Back to Journals » Journal of Inflammation Research » Volume 17
Microglia in Ischemic Stroke: Pathogenesis Insights and Therapeutic Challenges
Authors Shui X, Chen J ![]() , Fu Z, Zhu H, Tao H, Li Z
, Fu Z, Zhu H, Tao H, Li Z ![]()
Received 20 February 2024
Accepted for publication 14 May 2024
Published 22 May 2024 Volume 2024:17 Pages 3335—3352
DOI https://doi.org/10.2147/JIR.S461795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xinyao Shui,1,* Jingsong Chen,2– 4,* Ziyue Fu,1 Haoyue Zhu,1 Hualin Tao,2– 4 Zhaoyinqian Li2– 4
1Clinical Medical College, Southwest Medical University, Luzhou, People’s Republic of China; 2Department of Laboratory Medicine, the Affiliated Hospital of Southwest Medical University, Luzhou, People’s Republic of China; 3Sichuan Province Engineering Technology Research Center of Molecular Diagnosis of Clinical Diseases, Luzhou, People’s Republic of China; 4Molecular Diagnosis of Clinical Diseases Key Laboratory of Luzhou, Luzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhaoyinqian Li, Department of Laboratory Medicine, the Affiliated Hospital of Southwest Medical University, No. 25, Taiping Street, Jiangyang District, Luzhou, Sichuan, 646000, People’s Republic of China, Email [email protected]
Abstract: Ischemic stroke is the most common type of stroke, which is the main cause of death and disability on a global scale. As the primary immune cells in the brain that are crucial for preserving homeostasis of the central nervous system microenvironment, microglia have been found to exhibit dual or even multiple effects at different stages of ischemic stroke. The anti-inflammatory polarization of microglia and release of neurotrophic factors may provide benefits by promoting neurological recovery at the lesion in the early phase after ischemic stroke. However, the pro-inflammatory polarization of microglia and secretion of inflammatory factors in the later phase of injury may exacerbate the ischemic lesion, suggesting the therapeutic potential of modulating the balance of microglial polarization to predispose them to anti-inflammatory transformation in ischemic stroke. Microglia-mediated signaling crosstalk with other cells may also be key to improving functional outcomes following ischemic stroke. Thus, this review provides an overview of microglial functions and responses under physiological and ischemic stroke conditions, including microglial activation, polarization, and interactions with other cells. We focus on approaches that promote anti-inflammatory polarization of microglia, inhibit microglial activation, and enhance beneficial cell-to-cell interactions. These targets may hold promise for the creation of innovative therapeutic strategies.
Keywords: microglia, ischemic stroke, phagocytosis, polarization, crosstalk, anti-inflammatory, therapeutic targets
Introduction
Stroke ranks as one of the top causes of death and disability globally.1 Over the past thirty years, there has been a 70% rise in the global incidence of stroke, a 43% increase in its mortality rate, and at the same time, a 32% increase in disability-adjusted life years.2 It is predicted that 12 million people will die from stroke by 2030.3 Stroke is divided into two types. Most strokes are essentially ischemic strokes due to blood clots or lumps blocking blood vessels, while a few strokes are hemorrhagic strokes caused by blood vessel rupture.4 The latter includes intracerebral hemorrhage and subarachnoid hemorrhage.5 Stroke has a significant impact on both physical and mental health, disrupting regular daily activities and leading to economic hardship for families and society. Numerous factors have been identified as controllable risk factors for stroke, including hyperlipidemia, alcohol and drug abuse, smoking, unhealthy diet, and diabetes.6
Even though ischemic stroke has a high incidence rate, the treatment options remain limited. Commonly used treatments include thrombolysis and surgical treatments. Thrombolysis includes intravenous thrombolysis (IVT),7 arterial thrombolysis (IAT),8 and bridging therapy (IVT combined with IAT).9 When it comes to thrombolytic drugs, the FDA only approves thrombolysis with tissue plasminogen activator. The surgical treatment for stroke is mechanical thrombectomy.10 Further research is being conducted on treatments such as antiplatelet therapy, neuroprotective therapy, stem cell therapy, and rehabilitation therapy, yet their therapeutic effects remain limited.6 The significance of inflammation in stroke has been emphasized more and more in recent studies. Moreover, the promotion of effective neurogenesis is increasingly being recognized as a crucial therapeutic approach for stroke treatment. The response of microglia post-stroke is intricate and closely linked to the development and prognosis of the stroke. Microglia, functioning as resident immune cells, rapidly activate after an ischemic injury, displaying both pro-inflammatory and anti-inflammatory characteristics and regulating neuroinflammation through polarization and secretion of various chemokines.11 Microglia-targeted therapy for ischemic stroke has significant potential due to their ability to clear away debris and secrete deleterious factors and trophic cytokines to support tissue regeneration and remodeling.12 Examining the basic roles of microglia in both physiological and ischemic brains, along with their communication with neurons and other glial cells, this review summarizes potential therapeutic targets for ischemic stroke that have been linked to them in the last decade. The article highlights potential therapeutic targets for ischemic stroke associated with microglia over the past ten years.
Physiological Functions of Microglia
Microglia, accounting for up to 10% of the cells in the brain,13 play a crucial role in protecting the central nervous system (CNS) from damage.14 Controversy remains regarding the exact origin of microglial progenitors, which are usually thought to originate from primitive yolk sac macrophages and infiltrate the CNS in the early stages of development.15 Most human microglia are situated in the gray matter, and they typically have a morphology characterized by cells with numerous short, fine processes.16 Microglia also express a number of specific and non-specific molecular markers that manifest different functions in different physio-pathological processes, respectively (Table 1).17 Microglia are typically referred to as “resting” in physiological conditions, whereas their reactive morphology under pathological conditions is referred to as “activated”. However, this terminology has been pointed out to be imprecise, since microglia are constantly responding to changes in their CNS environment (the adjacent tissue cells and the various factors they secrete) in different ways (by continually retracting and extending their ramified processes), even in normal physiological circumstances.18 Microglia also play a unique “sentinel” surveillance role,13 monitoring the quantity of synapses, which are neuronal structures that rapidly transmit information between neurons and transform this information during transmission.19 They also promote synapse formation, prune synapses, phagocytose cellular synapses, control neuronal firings, and tidy away debris, thereby preserving homeostasis in the CNS.20,21 So there is no sudden shift from “resting” to “activated” in response to disease, injury, or other pathological conditions. Therefore, many scholars prefer to rename resting microglia as “homeostatic” and to use “reactive to” or “responding to” (ie, the context) to describe microglial activation.22 Despite the variety of names, the traditional terms “resting” and “activated” are still widely used.
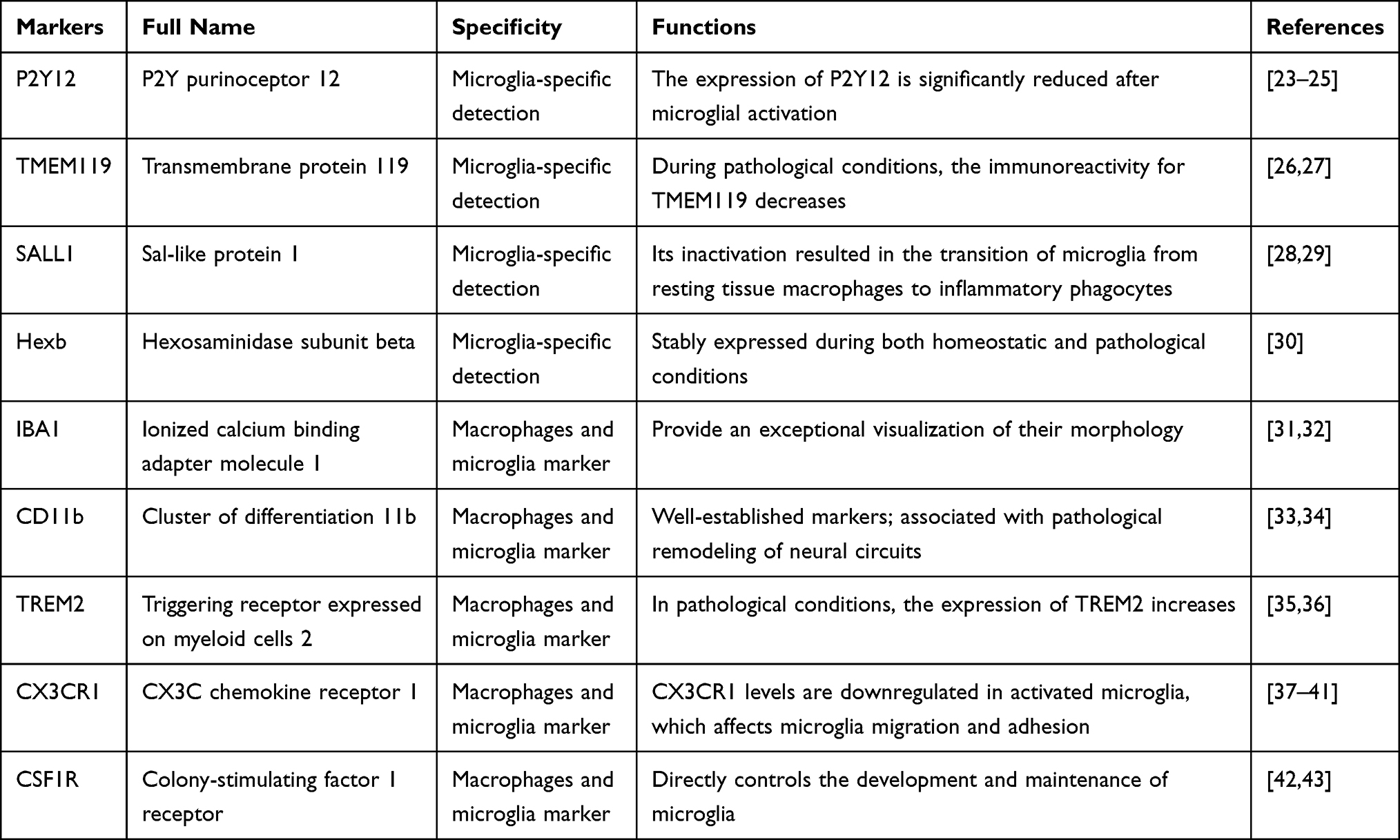 |
Table 1 Major Markers for Specific Identification of Microglia |
Microglia and Phagocytosis
The primary role of microglia is the destruction of entire cells or cellular substructures, particularly synapses.44 When brain tissue is infected or brain cells perish, the parenchyma is left with dead cells and aggregated proteins. Failure to remove these substances can lead to tissue damage as they release reactive oxygen species (ROS) and inflammatory mediators.45 In cases of brain tissue infection or cell death, the parenchyma may accumulate dead cells and aggregated proteins. Therefore, under physiological conditions, microglial phagocytosis is the main way of getting rid of unwanted materials in the parenchyma.46 During development, nucleotides (adenosine triphosphate (ATP), adenosine diphosphate (ADP), or uridine triphosphate (UTP)) released by neurons are involved in synaptic pruning through activation of P2Y12 receptors on microglia. These receptors are upregulated, particularly during ischemic brain injury.47,48 During pathological conditions, neurons also release nucleotides that act as “find-me” signals, directing microglia to the site of damage and promoting their activation.46 Phosphatidylserine (PS), the primary phospholipid in the inner leaflet of the neuronal plasma membrane,49 acts as a crucial “eat-me” signal. Nevertheless, it remains unclear whether the presence of PS alone is adequate to trigger phagocytosis. During cell death/dying, the irreversible translocation of PS to the cell surface can be caused by ATP depletion, intracellular calcium accumulation, DNA damage, and oxidative stress.50,51 This is recognized either directly or indirectly (via bridging proteins that interact with PS) by microglial receptors, which then induce its phagocytosis.52 Several receptors directly recognize PS, including brain angiogenesis inhibitor I, Stabilin2,53 members of the T-cell immunoglobulin and mucin family,52,53 and the phosphatidylserine receptor. Bridging proteins, such as growth arrest-specific gene 6 and milk fat globule-EGF factor 8, bind to PS to facilitate indirect recognition.54 Interacting with low-density lipoprotein receptor-related protein 1 on microglia, cell surface calreticulin serves as an “eat-me” signal to initiate phagocytosis.50,55 Microglia are possibly the sole cells in the brain that express various crucial complement components, including complement component C1q (C1q), complement receptor 3 (CR3), and CR5.56 By binding to cell debris or cells that are meant to be phagocytosed, complement can enhance phagocytosis. C1q, for instance, induces phagocytosis by binding to calreticulin or cell-surface PS to enhance signaling recognition, or directly to the functional phagocytosis CR3 on the surface of microglia.57
The Role of Microglia in Ischemic Brain
Ischemic strokes are typically divided into three stages: the acute phase (within 3 days), the subacute phase (7–10 days), and the chronic phase (weeks to months). The ischemic-damaged brain tissue is comprised of two main regions: the ischemic core and the penumbra. With minimal blood flow in the ischemic core leading to instant neuronal death, the penumbra, accounting for half of the lesion volume, is a hypoperfused area on the periphery of the core.58 Over time, this area gradually transforms into irreversibly damaged tissue, becoming the ischemic core. However, with rapid reperfusion, the penumbra can be salvaged and normal function can be restored.59 We have summarized the manifestations of microglia in the ischemic brain into three main areas: activation, polarization, and crosstalk with other cells (Figure 1).
 |
Figure 1 Microglial behavior after ischemia and their crosstalk with other cells. After ischemic stroke, complement factors are released at the site of the pathology, directing microglia to the lesioned or damaged area. Damage-associated molecular pattern molecules (DAMPs) are released from damaged or dying cells, which activate microglia. In addition to morphological changes, activated microglia polarize in response to different factors towards an anti-inflammatory phenotype that secretes neurotrophic factors and acts as a neuroprotectant, and a pro-inflammatory phenotype that releases deleterious factors that not only injure oligodendrocytes, but also induce A1-responsive astrocytes. At the same time, signals from these cells can, in turn, modulate microglial responses. Abbreviations: TNF, tumor necrosis factor; IL, interleukin; NO, nitric oxide; ROS, reactive oxygen species; MMPs, matrix metalloproteinases; IFN, interferon; iNOS, inducible nitric oxide synthase; TGF, transforming growth factor; IGF, insulin-like growth factor; NGF, nerve growth factor; VEGF, vascular endothelial growth factor; GLT, glutamate transporter; GDNF, glial-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; BDNF, brain-derived neurotrophic factor; NT, neurotrophins; C, complement; HMGB1, damage-associated molecular pattern molecules; ATP, adenosine triphosphate; PRX, peroxiredoxin; LPS, lipopolysaccharide; IRF, interferon regulatory factor; Glu, glutamate; UTP, uridine triphosphate; HSPs, heat shock proteins. |
Microglia and Activation
When cerebral ischemia occurs, neurons in the vicinity of the affected arteries tend to perish rapidly, accompanied by cerebral oedema, blood-brain barrier destruction, and neuroinflammation.60 When cells are under stress and are dying, they begin to emit molecules of danger that have an immediate effect on microglia including ATP, UTP, and damage-associated molecular pattern molecules (DAMPs).61 Since complement factors such as C5a are released at pathological sites, the motility of microglia can be enhanced and directed to the diseased or damaged area through G-protein-dependent pathways and rearrangement of the actin cytoskeleton.62 Purinergic receptors P2X7 and P2Y12 are expressed in microglia and act as key sensors of brain injury, triggering microglia activation upon stimulation.63 They can both be activated by ATP, with activation of the P2X7 receptor triggering microglial proliferation,64 superoxide production,65 and release of interleukin 1β (IL-1β)66 and tumor necrosis factor (TNF)-α.67 Upon activation of the P2Y12 receptor, microglia undergo process extension and then migrate towards the stimulus by interacting with integrin-β1.25,68 Microglia possess Toll-like receptors (TLRs), a group of transmembrane proteins that are essential for detecting and defending against microbials. They can be stimulated by endogenous ligands and agonists, such as high-mobility group box 1 (HMGB1) protein,69 fibronectin, heparan sulfate, galectin-3,70 peroxiredoxin (PRX) family proteins,71 heat shock proteins (HSPs),72 and hyaluronic acid. This triggers the activation of microglia, which increases the expression of several pro-inflammatory genes.73 Prostaglandin E2, an inflammatory mediator, interacts with the G protein-coupled receptor EP2 expressed on microglia. Activation of the EP2 receptor exacerbates the induction of inflammatory mediators such as cyclooxygenase-2, IL-6, TNF-α, and IL-1β.74 The triggering receptor expressed on myeloid cell 2 (TREM2) is an immune-related receptor on the surface of microglia. One of its ligands, HSP60, stimulates TREM2 to activate microglial phagocytosis, and the absence of TREM2 promotes inflammation.75,76 In addition, zinc released from neurons after injury has been shown to trigger microglial activation by sequentially activating nicotinamide adenine dinucleotide phosphate oxidase, poly (ADP-ribose) polymerase-1 (PARP-1), and nuclear factor kappa B (NF-κB).77 However, when microglia become over-activated, it can result in neuronal dysfunction, oxidative stress, and neuroinflammation. This is caused by the excessive production of various cytotoxic factors including ROS, IL-1β, TNF-α, and nitric oxide (NO).78,79 Microglial activation is the initial stage of the inflammatory response triggered by ischemic brain injury, followed by infiltration of various immune cells, such as T-cells, natural killer cells, and neutrophils, all of which exacerbate the inflammatory process.80,81
There is a connection between the morphology and function of microglia as well. Ramified microglia are associated with a homeostatic state, while activated microglia have rounded cell bodies and usually have fewer and shorter processes.22 After stroke, microglia undergo four distinct morphological stages that signify a rising level of activation: ramified, intermediate, amoeboid, and round.82 In the uninjured area, microglia show a ramified pattern, while in the peri-infarct region, intermediate and amoeboid cells can be seen.83 When microglia take on a rounded form, it is a sign of a highly activated state, usually found in the core of an infarct.84 In the acute phase of ischemic stroke, neurons in the ischemic core region experience a lack of glucose and hypoxia, causing rapid activation of microglia within a few minutes.85 At 24 hours, activated microglia can be found in the ischemic core and the boundary region, with their presence increasing in the ischemic core after 72 hours.86 Simultaneously, activated microglia release various inflammatory factors that play a role in a robust inflammatory response.87 Microglial activation reaches its peak on Day 2 or 3 and lasts for several weeks thereafter.60 In addition to the infarct site, activated microglia can be observed in areas further away during the chronic phase.86 In the ischemic core, microglia are primarily activated due to excitotoxic cascade signals,88 while in the peri-infarct regions, their activation is initiated by DAMPs.89 Activated microglia remain in and around the area of the residual infarct for a period of 30 days.90
Microglia and Polarization
Microglia not only undergo morphological changes, but also exhibit different patterns of gene expression in different contexts, species, sex, time, and space, as well as genetic transcriptional profiles, leading to differentiation of cells into different phenotypic groups (including motility, morphology, and ultrastructure).22 Many current studies have used integrative analyses of gene and protein expression, single-cell technologies, and multi-omics to propose new nomenclatures based on the different microglia states observed in various species and models. For example, the disease-associated microglia (DAMs) in Alzheimer’s disease (AD) pathology models;23 microglial neurodegenerative phenotype (MGnD) in several disease models;91 activated response microglia (ARMs) and interferon-responsive microglia (IRMs) in an AD pathology mouse model;92 human AD microglia (HAMs).93 For more information on microglia nomenclature, we refer the reader to the distinguished review “Microglia States and Nomenclature: A Field at Its Crossroads”. The application of this nomenclature is somewhat controversial, and there is insufficient evidence to suggest that “DAM” applies to microglia in all cases of injury. Previous studies have classified microglia into anti-inflammatory and pro-inflammatory phenotypes based primarily on their responses, including pro- and anti-inflammatory characteristics of individual cells and small populations in the vicinity.94 Upon stimulation of TLR or ATP receptors by DAMPs, lipopolysaccharide (LPS) and interferon (IFN)-γ can induce activated microglia to the pro-inflammatory phenotype, resulting in the secretion of numerous pro-inflammatory mediators.95,96 In contrast, fractalkine 16 (CXCL16), IL-4, and IL-13 can trigger a shift in microglia towards an anti-inflammatory phenotype, leading to the secretion of anti-inflammatory cytokines that inhibit neuroinflammation mediated by the pro-inflammatory phenotype of microglia.96–98 The molecular processes underlying microglial polarization are still unclear, though.99 Early in ischemic stroke, microglia and recruited macrophages exhibit an anti-inflammatory phenotype that protects neurons from oxygen-glucose deprivation (OGD).100,101 However, as the stroke progresses, these cells gradually change into a pro-inflammatory phenotype in the peri-infarct regions, which exacerbates the damage that caused by OGD to neurons.100 Hence, it is important to consider the equilibrium between proinflammatory and anti-inflammatory reactions in order to predict the outcome of a stroke.102
Microglia and Secretion
It is thought that microglia, primarily of the pro-inflammatory phenotype, contribute to the advancement or escalation of neuronal degeneration and inflammation in numerous brain diseases. This is a result of their generation of damaging elements such as inflammatory cytokines, NO, and superoxide anions.80 First, in response to an inflammatory stimulus, pro-inflammatory phenotype microglia secrete inflammatory molecules like inducible nitric oxide synthase (iNOS), IL-1β, TNF-α, and IL-6 to trigger a robust inflammatory response.103–105 Nonetheless, data is accumulating that microglial cells, predominantly of the anti-inflammatory phenotype, are able to protect nerve tissue from neuroinflammation by releasing substances that have anti-inflammatory properties.106 For example, CD11c+ microglia appear after behavioral pain hypersensitivity following nerve injury and express insulin-like growth factor 1 (IGF-1), which contributes to pain recovery.107 Two anti-inflammatory molecules, brain-derived neurotrophic factor (BDNF) and amylase 1, can be released by microglia when they are activated by astrocytes during tissue healing.108 In the normal brain, microglial subtypes express neurotrophins from the nerve growth factor (NGF) gene family, which play a role in promoting the development and normal function of neurons and glia. Additionally, they express neurotrophin 3 (NT-3), which encourages phagocytic function and microglial growth.109 Extracellular vesicles (EVs) secreted by microglia in the ischemic cerebral environment are enriched with transforming growth factor β1 (TGF-β1). This molecule stimulates the polarization of microglia into anti-inflammatory phenotypes, thereby aiding in the anti-inflammatory response.110 Furthermore, in response to inflammatory stimuli, microglia express higher levels of neuregulin-1, glial cell-derived neurotrophic factor (GDNF), and their receptors.111
Microglial Crosstalk with Other Cells
Microglia are essential for interactions with different types of cells in the CNS and are in charge of a number of developmental and functional activities, such as the removal of dead neurons and synaptic pruning.112 Acute damage and recovery after stroke can be impacted by the dynamic interactions that occur between microglia and neurons, as well as between other glial cells.113
Microglia and Neurons
Following ischemic stroke, neurons are severely damaged by the effects of ischemia and hypoxia.114 When neurons run out of energy, lactic acid builds up and causes acidosis. By raising H+ concentrations and speeding up the conversion of superoxide anion (O2−) to hydrogen peroxide (H2O2) or the more reactive hydroperoxyl radical (HO2), an acidic environment encourages a pro-oxidant effect.115 ROS are naturally occurring byproducts of oxygen metabolism, including peroxides, free radicals, and oxygen ions.116 One of the main causes of neuronal malfunction and death is excess ROS, which also causes oxidative stress by oxidizing proteins, DNA, and RNA. Lipid peroxidation is another impact of excess ROS.117 As the primary excitatory transmitter in neuron-to-neuron communication, glutamate quickly rises in the ischemic brain as soon as ischemia occurs, with activated microglia being a source.118,119 Ionotropic glutamate receptor overactivation causes excitotoxicity, resulting in the death of neurons.120 By releasing “On” and “Off” signals, neurons can regulate microglial activity based on the normal or pathological conditions.121 Interactions between neurons and microglia involve a range of ligands and receptors.122 For example, CX3CL1 is a neuronally expressed chemokine, and its G-protein-coupled receptor, CX3CR, is predominantly expressed on microglia.123,124 In reaction to pro-inflammatory stimuli, CX3CL1 is secreted and its levels are increased, contributing to neuron-microglia communication. Mice that lack CX3CL1 show smaller infarct sizes and experience better functional outcomes.125 The pyrin domain-containing 3 (NLRP3), which has recently attracted the attention of many researchers, has the potential to induce inflammatory responses and trigger a number of inflammatory pathways.126 Research indicates that CX3CR1 adversely regulates NLRP3 signaling.127 Neurons have the capacity to affect microglial functioning, and a recent study revealed that lipocalin-2 secretion from damaged neurons can cause microglia to shift towards an anti-inflammatory phenotype. In addition, Lipocalin-2 stimulates BBB breakdown, white matter damage, neuronal death, and neutrophil infiltration. It also intensifies inflammation following a stroke.128 Furthermore, ischemia-induced damaged neurons can trigger the release of IL-4, which may then induce microglia to express the IL-4 receptor more strongly. Subsequently, this triggers the transformation of microglia into an anti-inflammatory phenotype.129 Moreover, the role of IGF-1 derived from microglia in maintaining neuronal life has been demonstrated. By encouraging angiogenesis and enhancing cerebrovascular function in ischemic regions, the IGF-1/insulin-like growth factor 1 receptor (IGF1R) axis mitigates ischemia-induced harm by supporting cerebral angiogenesis and neurogenesis.130
Microglia and Astrocytes
Within the CNS, astrocytes are not only the most abundant cell type, but also an integral part of the brain’s innate immune system.131 It has been shown that activated astrocytes exhibit two polarization states: a neurotoxic or pro-inflammatory phenotype (A1) and a neuroprotective or anti-inflammatory phenotype (A2).132 (Similar to the microglia paradigm) A1 astrocytes are considered deleterious due to the up-regulation of synaptically detrimental genes and secretion of toxins, whereas A2 astrocytes up-regulate neurotrophic or anti-inflammatory genes and promote reparative functions, suggesting that they are protective.132,133 Recently, astrocyte subtypes with different expression patterns have been identified using single-nuclei and single-cell studies in different species, diseases and injury models, such as Alzheimer’s disease,134 amyotrophic lateral sclerosis,135 HIV infection,136 and spinal cord injury.137 The emergence of new typing patterns further illustrates the complexity of astrocyte expression in disease development. Microglia are the first responders to injury, and they can trigger astrocyte activation. It has been established that microglial activation is able to induce A1 reactive astrocytes both in vitro and in vivo conditions. This is due to the release of three cytokines: IL-1α, TNF-α, and C1q.132 A1 astrocytes lose their phagocytic function and fail to support neurite growth and synaptogenesis. Instead, they trigger the death of neurons and oligodendrocytes. In contrast, microglia induce the A2 phenotype in astrocytes by downregulating the astrocytic P2Y1 purinergic receptors and forming an astrocytic scar for neuroprotective and repair functions.138 By inducing intracellular signal transduction in astrocytes, activated microglia secrete various pro-inflammatory mediators that either directly initiate or promote astrocytic responses. These mediators include NF-κB, signal transducer and activator of transcription (STATs), and mitogen-activated protein kinase pathways.139 These responses include the formation of scars to exert a protective effect or the release of cytokines to exacerbate inflammatory responses.140 Further activation of distant microglia by astrocytes results in heightened secretion of specific inflammatory cytokines.141 Furthermore, astrocyte-derived IL-33 signals primarily to microglia in physiological circumstances and encourages them to engulf synapses.142 Pathologically, by increasing the production of heme-oxygenase (HO)-1 and lowering intracellular ROS levels in microglia, astrocytes can further regulate the inflammatory response of microglia, thereby preventing excessive inflammatory responses in the brain.143 The extracellular release of C3 is initiated by the activation of the NF-κB pathway, an inflammatory pathway in astrocytes. C3 then interacts with microglial C3aR to facilitate microglial phagocytosis.144
Microglia and Oligodendrocytes
Oligodendrocytes, which form the myelin sheath in the CNS, are particularly susceptible to ischemia.145 After merely 30 minutes of ischemia, there was obvious swelling of oligodendrocytes. By the time 3 hours had passed, a large number of oligodendrocytes had been fatally damaged, occurring before the neurons in the ischemic region met their demise.146 Various mechanisms have been identified as potentially causing harm to oligodendrocytes in ischemic conditions, including a deficiency of trophic factors, oxidative stress, and activation of apoptotic pathways.147 Following hypoxic injury, microglia produce inflammatory cytokines such as TNF-α and IL-1β via the mitogen-activated protein kinase signaling pathway, leading to oligodendrocyte loss.148 In addition, the expression of N-methyl D-aspartate receptor subunits on microglia after hypoxia can lead to oligodendrocyte death by mediating nitric oxide production through the NF-κB signaling pathway.149 Microglia-generated ROS can be damaging to both neurons and oligodendrocytes.150 However, evidence has come to light that microglia have a beneficial effect on oligodendrocytes/oligodendrocyte pro-genitor cells, with the anti-inflammatory phenotype polarization of microglia being the primary source of its protective function. This can drive the differentiation of oligodendrocytes and promote myelin regeneration during myelin regeneration after stroke.151 The pro-inflammatory phenotype of microglia exacerbates oxygen glucose deprivation-induced oligodendrocyte death.152
Treatment Targets for Microglia in Ischemic Stroke
A wave of studies on microglia has been conducted in the last ten years, revealing possible targets for treatment. Overall, we divide them into three categories: adjusting the polarization of microglia to promote anti-inflammatory effects, inhibiting their activation, and controlling how other cells interact with microglia (Table 2).
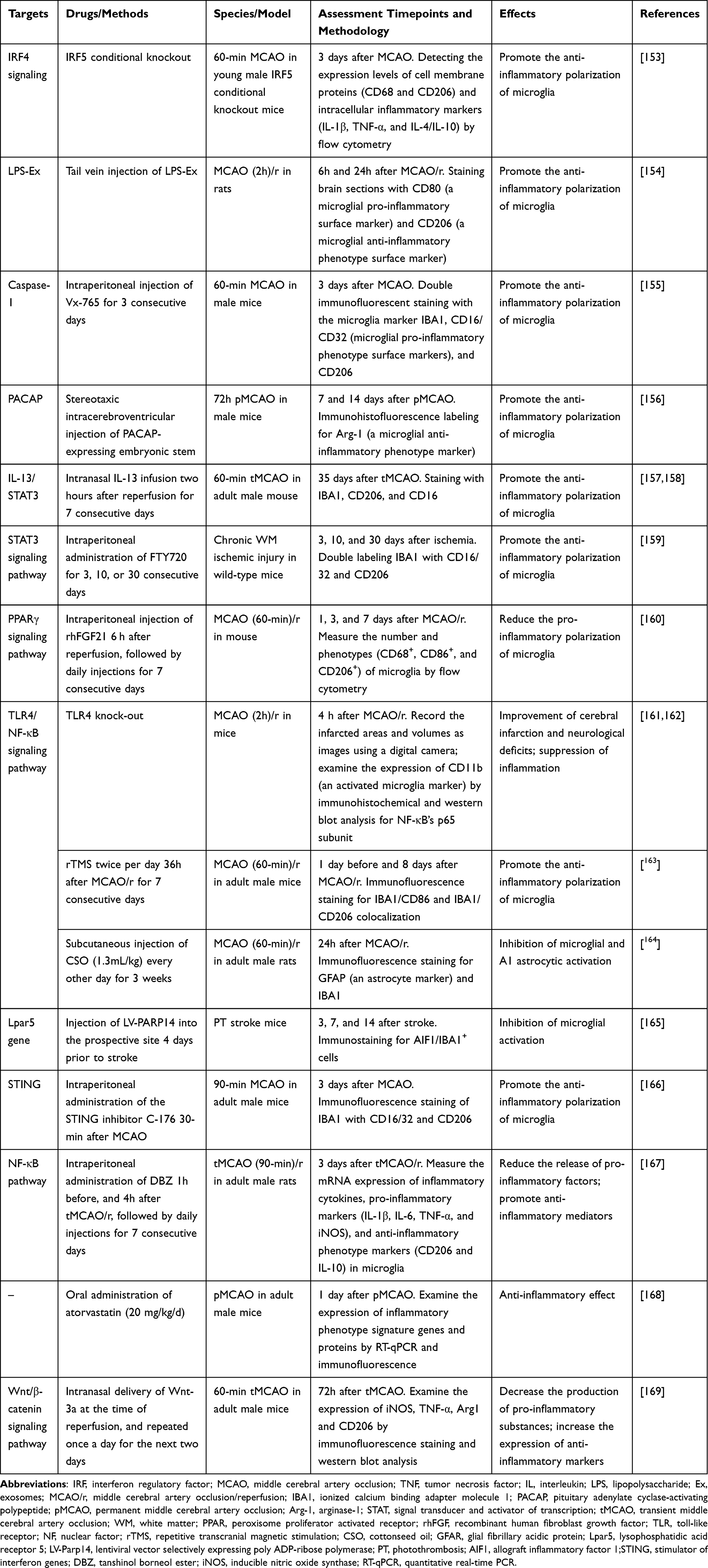 |
Table 2 Therapeutic Targets for Ischemic Stroke Associated with Microglia |
Promoting Anti-Inflammatory Microglial Polarization
Since microglia show a dynamic response to ischemic injury,100 it has been proposed that inhibiting the pro-inflammatory phenotype may be a plausible therapeutic strategy for cerebral ischemia. Different inducers have been successfully used in in vitro experiments to analyze and regulate the polarization of microglia. Tanshinol borneol ester (DBZ) is a newly developed synthetic compound that has anti-inflammatory and anti-atherosclerotic characteristics. Research has demonstrated that in both LPS-stimulated BV2 cells and mouse primary microglia cells, NF-κB activity is dramatically inhibited by DBZ, which also reduces the production of pro-inflammatory molecules and increases the expression of mediators of anti-inflammatory phenotypes.167 A series of studies on mice with permanent middle cerebral artery occlusion (pMCAO) by Zhang et al discovered that atorvastatin had a positive effect on the defects in sensorimotor function, as well as reducing microglia-induced neuroinflammation through the suppression of proinflammatory polarization of microglia in the peri-infarct cortex.168 What’s more, through the peroxisome proliferator-activated receptor γ (PPARγ) signaling pathway, ginkgetin and recombinant human fibroblast growth factor 21 (rhFGF21) therapies have been shown to enhance the anti-inflammatory polarization of microglia, which ultimately leads to the suppression of neuroinflammation and the enhancement of neurological function recovery in individuals suffering from ischemic stroke.160,170 In a mouse model of acute ischemic stroke (AIS), Chuanzhitongluo (CZTL), an ancient Chinese medicine mixture, was found to attenuate the inflammatory response by blocking the activation of the NLRP3 inflammasome and promoting a shift of microglia from a pro-inflammatory phenotype to an anti-inflammatory one.171 Research utilizing the transient middle cerebral artery occlusion mouse model (tMCAO) has revealed that IL-13 can enhance the long-term prognosis of ischemic stroke by inducing microglia/macrophages to adopt an anti-inflammatory phenotype, possibly through the inhibition of STAT3 phosphorylation.157,158 The process of microglia/macrophage polarization towards the anti-inflammatory phenotype in cerebral ischemia/reperfusion (I/R) damage has also been demonstrated to be regulated by the janus-activated kinase (JAK2)/STAT3 signaling pathway.158 The study by Qin et al identified Fingolimod (FTY720) as an immune modulator that reduces microglia-mediated neuroinflammation after ischemia and uses the STAT3 pathway to polarize microglia into an anti-inflammatory state, stimulating the development of oligodendrocytes.159
In response to ischemic injury, the body spontaneously generates other anti-inflammatory and pro-inflammatory responses associated with microglia. These responses include the regulation of gene transcription and the production of various functional proteins and molecules. Our goal is to identify these regulators and guide them in a favorable direction to enhance endogenous brain repair. The interferon regulatory factor (IRF) family, particularly the IRF5-IRF4 regulatory axis, has recently been found to be significantly associated with neuroinflammation and the polarization of microglia following cerebral ischemia. Abdullah et al found that IRF4 suppresses inflammation and encourages the anti-inflammatory polarization of microglia, in contrast to IRF5, which induces pro-inflammatory polarization.172 An increasing amount of research has demonstrated that the impact of microRNAs (miRs) on the polarization or activation of microglia in CNS disorders is growing. For example, miR-124 and miR-Let7a have roles in regulating microglia polarization towards the anti-inflammatory phenotype.173,174 MiR-155 has been identified as a pro-inflammatory microRNA. The absence of miR-155 diminishes microglial polarization to the pro-inflammatory phenotype.175 Moreover, exosomes (Ex), which are released by macrophages, have been demonstrated in many studies to regulate inflammation by transitioning gene transcription.176 In contrast, exosomes produced by the LPS-stimulated macrophage RAW264.7 cell line (LPS-Ex) contained higher levels of anti-inflammatory factors and genes (miR-212, miR-21-3p, or miR-21#, and miR-126-5p),177–179 which promoted the polarization of microglial cells from a pro-inflammatory to an anti-inflammatory phenotype and exerted a more potent and neuroprotective effect.154
Knockout models or inhibitors of different inflammatory proteins, signaling receptors, and cytokines generated from microglia may be fully neuroprotective in ischemic brain injury.102 In the brain, innate immune cells (astrocytes, microglia, and oligodendrocytes) are the main sources of colony-stimulating factor 1 (CSF-1). Many CSF-1R inhibitors have been created and are widely used to deplete microglia, such as Dasatinib, PLX3397, and PLX5622.180 In an experiment using tMCAO (30 min) mice as models, investigators assessed the immunomodulatory effects of long-term administration of PLX5622 at different time periods after ischemia through in vivo multimodal imaging. They found that CSF-1R inhibition transiently reduced neuroinflammation within the infarct.181 Recently, continuous CSF-1R inhibition before and after injury has been found in other disease models (tMCAO and pMCAO rodent models, TBI mouse model) to instead exacerbate inflammation and neurological deficits during the first few days of the injury, highlighting the anti-inflammatory role of microglia in the early stage of brain damage.138,182,183 It is not yet clear when inhibition of microglia is most effective, and research into the optimal therapeutic window may become critical for this targeted therapy. In the wounded CNS, microglia selectively express the calcium-activated potassium channel K(Ca)3.1. This channel’s activity is linked to the activation of microglia, which promotes inflammation. By using Senicapoc, a K(Ca)3.1 inhibitor, it could lead to modulation of stroke outcome by interfering with the inflammatory cascade that occurs after I/R.184 Following middle cerebral artery occlusion (MCAO), the stimulator of interferon genes (STING) is activated by mitochondrial DNA (mtDNA) and mediates microglia polarization towards a pro-inflammatory phenotype through IRF3/NF-κB signaling. In a recent study, I/R-induced neuronal injury, edema, and cerebral infarction were reduced by intraperitoneal injection of the specific STING inhibitor C-176 into a mouse model of MCAO.166 Furthermore, after ischemia, there is an increase in TLR4 expression on microglial membranes, which activates NF-κB and leads to a pro-inflammatory response in microglia.185 The neuroprotective effects of TLR4 knock-out mice in focal cerebral ischemia further support the possibility that we may be able to offer a potential treatment strategy for ischemic stroke by altering microglial phenotypes through the TLR4/NF-κB signaling pathway.161,162 In addition, Vx-765, a small-molecule caspase-1 inhibitor, safeguards against MCAO injury and reduces microglia-induced neuroinflammation mainly by altering microglia polarization from a pro-inflammatory phenotype to an anti-inflammatory phenotype.155
There are a number of other manipulations in experiments that have also been suggested to be connected to the anti-inflammatory effects of microglia, and thus, may hold therapeutic promise. In a study, investigators implanted stem cells that produced pituitary adenylate cyclase-activating polypeptide (PACAP) into the brains of mice. After seven days of ischemia, the data demonstrated that this caused microglia to polarize towards an anti-inflammatory phenotype.156 What’s more, it has recently been discovered that cerebral I/R injury-induced neuronal pyroptosis and locomotor impairments can be lessened by the non-invasive neuromodulatory method of repetitive transcranial magnetic stimulation (rTMS). This is achieved by restraining the pro-inflammatory activation and promoting the anti-inflammatory activation of microglia in the peri-infarcted region.163
Inhibition of Microglia Activation
Impeding the activation of microglia after stroke can reduce neuroinflammation. When human umbilical cord blood mononuclear cells are administered systemically after a MCAO in rats, it leads to a reduction in activated microglia and results in a decrease in the infarct size.186 Blocking the cellular P2Y12 receptors could impede microglial activation, resulting in a decrease in proinflammatory cytokine levels. This suggests that P2Y12 inhibitors have additional neuroprotective and anti-inflammatory benefits against ischemic stroke.187 According to Ying et al’s study, photothrombotic (PT) stroke mice have significantly elevated levels of PARP14 (poly (ADP-ribose) polymerase family, member 14) in the proximity of the infarct zone. Overexpression of PARP14 inhibits the transcription of the lysophosphatidic acid receptor 5 (Lpar5) gene, thus inhibiting microglial activation post-stroke.165 Other inhibitors of microglial activation, such as minocycline, ginsenoside Rd, and calycosin, have shown possible beneficial effects.188–190 Researchers combined the histone deacetylase inhibitor MS-275 and resveratrol in the pMCAO mouse model and found that it had a primary direct effect on inhibiting microglia and macrophage activation.191 MiRs can also affect microglial activation, in addition to their involvement in microglial polarization in neurological diseases. For instance, overexpression of let-7c-5p, a highly conserved miRNA, was observed to inhibit microglial activation in mice with experimental stroke, correlating with a decrease in infarction volume and improved neurological outcomes.192 As numerous signaling events are linked to microglial activity during ischemia, it is difficult to fully deactivate microglial activation.141
Regulating the Interaction Between Microglia and Other Cells
Controlling the exchange of information between microglia and other cells more precisely could open up possibilities for new treatments for stroke. Microglia express CX3CR1 as described previously. A deficiency in CX3CR1 has been shown to dysregulate microglial responses in three distinct in vivo models, leading to neurotoxicity. Consequently, enhancing CX3CR1 signaling could serve to protect against microglial neurotoxicity.193 Furthermore, the collaboration between microglia and astrocytes is thought to be a part of the process of forming a glial scar after stroke. In studies conducted in vivo and in vitro, treatment with anti-inflammatory microglial small EVs was observed to inhibit astrocyte proliferation, leading to a decrease in glial scar formation and an improvement in recovery after stroke.110,194 Yun et al reported that NLY01, a potent glucagon-like peptide-1 receptor agonist, can protect neurons by directly blocking microglia-mediated astrocyte transition into the neurotoxic A1 phenotype.195 What’s more, it has been demonstrated that cottonseed oil (CSO) guards against peripheral tissue damage, such as inflammatory bowel disease, through its anti-inflammatory effects.196 To investigate the anti-inflammatory effects of CSOs in stroke, it has recently been discovered that the use of CSO can reduce the severity of ischemic stroke damage by preventing microglial and astrocyte activation and inflammation. This is associated with a decreased activation of the neurotoxic A1 phenotype astrocytes and an inhibition of the TLR4/NF-κB pathway.164 In addition, Wnt-3a, a newly discovered Wnt protein, has been found to specifically enhance the Wnt/β-catenin signaling pathway, influencing cell proliferation and apoptosis. Results from research conducted using the tMCAO mouse model revealed that intranasal Wnt-3a could reduce the toxic effects of microglia/macrophages and astrocytes in ischemic brain injury.169
Conclusion
With the prevalence of ischemic stroke across the world, researchers are dedicating more resources to investigating treatments for stroke, as the major available treatments remain limited. This review gives a quick rundown of the physiological roles played by microglia and how they appear in the ischemic brain, including phagocytosis by microglia, microglial activation and polarization processes, and crosstalk with other glial cells. We summarize the therapeutic targets of microglia to prevent deterioration of the affected area and attenuate the inflammatory response by intervening at different stages of stroke, ultimately improving the prognosis and providing neuroprotection against stroke. With the increasing identification of substances and signaling pathways implicated in the intricate process of microglial activation and polarization following a stroke, it highlights the potential of microglial-targeted therapy. However, the modulation of microglial function following a stroke remains a topic of debate, and many therapies targeting microglia, such as immune regulatory strategies and anti-inflammatory treatments, have only been successful in animal models, such as mice. Because of the many differences in the physiological systems of rodents and humans, and the controversy over whether the microglial cell state found in mice exists in humans, it is essential to study the differences in the types and functions of microglia in humans and animal models. Because of the many differences in the physiological systems of rodents and humans, and the controversy over whether the microglial cell state found in mice exists in humans, further investigation is required before attempting to utilize microglia for clinical applications.
Funding
This study was funded by the College Students’ Innovative Entrepreneurial Training Plan Program (2022183).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Owolabi MO, Thrift AG, Mahal A, et al. Primary stroke prevention worldwide: translating evidence into action. Lancet Public Health. 2022;7(1):e74–e85. doi:10.1016/s2468-2667(21)00230-9
2. Campos Nonato Ir. Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021;20(10):795–820. doi:10.1016/s1474-4422(21)00252-0
3. Feigin VL, Forouzanfar MH, Krishnamurthi R, et al. Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet. 2014;383(9913):245–254. doi:10.1016/s0140-6736(13)61953-4
4. Virani SS, Alonso A, Benjamin EJ, et al. Heart Disease and Stroke Statistics-2020 Update: a Report From the American Heart Association. Circulation. 2020;141(9):e139–e596. doi:10.1161/cir.0000000000000757
5. Campbell BCV, De Silva DA, Macleod MR, et al. Ischaemic stroke. Nat Rev Dis Primers. 2019;5(1):70. doi:10.1038/s41572-019-0118-8
6. Kuriakose D, Xiao Z. Pathophysiology and treatment of stroke: present status and future perspectives. Int J Mol Sci. 2020;21:20.
7. Chapman SN, Mehndiratta P, Johansen MC, McMurry TL, Johnston KC, Southerland AM. Current perspectives on the use of intravenous recombinant tissue plasminogen activator (tPA) for treatment of acute ischemic stroke. Vasc Health Risk Manage. 2014;10:75–87. doi:10.2147/vhrm.S39213
8. Segura T, Calleja S, Jordan J. Recommendations and treatment strategies for the management of acute ischemic stroke. Exp Opin Pharmacother. 2008;9(7):1071–1085. doi:10.1517/14656566.9.7.1071
9. Jung S, Stapf C, Arnold M. Stroke unit management and revascularisation in acute ischemic stroke. Eur Neurol. 2015;73(1–2):98–105. doi:10.1159/000365210
10. Lambrinos A, Schaink AK, Dhalla I, et al. Mechanical thrombectomy in acute ischemic stroke: a systematic review. Can J Neurol Sci. 2016;43(4):455–460. doi:10.1017/cjn.2016.30
11. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nature Med. 2011;17(7):796–808. doi:10.1038/nm.2399
12. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–774. doi:10.1038/nri3070
13. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi:10.1126/science.1110647
14. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–312. doi:10.1038/nrn3722
15. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:6005):841–5. doi:10.1126/science.1194637
16. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi:10.1016/0306-4522(90)90229-w
17. Andoh M, Koyama R. Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells. 2021;10:1.
18. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi:10.1038/nn1997
19. Südhof TC. The cell biology of synapse formation. J Cell Biol. 2021;220:7).
20. Vainchtein ID, Molofsky AV. Astrocytes and Microglia: in Sickness and in Health. Trends Neurosci. 2020;43(3):144–154. doi:10.1016/j.tins.2020.01.003
21. Wang K, Li J, Zhang Y, et al. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci. Ther. 2021;27(5):528–539. doi:10.1111/cns.13619
22. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458–3483. doi:10.1016/j.neuron.2022.10.020
23. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi:10.1016/j.cell.2017.05.018
24. Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:6286):712–716. doi:10.1126/science.aad8373
25. Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9(12):1512–1519. doi:10.1038/nn1805
26. van Wageningen TA, Vlaar E, Kooij G, Jongenelen CAM, Geurts JJG, van Dam AM. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol Commun. 2019;7(1):206. doi:10.1186/s40478-019-0850-z
27. Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. 2016;113(12):E1738–46. doi:10.1073/pnas.1525528113
28. Scott EP, Breyak E, Nishinakamura R, Nakagawa Y. The zinc finger transcription factor Sall1 is required for the early developmental transition of microglia in mouse embryos. Glia. 2022;70(9):1720–1733. doi:10.1002/glia.24192
29. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19(10):622–635. doi:10.1038/s41583-018-0057-5
30. Masuda T, Amann L, Sankowski R, et al. Novel Hexb-based tools for studying microglia in the CNS. Nat Immunol. 2020;21(7):802–815. doi:10.1038/s41590-020-0707-4
31. Tischer J, Krueger M, Mueller W, et al. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia. 2016;64(9):1562–1572. doi:10.1002/glia.23024
32. Shapiro LA, Perez ZD, Foresti ML, Arisi GM, Ribak CE. Morphological and ultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res. 2009;1266:29–36. doi:10.1016/j.brainres.2009.02.031
33. Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88(16):7438–7442. doi:10.1073/pnas.88.16.7438
34. Bisht K, Sharma KP, Lecours C, et al. Dark microglia: a new phenotype predominantly associated with pathological states. Glia. 2016;64(5):826–839. doi:10.1002/glia.22966
35. Chertoff M, Shrivastava K, Gonzalez B, Acarin L, Giménez-Llort L. Differential modulation of TREM2 protein during postnatal brain development in mice. PloS one. 2013;8(8):e72083. doi:10.1371/journal.pone.0072083
36. Böttcher C, Schlickeiser S, Sneeboer MAM, et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci. 2019;22(1):78–90. doi:10.1038/s41593-018-0290-2
37. Plemel JR, Stratton JA, Michaels NJ, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv. 2020;6(3):eaay6324. doi:10.1126/sciadv.aay6324
38. Vasek MJ, Garber C, Dorsey D, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:7608):538–43. doi:10.1038/nature18283
39. Garcia-Bonilla L, Faraco G, Moore J, et al. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J Neuroinflammation. 2016;13(1):285. doi:10.1186/s12974-016-0750-0
40. Carrillo GL, Ballard VA, Glausen T, et al. Toxoplasma infection induces microglia-neuron contact and the loss of perisomatic inhibitory synapses. Glia. 2020;68(10):1968–1986. doi:10.1002/glia.23816
41. Jones BA, Beamer M, Ahmed S. Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol interv. 2010;10(5):263–270. doi:10.1124/mi.10.5.3
42. Sasmono RT, Oceandy D, Pollard JW, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101(3):1155–1163. doi:10.1182/blood-2002-02-0569
43. Chitu V, Gokhan Ş, Nandi S, Mehler MF, Stanley ER. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016;39(6):378–393. doi:10.1016/j.tins.2016.03.005
44. Kreutzberg GW. Microglia, the first line of defence in brain pathologies. Arzneimittel-Forschung. 1995;45:3.
45. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nature Med. 2017;23(2):1–13. doi:10.1038/nm.4269
46. Galloway DA, Phillips AEM, Owen DRJ, Moore CS. Phagocytosis in the Brain: homeostasis and Disease. Front Immunol. 2019;10:790. doi:10.3389/fimmu.2019.00790
47. Sipe GO, Lowery RL, Tremblay M, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun. 2016;7:10905. doi:10.1038/ncomms10905
48. Zhang W, Zhao J, Wang R, et al. Macrophages reprogram after ischemic stroke and promote efferocytosis and inflammation resolution in the mouse brain. CNS Neurosci. Ther. 2019;25(12):1329–1342. doi:10.1111/cns.13256
49. Kim HY, Huang BX, Spector AA. Phosphatidylserine in the brain: metabolism and function. Prog Lipid Res. 2014;56:1–18. doi:10.1016/j.plipres.2014.06.002
50. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15(4):209–216. doi:10.1038/nrn3710
51. Zhang Y, Li H, Li X, et al. TMEM16F aggravates neuronal loss by mediating microglial phagocytosis of neurons in a rat experimental cerebral ischemia and reperfusion model. Front Immunol. 2020;11:1144. doi:10.3389/fimmu.2020.01144
52. Hu X, Liou AK, Leak RK, et al. Neurobiology of microglial action in CNS injuries: receptor-mediated signaling mechanisms and functional roles. Prog Neurobiol. 2014;119–120:60–84. doi:10.1016/j.pneurobio.2014.06.002
53. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:7168):435–9. doi:10.1038/nature06307
54. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:6885):182–7. doi:10.1038/417182a
55. Cockram TOJ, Dundee JM, Popescu AS, Brown GC. The Phagocytic Code Regulating Phagocytosis of Mammalian Cells. Front Immunol. 2021;12:629979. doi:10.3389/fimmu.2021.629979
56. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi:10.1146/annurev-neuro-061010-113810
57. Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. 2012;32(3):946–952. doi:10.1523/jneurosci.3830-11.2012
58. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–470. doi:10.1111/j.1747-4949.2009.00387.x
59. Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36(4):557–565. doi:10.1002/ana.410360404
60. Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27(12):1941–1953. doi:10.1038/sj.jcbfm.9600495
61. Berchtold D, Priller J, Meisel C, Meisel A. Interaction of microglia with infiltrating immune cells in the different phases of stroke. Brain Pathol. 2020;30(6):1208–1218. doi:10.1111/bpa.12911
62. Nolte C, Möller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73(4):1091–1107. doi:10.1016/0306-4522(96)00106-6
63. Sperlágh B, Illes P. Purinergic modulation of microglial cell activation. Puriner Signal. 2007;3(1–2):117–127. doi:10.1007/s11302-006-9043-x
64. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29(12):3781–3791. doi:10.1523/jneurosci.5512-08.2009
65. Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem. 2003;278(15):13309–13317. doi:10.1074/jbc.M209478200
66. Takenouchi T, Sugama S, Iwamaru Y, Hashimoto M, Kitani H. Modulation of the ATP-lnduced release and processing of IL-1beta in microglial cells. Crit Rev Immunol. 2009;29(4):335–345. doi:10.1615/critrevimmunol.v29.i4.40
67. Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24(1):1–7. doi:10.1523/jneurosci.3792-03.2004
68. Ohsawa K, Irino Y, Sanagi T, et al. P2Y12 receptor-mediated integrin-beta1 activation regulates microglial process extension induced by ATP. Glia. 2010;58(7):790–801. doi:10.1002/glia.20963
69. Faraco G, Fossati S, Bianchi ME, et al. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103(2):590–603. doi:10.1111/j.1471-4159.2007.04788.x
70. Burguillos MA, Svensson M, Schulte T, et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015;10(9):1626–1638. doi:10.1016/j.celrep.2015.02.012
71. Shichita T, Hasegawa E, Kimura A, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nature Med. 2012;18(6):911–917. doi:10.1038/nm.2749
72. Lehnardt S, Schott E, Trimbuch T, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci. 2008;28(10):2320–2331. doi:10.1523/jneurosci.4760-07.2008
73. Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7(4):378–391. doi:10.1016/j.nurt.2010.07.005
74. Rojas A, Banik A, Chen D, Flood K, Ganesh T, Dingledine R. Novel Microglia Cell Line Expressing the Human EP2 Receptor. ACS Chem. Neurosci. 2019;10(10):4280–4292. doi:10.1021/acschemneuro.9b00311
75. Wang H, Li X, Wang Q, Ma J, Gao X, Wang M. TREM2, microglial and ischemic stroke. J Neuroimmunol. 2023;381:578108. doi:10.1016/j.jneuroim.2023.578108
76. Stefano L, Racchetti G, Bianco F, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110(1):284–294. doi:10.1111/j.1471-4159.2009.06130.x
77. Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci. 2008;28(22):5827–5835. doi:10.1523/jneurosci.1236-08.2008
78. Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Euro J Neurosci. 2001;13(3):529–538. doi:10.1046/j.1460-9568.2001.01418.x
79. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi:10.1038/nrn2038
80. Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematological Disord. 2004;4(1):65–84. doi:10.2174/1568006043481284
81. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. doi:10.1189/jlb.1109766
82. Thored P, Heldmann U, Gomes-Leal W, et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57(8):835–849. doi:10.1002/glia.20810
83. Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J Comparat Neurol. 1997;386(3):461–476.
84. Anttila JE, Whitaker KW, Wires ES, Harvey BK, Airavaara M. Role of microglia in ischemic focal stroke and recovery: focus on Toll-like receptors. Prog Neuro Psychopharmacol Biol Psychiatry. 2017;79(Pt A):3–14. doi:10.1016/j.pnpbp.2016.07.003
85. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clinic Develop Immunol. 2013;2013:746068. doi:10.1155/2013/746068
86. Shi QJ, Wang H, Liu ZX, et al. HAMI 3379, a CysLT2R antagonist, dose- and time-dependently attenuates brain injury and inhibits microglial inflammation after focal cerebral ischemia in rats. Neuroscience. 2015;291:53–69. doi:10.1016/j.neuroscience.2015.02.002
87. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32(9):1677–1698. doi:10.1038/jcbfm.2012.88
88. Murugan M, Ling EA, Kaur C. Glutamate receptors in microglia. CNS Neurol Disorders Drug Targ. 2013;12(6):773–784. doi:10.2174/18715273113126660174
89. Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci USA. 2013;110(43):E4098–107. doi:10.1073/pnas.1308679110
90. Gorlamandala N, Parmar J, Craig AJ, et al. Focal ischaemic infarcts expand faster in cerebellar cortex than cerebral cortex in a mouse photothrombotic stroke model. Transl Stroke Res. 2018;9(6):643–653. doi:10.1007/s12975-018-0615-1
91. Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi:10.1016/j.immuni.2017.08.008
92. Sala Frigerio C, Wolfs L, Fattorelli N, et al. The major risk factors for alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019;27(4):1293–1306.e6. doi:10.1016/j.celrep.2019.03.099
93. Srinivasan K, Friedman BA, Etxeberria A, et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020;31(13):107843. doi:10.1016/j.celrep.2020.107843
94. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 2014;6:13. doi:10.12703/p6-13
95. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
96. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi:10.1016/j.immuni.2010.05.007
97. Lepore F, D’Alessandro G, Antonangeli F, et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol. 2018;9:2750. doi:10.3389/fimmu.2018.02750
98. Liu X, Liu J, Zhao S, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke. 2016;47(2):498–504. doi:10.1161/strokeaha.115.012079
99. Qin C, Liu Q, Hu ZW, et al. Microglial TLR4-dependent autophagy induces ischemic white matter damage via STAT1/6 pathway. Theranostics. 2018;8(19):5434–5451. doi:10.7150/thno.27882
100. Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–3070. doi:10.1161/strokeaha.112.659656
101. Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (Review). Mole Med Rep. 2020;21(5):2006–2018. doi:10.3892/mmr.2020.11003
102. Lee Y, Lee SR, Choi SS, Yeo HG, Chang KT, Lee HJ. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int. 2014;2014:297241. doi:10.1155/2014/297241
103. Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14(11):1189–1197. doi:10.2174/092986707780597961
104. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–224. doi:10.1038/nrneurol.2014.38
105. Collmann FM, Pijnenburg R, Hamzei-Taj S, et al. Individual in vivo profiles of microglia polarization after stroke, represented by the genes iNOS and Ym1. Front Immunol. 2019;10:1236. doi:10.3389/fimmu.2019.01236
106. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619–643. doi:10.1146/annurev-physiol-022516-034406
107. Kohno K, Shirasaka R, Yoshihara K, et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science. 2022;376:6588):86–90. doi:10.1126/science.abf6805
108. Louveau A, Nerrière-Daguin V, Vanhove B, et al. Targeting the CD80/CD86 costimulatory pathway with CTLA4-Ig directs microglia toward a repair phenotype and promotes axonal outgrowth. Glia. 2015;63(12):2298–2312. doi:10.1002/glia.22894
109. Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16(8):2508–2521. doi:10.1523/jneurosci.16-08-02508.1996
110. Zhang L, Wei W, Ai X, et al. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell Death Dis. 2021;12(11):1068. doi:10.1038/s41419-021-04363-7
111. Kronenberg J, Merkel L, Heckers S, Gudi V, Schwab MH, Stangel M. Investigation of neuregulin-1 and glial cell-derived neurotrophic factor in rodent astrocytes and microglia. J Mol Neurosci. 2019;67(3):484–493. doi:10.1007/s12031-019-1258-8
112. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. 2018;362:6411):181–185. doi:10.1126/science.aat0473
113. Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disorders Drug Targ. 2013;12(3):302–315. doi:10.2174/1871527311312030004
114. Taoufik E, Probert L. Ischemic neuronal damage. Curr Pharm Des. 2008;14(33):3565–3573. doi:10.2174/138161208786848748
115. Saeed SA, Shad KF, Saleem T, Javed F, Khan MU. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp Brain Res. 2007;182(1):1–10. doi:10.1007/s00221-007-1050-9
116. Bergendi L, Benes L, Duracková Z, Ferencik M. Chemistry, physiology and pathology of free radicals. Life Sci. 1999;65(18–19):1865–1874. doi:10.1016/s0024-3205(99)00439-7
117. Chen H, Yoshioka H, Kim GS, et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14(8):1505–1517. doi:10.1089/ars.2010.3576
118. Nishizawa Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001;69(4):369–381. doi:10.1016/s0024-3205(01)01142-0
119. Takeuchi H, Jin S, Suzuki H, et al. Blockade of microglial glutamate release protects against ischemic brain injury. Exp Neurol. 2008;214(1):144–146. doi:10.1016/j.expneurol.2008.08.001
120. Greenwood SM, Connolly CN. Dendritic and mitochondrial changes during glutamate excitotoxicity. Neuropharmacology. 2007;53(8):891–898. doi:10.1016/j.neuropharm.2007.10.003
121. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247–272. doi:10.1016/j.pneurobio.2016.01.005
122. Correa FG, Hernangómez M, Guaza C. Understanding microglia-neuron cross talk: relevance of the microglia-neuron cocultures. Methods Mol Biol. 2013;1041:215–229. doi:10.1007/978-1-62703-520-0_20
123. Cotter R, Williams C, Ryan L, et al. Fractalkine (CX3CL1) and brain inflammation: implications for HIV-1-associated dementia. J Neurovirol. 2002;8(6):585–598. doi:10.1080/13550280290100950
124. Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M. Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Euro J Neurosci. 2002;15(10):1663–1668. doi:10.1046/j.1460-9568.2002.02007.x
125. Soriano SG, Coxon A, Wang YF, et al. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30(1):134–139. doi:10.1161/01.str.30.1.134
126. Song N, Li T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front Immunol. 2018;9:2305. doi:10.3389/fimmu.2018.02305
127. Ślusarczyk J, Trojan E, Wydra K, et al. Beneficial impact of intracerebroventricular fractalkine administration on behavioral and biochemical changes induced by prenatal stress in adult rats: possible role of NLRP3 inflammasome pathway. Biochem. Pharmacol. 2016;113:45–56. doi:10.1016/j.bcp.2016.05.008
128. Zhao RY, Wei PJ, Sun X, et al. Role of lipocalin 2 in stroke. Neurobiol Dis. 2023;179:106044. doi:10.1016/j.nbd.2023.106044
129. Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J Neurosci. 2015;35(32):11281–11291. doi:10.1523/jneurosci.1685-15.2015
130. Zhang J, Liu M, Huang M, et al. Ginsenoside F1 promotes angiogenesis by activating the IGF-1/IGF1R pathway. Pharmacol Res. 2019;144:292–305. doi:10.1016/j.phrs.2019.04.021
131. Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–120. doi:10.1016/j.pneurobio.2015.09.008
132. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:7638):481–487. doi:10.1038/nature21029
133. Liddelow SA, Barres BA. Reactive Astrocytes: production, Function, and Therapeutic Potential. Immunity. 2017;46(6):957–967. doi:10.1016/j.immuni.2017.06.006
134. Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:7761):332–337. doi:10.1038/s41586-019-1195-2
135. Liu W, Venugopal S, Majid S, et al. Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis. Neurobiol Dis. 2020;141:104877. doi:10.1016/j.nbd.2020.104877
136. Edara VV, Ghorpade A, Borgmann K. Insights into the gene expression profiles of active and restricted red/green-HIV(+) human astrocytes: implications for shock or lock therapies in the brain. J Virol. 2020;94:6.
137. Qian D, Li L, Rong Y, et al. Blocking Notch signal pathway suppresses the activation of neurotoxic A1 astrocytes after spinal cord injury. Cell Cycle. 2019;18(21):3010–3029. doi:10.1080/15384101.2019.1667189
138. Shinozaki Y, Shibata K, Yoshida K, et al. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y(1) receptor downregulation. Cell Rep. 2017;19(6):1151–1164. doi:10.1016/j.celrep.2017.04.047
139. Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA. 2012;109(4):E197–205. doi:10.1073/pnas.1111098109
140. Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi:10.1186/1742-2094-4-26
141. Qin C, Zhou LQ, Ma XT, et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull. 2019;35(5):921–933. doi:10.1007/s12264-019-00388-3
142. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–1273. doi:10.1126/science.aal3589
143. Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26(6):1880–1887. doi:10.1523/jneurosci.3696-05.2006
144. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer’s disease. J Neurosci. 2016;36(2):577–589. doi:10.1523/jneurosci.2117-15.2016
145. Back SA, Han BH, Luo NL, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22(2):455–463. doi:10.1523/jneurosci.22-02-00455.2002
146. Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27(9):1641–6; discussion 1647. doi:10.1161/01.str.27.9.1641
147. Matute C, Domercq M, Pérez-Samartín A, Ransom BR. Protecting white matter from stroke injury. Stroke. 2013;44(4):1204–1211. doi:10.1161/strokeaha.112.658328
148. Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008;18(3):387–400. doi:10.1111/j.1750-3639.2008.00138.x
149. Murugan M, Sivakumar V, Lu J, Ling EA, Kaur C. Expression of N-methyl D-aspartate receptor subunits in amoeboid microglia mediates production of nitric oxide via NF-κB signaling pathway and oligodendrocyte cell death in hypoxic postnatal rats. Glia. 2011;59(4):521–539. doi:10.1002/glia.21121
150. van der Goes A, Brouwer J, Hoekstra K, Roos D, van den Berg TK, Dijkstra CD. Reactive oxygen species are required for the phagocytosis of myelin by macrophages. J Neuroimmunol. 1998;92(1–2):67–75. doi:10.1016/s0165-5728(98)00175-1
151. Miron VE, Boyd A, Zhao JW, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. doi:10.1038/nn.3469
152. Wang G, Zhang J, Hu X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33(12):1864–1874. doi:10.1038/jcbfm.2013.146
153. Al Mamun A, Chauhan A, Yu H, Xu Y, Sharmeen R, Liu F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Euro J Neurosci. 2018;47(2):140–149. doi:10.1111/ejn.13778
154. Zheng Y, He R, Wang P, Shi Y, Zhao L, Liang J. Exosomes from LPS-stimulated macrophages induce neuroprotection and functional improvement after ischemic stroke by modulating microglial polarization. Biomater. Sci. 2019;7(5):2037–2049. doi:10.1039/c8bm01449c
155. Li Q, Dai Z, Cao Y, Wang L. Caspase-1 inhibition mediates neuroprotection in experimental stroke by polarizing M2 microglia/macrophage and suppressing NF-κB activation. Biochem. Biophys. Res. Commun. 2019;513(2):479–485. doi:10.1016/j.bbrc.2019.03.202
156. Brifault C, Gras M, Liot D, May V, Vaudry D, Wurtz O. Delayed pituitary adenylate cyclase-activating polypeptide delivery after brain stroke improves functional recovery by inducing m2 microglia/macrophage polarization. Stroke. 2015;46(2):520–528. doi:10.1161/strokeaha.114.006864
157. Chen D, Li J, Huang Y, et al. Interleukin 13 promotes long-term recovery after ischemic stroke by inhibiting the activation of STAT3. J Neuroinflammation. 2022;19(1):112. doi:10.1186/s12974-022-02471-5
158. Zhong Y, Gu L, Ye Y, et al. JAK2/STAT3 axis intermediates microglia/macrophage polarization during cerebral ischemia/reperfusion injury. Neuroscience. 2022;496:119–128. doi:10.1016/j.neuroscience.2022.05.016
159. Qin C, Fan WH, Liu Q, et al. Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke. 2017;48(12):3336–3346. doi:10.1161/strokeaha.117.018505
160. Wang D, Liu F, Zhu L, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. 2020;17(1):257. doi:10.1186/s12974-020-01921-2
161. Li R, Zhou Y, Zhang S, Li J, Zheng Y, Fan X. The natural (poly)phenols as modulators of microglia polarization via TLR4/NF-κB pathway exert anti-inflammatory activity in ischemic stroke. Eur. J. Pharmacol. 2022;914:174660. doi:10.1016/j.ejphar.2021.174660
162. Hyakkoku K, Hamanaka J, Tsuruma K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171(1):258–267. doi:10.1016/j.neuroscience.2010.08.054
163. Luo L, Liu M, Fan Y, et al. Intermittent theta-burst stimulation improves motor function by inhibiting neuronal pyroptosis and regulating microglial polarization via TLR4/NFκB/NLRP3 signaling pathway in cerebral ischemic mice. J Neuroinflammation. 2022;19(1):141. doi:10.1186/s12974-022-02501-2
164. Liu M, Xu Z, Wang L, et al. Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J Neuroinflammation. 2020;17(1):270. doi:10.1186/s12974-020-01946-7
165. Tang Y, Liu J, Wang Y, et al. PARP14 inhibits microglial activation via LPAR5 to promote post-stroke functional recovery. Autophagy. 2021;17(10):2905–2922. doi:10.1080/15548627.2020.1847799
166. Kong L, Li W, Chang E, et al. mtDNA-STING Axis Mediates Microglial Polarization via IRF3/NF-κB Signaling After Ischemic Stroke. Front Immunol. 2022;13:860977. doi:10.3389/fimmu.2022.860977
167. Liao S, Wu J, Liu R, et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 2020;36:101644. doi:10.1016/j.redox.2020.101644
168. Zhang P, Zhang X, Huang Y, et al. Atorvastatin alleviates microglia-mediated neuroinflammation via modulating the microbial composition and the intestinal barrier function in ischemic stroke mice. Free Radic Biol Med. 2021;162:104–117. doi:10.1016/j.freeradbiomed.2020.11.032
169. Zhang D, Lu Z, Man J, et al. Wnt-3a alleviates neuroinflammation after ischemic stroke by modulating the responses of microglia/macrophages and astrocytes. Int Immunopharmacol. 2019;75:105760. doi:10.1016/j.intimp.2019.105760
170. Tang T, Wang X, Qi E, Li S, Sun H. Ginkgetin Promotes M2 Polarization of Microglia and Exert Neuroprotection in Ischemic Stroke via Modulation of PPARγ Pathway. Neurochem Res. 2022;47(10):2963–2974. doi:10.1007/s11064-022-03583-3
171. Wang Q, Han B, Man X, Gu H, Sun J. Chuanzhitongluo regulates microglia polarization and inflammatory response in acute ischemic stroke. Brain Res. Bull. 2022;190:97–104. doi:10.1016/j.brainresbull.2022.09.015
172. Al Mamun A, Chauhan A, Qi S, et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc Natl Acad Sci USA. 2020;117(3):1742–1752. doi:10.1073/pnas.1914742117
173. Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61(1):91–103. doi:10.1002/glia.22363
174. Cho KJ, Song J, Oh Y, Lee JE. MicroRNA-Let-7a regulates the function of microglia in inflammation. Mole Cell Neurosci. 2015;68:167–176. doi:10.1016/j.mcn.2015.07.004
175. Butovsky O, Jedrychowski MP, Cialic R, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77(1):75–99. doi:10.1002/ana.24304
176. McDonald MK, Tian Y, Qureshi RA, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. 2014;155(8):1527–1539. doi:10.1016/j.pain.2014.04.029
177. Stoorvogel W. Functional transfer of microRNA by exosomes. Blood. 2012;119(3):646–648. doi:10.1182/blood-2011-11-389478
178. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103(33):12481–12486. doi:10.1073/pnas.0605298103
179. Hsu SD, Lin FM, Wu WY, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–9. doi:10.1093/nar/gkq1107
180. Barca C, Foray C, Hermann S, et al. The colony stimulating factor-1 receptor (CSF-1R)-mediated regulation of microglia/macrophages as a target for neurological disorders (Glioma, Stroke). Front Immunol. 2021;12:787307. doi:10.3389/fimmu.2021.787307
181. Barca C, Kiliaan AJ, Foray C, et al. A Longitudinal PET/MRI study of colony-stimulating factor 1 receptor-mediated microglia depletion in experimental stroke. J Nucl Med. 2022;63(3):446–452. doi:10.2967/jnumed.121.262279
182. Szalay G, Martinecz B, Lénárt N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun. 2016;7:11499. doi:10.1038/ncomms11499
183. Jin WN, Shi SX, Li Z, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017;37(6):2224–2236. doi:10.1177/0271678x17694185
184. Staal RGW, Weinstein JR, Nattini M, Cajina M, Chandresana G, Möller T. Senicapoc: repurposing a Drug to Target Microglia K(Ca)3.1 in Stroke. Neurochem Res. 2017;42(9):2639–2645. doi:10.1007/s11064-017-2223-y
185. Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5(2):73–90.
186. Womble TA, Green S, Shahaduzzaman M, et al. Monocytes are essential for the neuroprotective effect of human cord blood cells following middle cerebral artery occlusion in rat. Mole Cell Neurosci. 2014;59:76–84. doi:10.1016/j.mcn.2014.01.004
187. Li F, Xu D, Hou K, Gou X, Li Y. The role of P2Y12 receptor inhibition in ischemic stroke on microglia, platelets and vascular smooth muscle cells. J Thromb Thrombol. 2020;50(4):874–885. doi:10.1007/s11239-020-02098-4
188. Zhang G, Xia F, Zhang Y, et al. Ginsenoside Rd Is Efficacious Against Acute Ischemic Stroke by Suppressing Microglial Proteasome-Mediated Inflammation. Mole Neurobiol. 2016;53(4):2529–2540. doi:10.1007/s12035-015-9261-8
189. Hsu CC, Kuo TW, Liu WP, Chang CP, Lin HJ. Calycosin preserves BDNF/TrkB signaling and reduces post-stroke neurological injury after cerebral ischemia by reducing accumulation of hypertrophic and TNF-α-containing microglia in rats. J Neuroimmune Pharmacol. 2020;15(2):326–339. doi:10.1007/s11481-019-09903-9
190. Yew WP, Djukic ND, Jayaseelan JSP, et al. Early treatment with minocycline following stroke in rats improves functional recovery and differentially modifies responses of peri-infarct microglia and astrocytes. J Neuroinflammation. 2019;16(1):6. doi:10.1186/s12974-018-1379-y
191. Mota M, Porrini V, Parrella E, et al. Neuroprotective epi-drugs quench the inflammatory response and microglial/macrophage activation in a mouse model of permanent brain ischemia. J Neuroinflammation. 2020;17(1):361. doi:10.1186/s12974-020-02028-4
192. Ni J, Wang X, Chen S, et al. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun. 2015;49:75–85. doi:10.1016/j.bbi.2015.04.014
193. Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–924. doi:10.1038/nn1715
194. Li Z, Song Y, He T, et al. M2 microglial small extracellular vesicles reduce glial scar formation via the miR-124/STAT3 pathway after ischemic stroke in mice. Theranostics. 2021;11(3):1232–1248. doi:10.7150/thno.48761
195. Yun SP, Kam TI, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nature Med. 2018;24(7):931–938. doi:10.1038/s41591-018-0051-5
196. Park JS, Choi J, Hwang SH, et al. Cottonseed oil protects against intestinal inflammation in dextran sodium sulfate-induced inflammatory bowel disease. J Med Food. 2019;22(7):672–679. doi:10.1089/jmf.2018.4323
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.