Back to Journals » Journal of Inflammation Research » Volume 15
Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases
Authors Wang M, Pan W ![]() , Xu Y, Zhang J, Wan J
, Xu Y, Zhang J, Wan J ![]() , Jiang H
, Jiang H
Received 8 December 2021
Accepted for publication 16 May 2022
Published 25 May 2022 Volume 2022:15 Pages 3083—3094
DOI https://doi.org/10.2147/JIR.S350109
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Monika Sharma
Menglong Wang,1– 3,* Wei Pan,1– 3,* Yao Xu,1– 3,* Jishou Zhang,1– 3 Jun Wan,1– 3 Hong Jiang1– 3
1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, 430060, People’s Republic of China; 2Cardiovascular Research Institute, Wuhan University, Wuhan, 430060, People’s Republic of China; 3Hubei Key Laboratory of Cardiology, Wuhan, 430060, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong Jiang; Jun Wan, Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, 430060, People’s Republic of China, Email [email protected]; [email protected]
Abstract: Microglia are tissue-resident macrophages of the central nervous system (CNS). In the CNS, microglia play an important role in the monitoring and intervention of synaptic and neuron-level activities. Interventions targeting microglia have been shown to improve the prognosis of various neurological diseases. Recently, studies have observed the activation of microglia in different cardiovascular diseases. In addition, different approaches that regulate the activity of microglia have been shown to modulate the incidence and progression of cardiovascular diseases. The change in autonomic nervous system activity after neuroinflammation may be a potential intermediate link between microglia and cardiovascular diseases. Here, in this review, we will discuss recent updates on the regulatory role of microglia in hypertension, myocardial infarction and ischemia/reperfusion injury. We propose that microglia serve as neuroimmune modulators and potential targets for cardiovascular diseases.
Keywords: neuroimmune, autonomic nervous system, central-peripheral crosstalk, sympathetic nervous system
Introduction
Microglia, commonly known as brain-resident immune cells, are ubiquitously present in the central nervous system (CNS)1 and participate in the monitoring of the microenvironment. Microglia are abundant within the brain and comprise up to approximately 20% of the total glial cells.2 They are present in the white and gray matter of the brain, but their distribution in the brain is uneven, and the cell density between different brain regions may vary substantially. The highest concentration was found in areas such as the hypothalamus and neostriatum, and the lowest densities of microglial cells were observed in the cerebellum, medulla oblongata and spinal cord.3 Recent findings have shown that microglia establish direct contact with different compartments of neurons. Microglia are involved in almost all brain diseases, including neurodegenerative diseases, traumatic brain injury, and mental illness. After activation, microglia can secrete pro-inflammatory and anti-inflammatory mediators and play a broad role during CNS injury.4
The autonomic nervous system, which comprises the sympathetic nervous system (SNS) and parasympathetic nervous system (PNS), contributes to the regulation of cardiac function.5 Sympathetic outflow is controlled by key cerebral nuclei and neural circuits in the CNS, predominantly the rostral ventrolateral medulla (RVLM),6 the nucleus tractus solitarius (NTS), and the hypothalamic paraventricular nucleus (PVN).7 The imbalance between the SNS and PNS, especially the continuous activation of the SNS, is one of the main contributors to pathological cardiac remodeling.8–10 However, the upstream regulators of SNS activity remain largely unknown. Recently, studies have shown that microglia may play an important role in regulating SNS activities and cardiovascular function by releasing various substances, including cytokines, chemokines, and growth factors.2 Given the importance of the SNS in cardiovascular function, this article mainly reviews the changes in the expression and activity of microglia in different cardiovascular diseases and how microglia contribute to the regulation of cardiovascular disease.
Origin and Functions of Microglia
Microglia are cells of mesodermal origin that populate the central nervous system during an early developmental stage. During early development, erythrocytes/myeloid progenitor cells in the yolk sac differentiate into tissue-resident macrophage progenitor cells. These cells with an amoeboid morphology migrate into the brain and reside in the brain tissue as microglial cells.11 By retaining the ability to divide and synthesize DNA, microglia are capable of self-renewal during inflammation in the case of cell depletion. In addition, when microglia are depleted and unable to self-replenish, bone marrow-derived monocytes are also capable of replenishing tissue-resident macrophages in the CNS.12
Microglial cells have been described as branched, tissue-resident macrophages in the brain. Researchers have attempted to construct a system defining the complexity of microglial activation, and the M1-M2 classification, which was applied to peripheral macrophages, divides the microglial activation state into classical activation (M1) or alternative activation (M2).13 According to the morphology and function of microglia, microglia are divided into three categories: M0, M1, and M2. The M0 phenotype represents microglia that are highly active in their presumed resting state, commonly known as the “resting” microglia phenotype, which monitors the presence of pathogens in the local environment and the changes in extracellular concentrations of constitutively expressed neurochemicals.14 In addition, highly dynamic synapses allow them to sense the microenvironment by interacting with blood vessels, neurons, ependymal cells, and other glial cells, such as astrocytes.15 The M1 phenotype is characterized by the production of pro-inflammatory cytokines (such as TNF-α, IL-6, and IL-1β), chemokines and reactive oxygen species (ROS), leading to an acute immune response. The M2 phenotype is characterized by the production of anti-inflammatory cytokines (such as IL-4 and IL-13), which promote tissue repair, debris removal, wound healing, and the restoration of brain homeostasis.16 As members of the mononuclear macrophage family, microglia also have the function of macrophages, including the identification and monitoring of dead cells, pathogenic microorganisms, and endogenous or exogenous compounds.
During development, microglia clear dead cells through “eat me signals”, which are produced by apoptotic cells and transmitted to microglia.17 The phagocytic activity of microglia also contributes to the homeostasis of synapses.18,19 When infection, tissue damage, or stimulatory signals are present in the microenvironment, microglia are activated and undergo phenotypic and morphological changes and migrate to the injury or stimulation site to produce an inflammatory response. Activated microglia, namely, the M1/M2 phenotypes, release neurochemicals with neuroprotective or neurotoxic effects.20,21
As mentioned above, the simplicity of classifying microglial polarization into the M0/M1/M2 concept is based on the classification of peripheral macrophages and is mainly applied to inflammatory reactions in diseased tissue. A series of reports have illuminated that the expression profiles, functions, survival and ultrastructural characteristics of microglia and monocyte-derived macrophages differ dramatically, even when their morphology and surface markers display similarities, in different immune microenvironments.22,23 These findings have drawn further attention to the classification of microglia and microphages.24,25
Recently, the emergence of novel single-cell techniques, such as cytometry by time-of-flight mass spectrometry (CyTOF) and single-cell RNA sequencing, revealed the heterogeneity of microglia and facilitated the understanding of microglial diversity.26 The resident macrophages in the CNS, according to the anatomical area, could further be divided into two major populations, microglia and CNS-associated macrophages (CAMs, also named border-associated macrophages [BAMs]).27 At E9.5, a phenotypically similar primitive macrophage population could be observed in the yolk sac. However, at E10.5, two macrophage populations distinguishable by the expression of CD206 were detected in the yolk sac. Later, at E12.5 and E14.5, CD206+ macrophages, the CAMs, mainly resided in the developing choroid plexus and the meninges, while CD206−/P2Y12+ macrophages were detected in the developing parenchyma, corresponding to microglia. These findings indicate early segregation of brain macrophages giving rise to CAMs and microglia.28
The Potential Role of Microglia in Cardiovascular Diseases
The Potential Role of Microglia in Hypertension
Increased neuroinflammation and sympathetic tone contribute to the incidence and maintenance of hypertension. Targeting the neuroinflammatory response with an anti-inflammatory reagent or overexpression of interleukin-10 in the brain attenuates hypertension.29,30 However, the cellular mechanisms by which neuroinflammation regulates blood pressure (BP) remain unclear. In a chronic systemic inflammation-induced hypertension model, sustained hypertension was induced after LPS infusion for 14 days, and the activation of microglia, increased IL-1β, IL-6 and TNF-α expression, and O2− production in the RVLM were observed. All of these changes were blunted by inhibiting microglial activation.31 In addition, the activation of microglia was observed in the PVN and motor cortex of both angiotensin II- and L-NG-nitro-l-arginine methyl ester-induced hypertension models.32 Targeted depletion of microglia significantly attenuated neuroinflammation in the PVN, the plasma vasopressin level, kidney norepinephrine concentration, and BP.32
Other studies using minocycline (50 mg/kg/day, oral administration), an inhibitor of microglial activation, to directly inhibit the activation of microglia reported the effective inhibition of sympathetic activity and the attenuation of hypertension both in spontaneously hypertensive rat (SHR) models (normal diet, duration of minocycline treatment 4–6 weeks) and in chronic angiotensin II (Ang II, 200 ng/kg/min)-infused rats (normal diet, duration of minocycline treatment 3–7 weeks).33 These results provide direct evidence that microglia are central to neuroinflammation and neuronal regulation of hypertension. However, other studies have reported that either systemic (25 mg/kg/day) or central administration of minocycline (0.5 µg/50 nL) into the PVN failed to decrease BP, although microglial activation was observed in the PVN in Ang II (high salt diet, 150 ng/kg/min, 2 weeks)-induced hypertensive rats and in stroke-prone spontaneously hypertensive rats (SHRSP) from 15 weeks old,34,35 which is possibly related to the diet (normal diet vs high salt diet), the animal model (SHRs vs SHRSP), the concentration and duration of Ang II used for modeling (200 ng/kg/min for 3–7 weeks vs 150 ng/kg/min for 2 weeks) and the dose/route of minocycline administration (50 mg/kg/day, oral administration vs 25 mg/kg/day or 0.5 µg/50 nL, systemic or central administration).
In addition to the pathological state discussed above, changes in microglia have been observed during a physiologically receptive state in acute hypertension and during the hypotension response.36 Increases and decreases in BP trigger alertness in the physiology of microglia in the brainstem region, inducing changes in the microglial spatial distribution and the number of synapses in contact with the microglial end processes. After 6 hours of acute hypertension, the number of synapses in contact with microglia increased by 30% in both regions of the brainstem, the CVLM and RVLM. Induction of acute hypotension for 6 hours caused microglia to reduce the number of synaptic contacts by >20% in both the CVLM and RVLM, However, these changes were not accompanied by characteristic morphological changes of the microglia, and the numbers of M1 or M2 microglia were not changed.36 This observation further indicates that the M1/M2 microglial classification cannot fully clarify the function of microglia.
Several key molecules that regulate hypertension by targeting microglia have been identified. The brain (pro)renin receptor (PRR) is a novel component of the renin-angiotensin system. The immunoreactivity of PRR is significantly correlated with systolic BP but not the use of antihypertensive drugs, suggesting that PRR might be a key initiator of the pathogenesis of hypertension.37,38 The subfornical organ (SFO) is one of seven circumventricular organs in the human and rodent brain that lacks a traditional blood–brain barrier (BBB), indicating that the SFO senses circulating factors such as Ang II or prorenin and plays a key role in the regulation of BP.39,40 In the SFO, most neurons and microglia, but not astrocytes, express PRR. At the same time, targeted knockdown of PRR attenuates the development of Ang II–induced hypertension in mice,41,42 while other work reported that minocycline could fully abolish the (pro)renin-elicited increases in pro-inflammatory cytokine expression in vitro,43 indicating that an intervention targeting PRR on microglia may be an effective method for the treatment of hypertension.
C-X3-C motif chemokine receptor 1 (CX3CR1), a microglial biomarker, is a chemokine receptor that binds to its ligand C-X3-C motif chemokine ligand 1 (CX3CL1). A previous study reported that CX3CL1 microinjection produces a cardiovascular response in the NTS of normal rats.44 Intracerebroventricular (ICV) administration of AZD8797, a CX3CR1 inhibitor, attenuates fructose-induced hypertension and the expression of pro-inflammatory cytokines.45
Kinins are considered potent vasoactive hormones and inflammatory mediators, and the expression of its extracellular amino terminal Kinin B1 receptor (B1R) is well documented on neurons, microglia, and astrocytes within the brain and spinal cord. B1R is markedly upregulated in the presence of inflammation or tissue injury,46 and its specific antagonist R715 (70 µg/kg/day) could reduce BP, decrease sympatho-excitation and exert a significant inhibitory effect on neuroinflammation in a DOCA-salt-induced hypertension mouse model.47 However, the effect of B1R antagonists on BP remains controversial. Acute injection of the B1R antagonist Leu8-des-Arg9-BK (12 nmol) into the fourth cerebral ventricle does not change the BP in Wistar Kyoto (WKY) rats or female SHRs.48 In contrast, the same B1R antagonist, Leu8-des-Arg9-BK (0.1–10 μg), infused into the lateral cerebral ventricle through an intracerebral guide cannula, was shown to reduce the BP and heart rate (HR) in male SHRs.49 Explanations for the conflicting results may be attributed to the differences in sex (female vs males) of the animals used in the two studies, the dose of pharmacological agents (12 nmol vs 0.1–10 µg) used and the route of agent administration (injection into the fourth cerebral ventricle vs the lateral cerebral ventricle). Nevertheless, a subsequent study reported that the B1R antagonist SSR240612 caused a pronounced antihypertensive effect in both SHRs and Ang II-treated rats,50 suggesting a potential role of B1R in the pathogenesis of hypertension.51
Triggering receptor expressed on myeloid cells 2 (TREM2) is a receptor that recognizes phospholipids, apoptotic cells and lipoproteins.52 Previous studies revealed that TREM2 deficiency exacerbates inflammatory cytokine release from activated M1 microglia and neuronal apoptosis, while TREM2 overexpression markedly attenuated inflammation and neuronal death in AD models.53,54 Recently, TREM2 was reported to be significantly upregulated in microglia in a hypertension model induced by Ang II infusion, and the overexpression of microglial TREM2 mitigated the microglial inflammatory response, suggesting its possible beneficial effects on BP regulation.55
Interventions targeting phenotypic changes in microglia also contribute to the progression of hypertension. High mobility group box protein 1 (HMGB1) is synthesized and released after the activation of microglia, functions as an alarming protein or damage-associated molecular pattern (DAMP) in response to neuroinflammation and is considered a potential mediator priming stress-induced microglia.56 Evidence has shown that the ablation of HMGB1 and the advanced glycation end-product receptor (RAGE) attenuates persistent chronic noise-induced M1-type microglial activation and hypertension,57 which theoretically suggests that reducing neuroinflammation and SNS activity in prehypertensive individuals may be a new strategy for the treatment of hypertension. In mice with Ang II–induced hypertension, supplementation with TGF-β significantly inhibited neuroinflammation and renal norepinephrine levels and increased BP. TGF-β regulates microglia to maintain brain homeostasis in response to hypertensive disorders, which shifts microglia to the immunosuppressive phenotype, namely, resting M0 microglia, and thus resists the increase in BP during the onset of hypertension.58 Based on these findings, TGF-β and its signal transduction pathway may be potential targets for controlling neurogenic hypertension, and resting microglia may play a key role in curbing neuroinflammation. Vitamin D (VitD), a generally recognized pleiotropic hormone, has been reported to possess anti-inflammatory, antioxidant and neuroprotective properties, in addition to its classic functions in calcium and phosphorus homeostasis.59 Although no significant difference in the trend of BP reduction was observed, chronic calcitriol treatment shifted microglial polarization from the pro-inflammatory M1 phenotype to the immunoregulatory M2 phenotype in SHRs, indicating the neuroprotective mechanisms of VitD in the hypertensive brain.60
TLR4, a pathogen recognition receptor, is expressed on leukocytes, cardiomyocytes, and endothelial cells and contributes to the activation of innate immunity. TLR4 is expressed primarily on microglia and sparsely on astrocytes and neurons.61,62 The binding of TLR4 to appropriate ligands activates microglia, induces a local inflammatory response and promotes the expression of pro-inflammatory cytokines.61 A previous study showed that exogenous Ang II stimulates TLR4 via Ang II type 1 receptor (AT1R), which could induce the activation of hypothalamic microglia ex vivo.63 Recently, it was demonstrated that TAK-242 (TLR4 inhibitor, 2 weeks) administration could abolish microglial activation and preserve BBB integrity in the PVN, RVLM, and NTS in SHRs.64 Moreover, TLR4 blockade attenuated the progression of MAP increases in SHRs and protected against autonomic dysfunction, suggesting that TLR4 is a viable alternative target in the treatment of hypertension.
Recently, the concept of an association between dysbiotic gut microbiota and hypertension has been established in both animal and human studies.65–67 A published study showed that intracerebroventricular administration of chemically modified tetracycline-3 (CMT-3), a tetracycline derivative with effective anti-inflammatory activity, could inhibit microglial activation and neuroinflammation in the PVN, decrease sympathetic activity and attenuate the increased mean arterial pressure in Ang II rats. In addition, the antihypertensive function of CMT-3 may be attributed to its regulatory effects on selective gut microbial communities and gut wall histopathology.68 Kefir, a probiotic obtained from the fermentation of milk by kefir grains, was shown to decrease BP and improve endothelial dysfunction in SHRs.69,70 One study indicated that the antihypertensive effects of kefir treatment, mediated at least in part through improved structural and functional integrity of the intestinal wall, abolished microglial activation and protection against neuroinflammation within the PVN and RVLM.71 These observations suggested the involvement of microglial activation in the regulation of selective gut microbiota and implicated these cells in BP control and brain-gut communication dysfunction in hypertension.
Additionally, aerobic training restores PVN autonomic nerve dysfunction, HMGB1 content, microglial activation, and inflammation to normal in SHRs. Aerobic training reduces microglial activation and the expression of pro-inflammatory cytokines, ultimately improving autonomic control and reducing BP and HR in SHRs.72 Some relevant clinical evidence also suggests the feasibility of aerobic training in attenuating hypertension.73 A comprehensive summary of these findings is shown in Table 1.
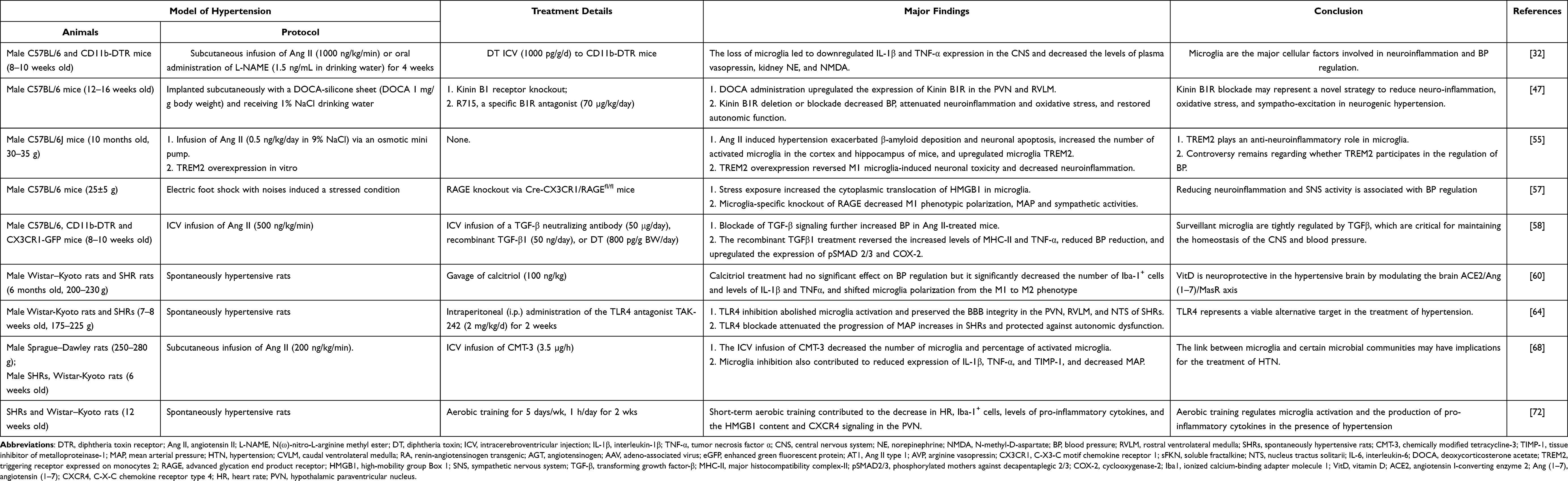 |
Table 1 The Potential Role of Microglia in Hypertension |
The Potential Role of Microglia in Myocardial Infarction
After myocardial infarction, microglial activation in the PVN of the hypothalamus has been observed,74 and increased levels of pro-inflammatory cytokines in the PVN then activate the hypothalamus-pituitary-adrenal axis, increase the activity of the sympathetic nervous system and contribute to the acute pro-inflammatory response in the myocardium after myocardial infarction.75 In addition, activated microglia were also detected in the RVLM, NTS and periaqueductal gray (PAG), regions known to have important cardiovascular regulatory functions.76 In a rat myocardial infarction model, the average number of microglia was not changed, but the proportion of activated microglia in the PVN was increased. The activation of microglia starts at 4 weeks and is sustained until 16 weeks after myocardial infarction.77 At 24 h and 1 week after myocardial infarction, a significant increase in the proportion of activated microglia was not observed. Furthermore, an ICV infusion of minocycline, beginning one week prior to infarction, significantly attenuated the increase in microglial activation by at least 50% in the PVN, RVLM, PAG and NTS, and neuronal activation was significantly reduced by 50% in the PVN and virtually abolished in the PAG, RVLM and NTS.76 Thus, myocardial infarction potentially induces microglial activation, and activated microglia contribute to increased neuronal activity.
P2X receptors are recognized as ligand-gated ion channels that respond to extracellular ATP.78 Among them, the P2X7 purinergic receptor (P2X7R) has been identified as a key mediator of inflammation.79 Based on accumulating evidence, P2X7R is involved in regulating cardiovascular activity both in peripheral and central regions.79,80 Colocalization of P2X7R with the microglial marker Iba-1 suggests that P2X7R is expressed on microglia rather than on neurons, and an intraperitoneal injection of P2X7R antagonists or P2X7 siRNA attenuates the increased levels of pro-inflammatory cytokines in the PVN and the augmented sympathetic nervous system activity after myocardial infarction, which may contribute to improved cardiac function.81
Macrophage-induced type C lectin (Mincle) is a key C-type lectin receptor that was originally discovered based on the potent induction of macrophages by inflammatory stimuli. It is rarely expressed under normal conditions but is strongly activated after stimulation with apoptotic fragments, necrotic cells, heat shock proteins, and nucleic acid fragments.82 Recently, Mincle expression was reported to be localized in microglia within the PVN, and its expression was markedly increased at 24 hours post-MI, together with sympathetic hyperactivity. Targeted knockdown of Mincle expression in the PVN attenuated microglial activation and sympathetic nerve activity, which contributed to decreased ventricular arrhythmia susceptibility post-MI.83 Furthermore, the NOD-like family NLRP3/IL-1β axis in the PVN mediates the cardioprotective effects of Mincle inhibition. Targeting the Mincle signaling pathway in the PVN represents a novel approach to reduce the sympathetic hyperactivity post-MI, likely limiting the complications associated with MI.
The effect of TLR4 on microglia was also reported for MI. In a rat model of MI, TLR4 was primarily localized in microglia, and its expression increased markedly within the PVN at 3 days post-MI. TLR4 knockdown via shRNA microinjection into the PVN resulted in a decreased degree of microglial activation, decreased activation of Fos protein (+) neurons in the PVN and ameliorated sympathoexcitation after MI.84 In addition, TLR4 knockdown in the PVN decreased the incidence of malignant ventricular arrhythmias following MI. However, another study showed that TLR4 colocalizes with GRP78, a marker of endoplasmic reticulum stress, in PVN neurons, and acute LPS treatment increases the expression of the TLR4 and TNF-α proteins in the PVN, which contributes to an increased HR and plasma norepinephrine concentration and decreased heart rate variability (HRV) and high frequency (HF) components of HRV.61 Further inhibition of TLR4 or endoplasmic reticulum stress attenuates LPS-induced microglial activation, indicating that TLR4 signaling promotes autonomic dysfunction, inflammation and microglial activation through neuronal ER stress in the PVN. Thus, the exact mechanisms by which central TLR4 regulates neuroinflammation and sympathetic activity require further research. A comprehensive summary of these findings is shown in Table 2.
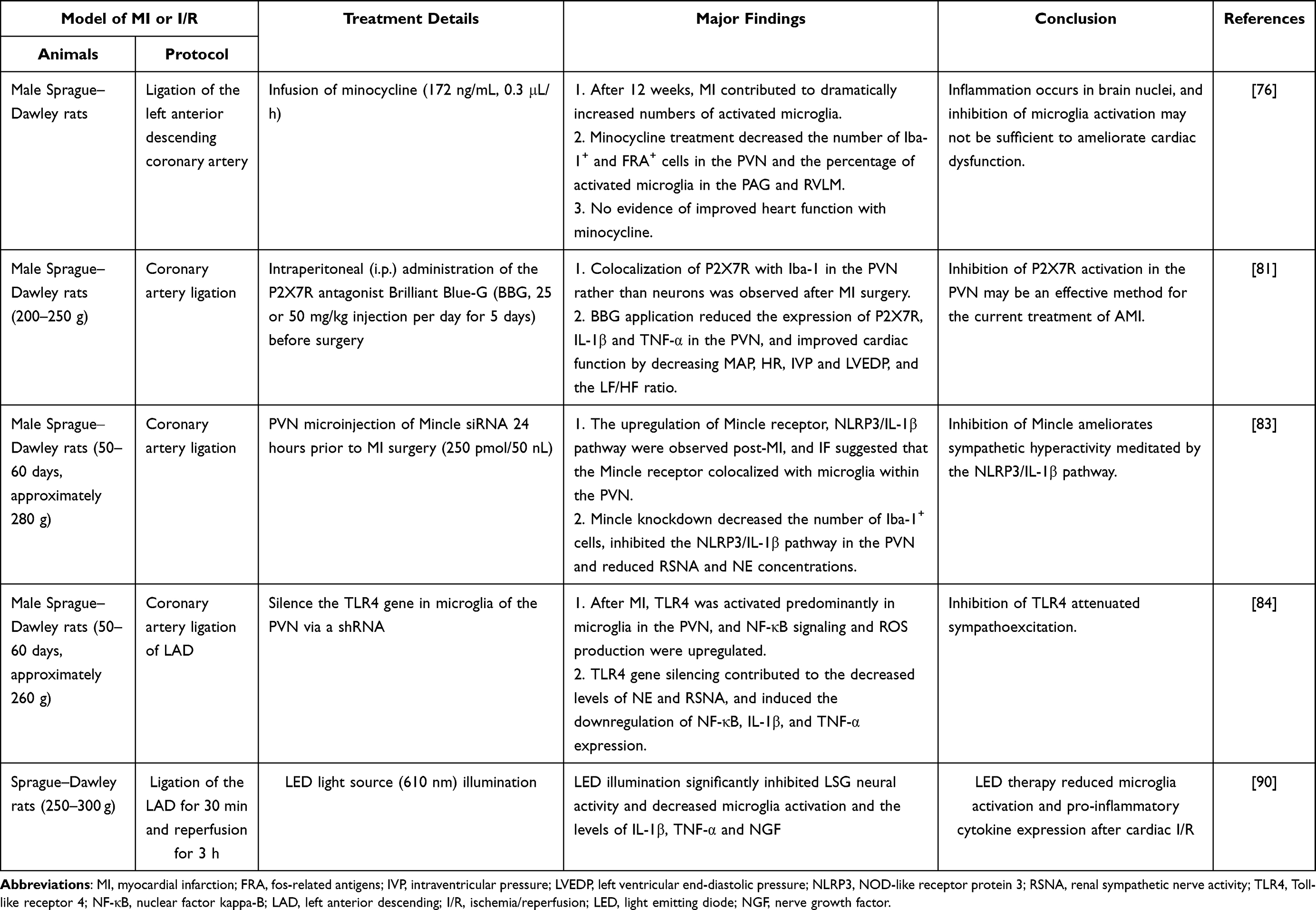 |
Table 2 The Potential Role of Microglia in Myocardial Infarction and Cardiac I/R Injury |
The Potential Role of Microglia in Cardiac Ischemia/Reperfusion Injuries
Studies have shown increased microglial activity in the caudate putamen and hippocampus after cardiac I/R injury.85,86 The timing of microglial activation following cardiac I/R was investigated. It was demonstrated that after coronary artery ligation for 30 min followed by various reperfusion durations, the level of microglial activation peaked at 3 days after reperfusion,87 suggesting a potential role of microglial activation in cardiac I/R injury.
Light-emitting diode (LED) therapy has been shown to attenuate neuroinflammatory responses by inhibiting the activation of microglia.88,89 Thus, LED therapy may protect against myocardial I/R injury by attenuating microglia and sympathetic activation. Recently, our studies showed that LED therapy (2.0 J/cm2, 610 nm) located at the skull surface of the hypothalamic PVN through the scalp and skull from 30 min before ischemia to 3 h after reperfusion could significantly attenuate the ischemia and infarct size following cardiac I/R.90 In addition, LED illumination significantly reduced the inducibility of ventricular arrhythmias after I/R injury. The attenuated activation of microglia and subsequently decreased peripheral sympathetic activity contribute to the protective effects of LED therapy against cardiac I/R injury.
Conclusions
In summary, microglia play an important role in the crosstalk between the CNS and the peripheral nervous system, and interventions targeting microglia may represent promising potential therapies for cardiovascular diseases, including hypertension, myocardial infarction, heart failure, cardiac ischemia/reperfusion and ventricular arrhythmias.
Data Sharing Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82100292, No. 82070436 and No. 81970287) and the Natural Science Foundation of Hubei Province (No. 2020CFB234).
Disclosure
The authors declare that they have no competing interests.
References
1. Kapoor K, Bhandare AM, Farnham MMJ, et al. Alerted microglia and the sympathetic nervous system: a novel form of microglia in the development of hypertension. Respir Physiol Neurobiol. 2016;226:51–62. doi:10.1016/j.resp.2015.11.015
2. Badoer E. Microglia: activation in acute and chronic inflammatory states and in response to cardiovascular dysfunction. Int J Biochem Cell Biol. 2010;42(10):1580–1585. doi:10.1016/j.biocel.2010.07.005
3. Savchenko VL, McKanna JA, Nikonenko IR, et al. Microglia and astrocytes in the adult rat brain: comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience. 2000;96(1):195–203. doi:10.1016/S0306-4522(99)00538-2
4. Wolf SA, Boddeke HWGM, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79(1):619–643. doi:10.1146/annurev-physiol-022516-034406
5. Levick SP, Murray DB, Janicki JS, et al. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension. 2010;55(2):270–276. doi:10.1161/HYPERTENSIONAHA.109.142042
6. Deng Y, Tan X, Li ML, et al. Angiotensin-converting enzyme 2 in the rostral ventrolateral medulla regulates cholinergic signaling and cardiovascular and sympathetic responses in hypertensive rats. Neurosci Bull. 2019;35(1):67–78. doi:10.1007/s12264-018-0298-3
7. Young CN, Davisson RL. Angiotensin-II, the brain, and hypertension: an update. Hypertension. 2015;66(5):920–926. doi:10.1161/HYPERTENSIONAHA.115.03624
8. Wang M, Li S, Zhou X, et al. Increased inflammation promotes ventricular arrhythmia through aggravating left stellate ganglion remodeling in a canine ischemia model. Int J Cardiol. 2017;248:286–293. doi:10.1016/j.ijcard.2017.08.011
9. Wang Y, Jiang W, Chen H, et al. Sympathetic nervous system mediates cardiac remodeling after myocardial infarction in a circadian disruption model. Front Cardiovasc Med. 2021;8:668387. doi:10.3389/fcvm.2021.668387
10. Coote JH, Chauhan RA. The sympathetic innervation of the heart: important new insights. Auton Neurosci. 2016;199:17–23. doi:10.1016/j.autneu.2016.08.014
11. Cronk JC, Kipnis J. Microglia – the brain’s busy bees. F1000Prime Rep. 2013;5. doi:10.12703/P5-53
12. Ajami B, Bennett JL, Krieger C, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. doi:10.1038/nn2014
13. Dubbelaar ML, Kracht L, Eggen BJL, et al. The kaleidoscope of microglial phenotypes. Front Immunol. 2018;9:1753. doi:10.3389/fimmu.2018.01753
14. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi:10.1126/science.1110647
15. von Bernhardi R, Heredia F, Salgado N, et al. Microglia function in the normal brain. Adv Exp Med Biol. 2016;949:67.
16. Cherry JD, Olschowka JA, Banion MO. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11(1):98. doi:10.1186/1742-2094-11-98
17. Marin-Teva JL, Dusart I, Colin C, et al. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–547. doi:10.1016/S0896-6273(04)00069-8
18. Roumier A. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. 2004;24(50):11421–11428. doi:10.1523/JNEUROSCI.2251-04.2004
19. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi:10.1126/science.1202529
20. Butovsky O, Talpalar AE, Ben-Yaakov K, et al. Activation of microglia by aggregated β-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Mol Cell Neurosci. 2005;29(3):381–393. doi:10.1016/j.mcn.2005.03.005
21. Lambertsen KL, Clausen BH, Babcock AA, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29(5):1319–1330. doi:10.1523/JNEUROSCI.5505-08.2009
22. Yamasaki R, Lu H, Butovsky O, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211(8):1533–1549. doi:10.1084/jem.20132477
23. Ajami B, Bennett JL, Krieger C, et al. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14(9):1142–1149. doi:10.1038/nn.2887
24. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–991. doi:10.1038/nn.4338
25. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
26. Masuda T, Sankowski R, Staszewski O, et al. Microglia heterogeneity in the single-cell era. Cell Rep. 2020;30(5):1271–1281. doi:10.1016/j.celrep.2020.01.010
27. Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell. 2019;179(2):292–311. doi:10.1016/j.cell.2019.08.053
28. Utz SG, See P, Mildenberger W, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. 2020;181(3):557–573 e18. doi:10.1016/j.cell.2020.03.021
29. Segiet A, Smykiewicz P, Kwiatkowski P, et al. Tumour necrosis factor and interleukin 10 in blood pressure regulation in spontaneously hypertensive and normotensive rats. Cytokine. 2019;113:185–194. doi:10.1016/j.cyto.2018.07.003
30. Shi P, Diez-Freire C, Jun JY, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56(2):297–303. doi:10.1161/HYPERTENSIONAHA.110.150409
31. Wu KLH, Chan SHH, Chan JYH. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9(1):212. doi:10.1186/1742-2094-9-212
32. Shen XZ, Li Y, Li L, et al. Microglia participate in neurogenic regulation of hypertension. Hypertension. 2015;66(2):309–316. doi:10.1161/HYPERTENSIONAHA.115.05333
33. Santisteban MM, Ahmari N, Carvajal JM, et al. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ Res. 2015;117(2):178–191. doi:10.1161/CIRCRESAHA.117.305853
34. Takesue K, Kishi T, Hirooka Y, et al. Activation of microglia within paraventricular nucleus of hypothalamus is NOT involved in maintenance of established hypertension. J Cardiol. 2017;69(1):84–88. doi:10.1016/j.jjcc.2016.01.004
35. Bardgett ME, Holbein WW, Herrera-Rosales M, et al. Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertension. 2014;63(3):527–534. doi:10.1161/HYPERTENSIONAHA.113.02429
36. Kapoor K, Bhandare AM, Nedoboy PE, et al. Dynamic changes in the relationship of microglia to cardiovascular neurons in response to increases and decreases in blood pressure. Neuroscience. 2016;329:12–29. doi:10.1016/j.neuroscience.2016.04.044
37. Burcklé C, Bader M. Prorenin and its ancient receptor. Hypertension. 2006;48(4):549–551.
38. Xu Q, Jensen DD, Peng H, et al. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol Ther. 2016;164:126–134. doi:10.1016/j.pharmthera.2016.04.006
39. McKinley MJ, Allen AM, Burns P, et al. Interaction of circulating hormones with the brain: the roles of the subfornical organ and the organum vasculosum of the lamina terminalis. Clin Exp Pharmacol Physiol Suppl. 1998;25(S1):S61–7. doi:10.1111/j.1440-1681.1998.tb02303.x
40. Osborn JW, Fink GD, Sved AF, et al. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep. 2007;9(3):228–235. doi:10.1007/s11906-007-0041-3
41. Cooper SG, Trivedi DP, Yamamoto R, et al. Increased (pro)renin receptor expression in the subfornical organ of hypertensive humans. Am J Physiol Heart Circ Physiol. 2018;314(4):H796–H804. doi:10.1152/ajpheart.00616.2017
42. Li W, Peng H, Cao T, et al. Brain-targeted (pro)renin receptor knockdown attenuates angiotensin II-dependent hypertension. Hypertension. 2012;59(6):1188–1194. doi:10.1161/HYPERTENSIONAHA.111.190108
43. Shi P, Grobe JL, Desland FA, et al. Direct pro-inflammatory effects of prorenin on microglia. PLoS One. 2014;9(10):e92937–e92937. doi:10.1371/journal.pone.0092937
44. Ruchaya PJ, Paton JFR, Murphy D, et al. A cardiovascular role for fractalkine and its cognate receptor, Cx3cr1, in the rat nucleus of the solitary tract. Neuroscience. 2012;209:119–127. doi:10.1016/j.neuroscience.2012.02.018
45. Ho CY, Sun GC, Tse J, et al. CX3CR1-microglia mediates neuroinflammation and blood pressure regulation in the nucleus tractus solitarii of fructose-induced hypertensive rats. J Neuroinflammation. 2020;17(1):1784.
46. Marceau F, Lussier A, Regoli D, et al. Pharmacology of kinins - their relevance to tissue-injury and inflammation. Gen Pharmacol. 1983;14(2):209–229. doi:10.1016/0306-3623(83)90001-0
47. Sriramula S, Lazartigues E. Kinin B1 receptor promotes neurogenic hypertension through activation of centrally mediated mechanisms. Hypertension. 2017;70(6):1122–1131. doi:10.1161/HYPERTENSIONAHA.117.09744
48. Martins DTO, Fior DR, Nakaie CR, et al. Kinin receptors of the central-nervous-system of spontaneously hypertensive rats related to the pressor-response to bradykinin. Br J Pharmacol. 1991;103(4):1851–1856. doi:10.1111/j.1476-5381.1991.tb12341.x
49. Alvarez AL, Delorenzi A, Santajuliana D, et al. Central bradykininergic system in normotensive and hypertensive rats. Clin Sci. 1992;82(5):513–519. doi:10.1042/cs0820513
50. De Brito Gariepy H, Carayon P, Ferrari B, et al. Contribution of the central dopaminergic system in the anti-hypertensive effect of kinin B1 receptor antagonists in two rat models of hypertension. Neuropeptides. 2010;44(2):191–198. doi:10.1016/j.npep.2009.12.011
51. Sriramula S. Kinin B1 receptor: a target for neuroinflammation in hypertension. Pharmacol Res. 2020;155:104715. doi:10.1016/j.phrs.2020.104715
52. Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem. 2015;290(43):26033–26042. doi:10.1074/jbc.M115.677286
53. Jiang T, Tan L, Zhu X-C, et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2014;39(13):2949–2962. doi:10.1038/npp.2014.164
54. Jay TR, Miller CM, Cheng PJ, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–295. doi:10.1084/jem.20142322
55. Xu X, Du L, Jiang J, et al. Microglial TREM2 mitigates inflammatory responses and neuronal apoptosis in angiotensin II-induced hypertension in middle-aged mice. Front Aging Neurosci. 2021;13:716917. doi:10.3389/fnagi.2021.716917
56. Weber MD, Frank MG, Tracey KJ, et al. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 2015;35(1):316–324. doi:10.1523/JNEUROSCI.3561-14.2015
57. Zhang ST, Hu L, Jiang J, et al. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation. 2020;17(1). doi:10.1186/s12974-019-1673-3
58. Li Y, Shen XZ, Li L, et al. Brain transforming growth factor-β resists hypertension via regulating microglial activation. Stroke. 2017;48(9):2557–2564. doi:10.1161/STROKEAHA.117.017370
59. Jiang P, Zhang W-Y, Li H-D, et al. Stress and vitamin D: altered vitamin D metabolism in both the hippocampus and myocardium of chronic unpredictable mild stress exposed rats. Psychoneuroendocrinology. 2013;38(10):2091–2098. doi:10.1016/j.psyneuen.2013.03.017
60. Cui C, Xu P, Li G, et al. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: role of renin-angiotensin system. Redox Biol. 2019;26:101295. doi:10.1016/j.redox.2019.101295
61. Masson GS, Nair AR, Dange RB, et al. Toll-like receptor 4 promotes autonomic dysfunction, inflammation and microglia activation in the hypothalamic paraventricular nucleus: role of endoplasmic reticulum stress. PLoS One. 2015;10(3):e0122850. doi:10.1371/journal.pone.0122850
62. Lee H, Lee S, Cho IH, et al. Toll-like receptors: sensor molecules for detecting damage to the nervous system. Curr Protein Pept Sci. 2013;14(1):33–42. doi:10.2174/1389203711314010006
63. Biancardi VC, Stranahan AM, Krause EG, et al. Cross talk between AT 1 receptors and Toll-like receptor 4 in microglia contributes to angiotensin II-derived ROS production in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol. 2016;310(3):H404–H415. doi:10.1152/ajpheart.00247.2015
64. Mowry FE, Peaden SC, Stern JE, et al. TLR4 and AT1R mediate blood-brain barrier disruption, neuroinflammation, and autonomic dysfunction in spontaneously hypertensive rats. Pharmacol Res. 2021;174:105877. doi:10.1016/j.phrs.2021.105877
65. Li J, Zhao F, Wang Y, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14. doi:10.1186/s40168-016-0222-x
66. Yang T, Santisteban MM, Rodriguez V, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65(6):1331–1340. doi:10.1161/HYPERTENSIONAHA.115.05315
67. Richards EM, Li J, Stevens BR, et al. Gut microbiome and neuroinflammation in hypertension. Circ Res. 2022;130(3):401–417. doi:10.1161/CIRCRESAHA.121.319816
68. Sharma RK, Yang T, Oliveira AC, et al. Microglial cells impact gut microbiota and gut pathology in angiotensin II-induced hypertension. Circ Res. 2019;124(5):727–736. doi:10.1161/CIRCRESAHA.118.313882
69. Rosa DD, Dias MMS, Grześkowiak ŁM, et al. Milk kefir: nutritional, microbiological and health benefits. Nutr Res Rev. 2017;30(1):82–96. doi:10.1017/S0954422416000275
70. Friques AG, Arpini CM, Kalil IC, et al. Chronic administration of the probiotic kefir improves the endothelial function in spontaneously hypertensive rats. J Transl Med. 2015;13(1):1–16. doi:10.1186/s12967-015-0759-7
71. de Almeida Silva M, Mowry FE, Peaden SC, et al. Kefir ameliorates hypertension via gut–brain mechanisms in spontaneously hypertensive rats. J Nutr Biochem. 2020;77:108318. doi:10.1016/j.jnutbio.2019.108318
72. Masson GS, Nair AR, Silva Soares PP, et al. Aerobic training normalizes autonomic dysfunction, HMGB1 content, microglia activation and inflammation in hypothalamic paraventricular nucleus of SHR. Am J Physiol Heart Circ Physiol. 2015;309(7):H1115–H1122. doi:10.1152/ajpheart.00349.2015
73. Kiviniemi AM, Tulppo MP, Eskelinen JJ, et al. Cardiac autonomic function and high-intensity interval training in middle-age men. Med Sci Sports Exerc. 2014;46(10):1960–1967. doi:10.1249/MSS.0000000000000307
74. Rana I, Stebbing M, Kompa A, et al. Microglia activation in the hypothalamic PVN following myocardial infarction. Brain Res. 2010;1326:96–104. doi:10.1016/j.brainres.2010.02.028
75. Francis J, Chu Y, Johnson AK, et al. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol. 2004;286(6):H2264–71. doi:10.1152/ajpheart.01072.2003
76. Dworak M, Stebbing M, Kompa AR, et al. Attenuation of microglial and neuronal activation in the brain by ICV minocycline following myocardial infarction. Auton Neurosci. 2014;185:43–50. doi:10.1016/j.autneu.2014.03.007
77. Dworak M, Stebbing M, Kompa AR, et al. Sustained activation of microglia in the hypothalamic PVN following myocardial infarction. Auton Neurosci. 2012;169(2):70–76. doi:10.1016/j.autneu.2012.04.004
78. Banfi C, Ferrario S, De Vincenti O, et al. P2 receptors in human heart: upregulation of P2X6 in patients undergoing heart transplantation, interaction with TNFalpha and potential role in myocardial cell death. J Mol Cell Cardiol. 2005;39(6):929–939. doi:10.1016/j.yjmcc.2005.09.002
79. Zhou J, Tian G, Quan Y, et al. Inhibition of P2X7 purinergic receptor ameliorates cardiac fibrosis by suppressing NLRP3/IL-1 β pathway. Oxid Med Cell Longev. 2020;2020:7956274. doi:10.1155/2020/7956274
80. Zempo H, Sugita Y, Ogawa M, et al. A P2X7 receptor antagonist attenuates experimental autoimmune myocarditis via suppressed myocardial CD4+ T and macrophage infiltration and NADPH oxidase 2/4 expression in mice. Heart Vessels. 2015;30(4):527–533. doi:10.1007/s00380-014-0527-2
81. Du D, Jiang M, Liu M, et al. Microglial P2X(7) receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci Lett. 2015;587:22–28. doi:10.1016/j.neulet.2014.12.026
82. Miyake Y, Ishikawa E, Ishikawa T, et al. Self and nonself recognition through C-type lectin receptor, Mincle. Self Nonself. 2010;1(4):310–313. doi:10.4161/self.1.4.13736
83. Wang Y, Yin J, Wang C, et al. Microglial Mincle receptor in the PVN contributes to sympathetic hyperactivity in acute myocardial infarction rat. J Cell Mol Med. 2019;23(1):112–125. doi:10.1111/jcmm.13890
84. Wang Y, Hu H, Yin J, et al. TLR4 participates in sympathetic hyperactivity Post-MI in the PVN by regulating NF-kappaB pathway and ROS production. Redox Biol. 2019;24:101186. doi:10.1016/j.redox.2019.101186
85. Taguchi N, Nakayama S, Tanaka M. Single administration of soluble epoxide hydrolase inhibitor suppresses neuroinflammation and improves neuronal damage after cardiac arrest in mice. Neurosci Res. 2016;111:56–63. doi:10.1016/j.neures.2016.05.002
86. Frick T, Springe D, Grandgirard D, et al. An improved simple rat model for global cerebral ischaemia by induced cardiac arrest. Neurol Res. 2016;38(4):373–380. doi:10.1179/1743132815Y.0000000090
87. Yuan S, Zhang X, Bo Y, et al. The effects of electroacupuncture treatment on the postoperative cognitive function in aged rats with acute myocardial ischemia–reperfusion. Brain Res. 2014;1593:19–29. doi:10.1016/j.brainres.2014.10.005
88. Ghanbari A, Ghareghani M, Zibara K, et al. Light-emitting diode (LED) therapy improves occipital cortex damage by decreasing apoptosis and increasing BDNF-expressing cells in methanol-induced toxicity in rats. Biomed Pharmacother. 2017;89:1320–1330. doi:10.1016/j.biopha.2017.03.024
89. Lee HI, Lee S-W, Kim NG, et al. Low‐level light emitting diode (LED) therapy suppresses inflammasome‐mediated brain damage in experimental ischemic stroke. J Biophoton. 2017;10(11):1502–1513. doi:10.1002/jbio.201600244
90. Wang S, Luo Q, Chen H, et al. Light emitting diode therapy protects against myocardial ischemia/reperfusion injury through mitigating neuroinflammation. Oxid Med Cell Longev. 2020;2020:9343160. doi:10.1155/2020/9343160
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.