106b-5p ameliorates cerebral ischemia and reperfusion injury in rats via inhibiting apoptosis and oxidative stress. Mol Neurobiol. 2017;54(4):2901–2921. doi:10.1007/s12035-016-9842-1
18. Liu W, Chen X, Zhang Y. Effects of microRNA-21 and microRNA-24 inhibitors on neuronal apoptosis in ischemic stroke. Am J Transl Res. 2016;8(7):3179–3187.
19. Ghafouri-Fard S, Shoorei H, Taheri M. Non-coding RNAs participate in the ischemia-reperfusion injury. Biomed Pharmacother. 2020;129:110419. doi:10.1016/j.biopha.2020.110419
20. Sun P, Liu DZ, Jickling GC, Sharp FR, Yin KJ. MicroRNA-based therapeutics in central nervous system injuries. J Cereb Blood Flow Metab. 2018;38(7):1125–1148. doi:10.1177/0271678X18773871
21. Zhao Q, Sun H, Yin L, Wang L. miR126a5pDbp and miR31aCrot/Mrpl4 interaction pairs crucial for the development of hypertension and stroke. Mol Med Rep. 2019;20(5):4151–4167. doi:10.3892/mmr.2019.10679
22. Wang J, Smith SB. A novel mechanism of sigma 1 receptor neuroprotection: modulation of miR-214-3p. Adv Exp Med Biol. 2019;1185:463–467.
23. Liu NN, Dong ZL, Han LL. MicroRNA-410 inhibition of the TIMP2-dependent MAPK pathway confers neuroprotection against oxidative stress-induced apoptosis after ischemic stroke in mice. Brain Res Bull. 2018;143:45–57. doi:10.1016/j.brainresbull.2018.09.009
24. Liu P, Zhao H, Wang R, et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke. 2015;46(2):513–519. doi:10.1161/STROKEAHA.114.007482
25. Zhang Y, Zhang Y. lncRNA ZFAS1 improves neuronal injury and inhibits inflammation, oxidative stress, and apoptosis by sponging miR-582 and upregulating NOS3 expression in cerebral ischemia/reperfusion injury. Inflammation. 2020;43(4):1337–1350. doi:10.1007/s10753-020-01212-1
26. Chang L, Zhang W, Shi S, et al. microRNA-195 attenuates neuronal apoptosis in rats with ischemic stroke through inhibiting KLF5-mediated activation of the JNK signaling pathway. Mol Med. 2020;26(1):31. doi:10.1186/s10020-020-00150-w
27. Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42(3):341–348. doi:10.1016/j.nbd.2011.01.027
28. Liu Z, Tuo YH, Chen JW, et al. NADPH oxidase inhibitor regulates microRNAs with improved outcome after mechanical reperfusion. J Neurointerv Surg. 2017;9(7):702–706. doi:10.1136/neurintsurg-2016-012463
29. Tao W, Yu L, Shu S, et al. miR-204-3p/Nox4 mediates memory deficits in a mouse model of Alzheimer’s disease. Mol Ther. 2020. doi:10.1016/j.ymthe.2020.09.006
30. Quan B, Zhang H, Xue R. miR-141 alleviates LPS-induced inflammation injury in WI-38 fibroblasts by up-regulation of NOX2. Life Sci. 2019;216:271–278. doi:10.1016/j.lfs.2018.11.056
31. Liu H, Wang J, Chen Y, et al. NPC-EXs alleviate endothelial oxidative stress and dysfunction through the miR-210 downstream Nox2 and VEGFR2 pathways. Oxid Med Cell Longev. 2017;2017:9397631. doi:10.1155/2017/9397631
32. Wu Y, Yao J, Feng K. miR-124-5p/NOX2 axis modulates the ROS production and the inflammatory microenvironment to protect against the cerebral I/R injury. Neurochem Res. 2020;45(2):404–417. doi:10.1007/s11064-019-02931-0
33. Zhang C, Wang J, Ma X, et al. ACE2-EPC-EXs protect ageing ECs against hypoxia/reoxygenation-induced injury through the miR-18a/Nox2/ROS pathway. J Cell Mol Med. 2018;22(3):1873–1882. doi:10.1111/jcmm.13471
34. Mao L, Zuo ML, Wang AP, et al. Low expression of miR5323p contributes to cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Mol Med Rep. 2020;22(3):2415–2423. doi:10.3892/mmr.2020.11325
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.