Back to Journals » The Application of Clinical Genetics » Volume 9
Mevalonate kinase deficiency: current perspectives
Authors Favier L, Schulert G
Received 19 April 2016
Accepted for publication 10 May 2016
Published 20 July 2016 Volume 2016:9 Pages 101—110
DOI https://doi.org/10.2147/TACG.S93933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Leslie A Favier, Grant S Schulert
Division of Rheumatology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
Abstract: Mevalonate kinase deficiency (MKD) is a recessively inherited autoinflammatory disorder with a spectrum of manifestations, including the well-defined clinical phenotypes of hyperimmunoglobulinemia D and periodic fever syndrome and mevalonic aciduria. Patients with MKD have recurrent attacks of hyperinflammation associated with fever, abdominal pain, arthralgias, and mucocutaneous lesions, and more severely affected patients also have dysmorphisms and central nervous system anomalies. MKD is caused by mutations in the gene encoding mevalonate kinase, with the degree of residual enzyme activity largely determining disease severity. Mevalonate kinase is essential for the biosynthesis of nonsterol isoprenoids, which mediate protein prenylation. Although the precise pathogenesis of MKD remains unclear, increasing evidence suggests that deficiency in protein prenylation leads to innate immune activation and systemic hyperinflammation. Given the emerging understanding of MKD as an autoinflammatory disorder, recent treatment approaches have largely focused on cytokine-directed biologic therapy. Herein, we review the current genetic and pathologic understanding of MKD, its various clinical phenotypes, and the evolving treatment approach for this multifaceted disorder.
Keywords: mevalonic aciduria, hyperimmunoglobulinemia D and periodic fever syndrome, autoinflammatory, periodic fever syndrome
Introduction
Mevalonate kinase deficiency (MKD) encompasses a spectrum of rare autoinflammatory disorders caused by a recessively inherited defect in the isoprenoid biosynthetic pathway. Once thought to be two disparate entities, mevalonic aciduria (MA) and hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) were discovered within 1 year of each other in the mid-1980s.1,2 These two clinically described phenotypes were subsequently linked in 1999 with the discovery of a shared genetic lesion in the MVK gene on chromosome 12q24.3 These gene defects not only interfere in the mevalonate pathway of cholesterol synthesis but also lead to episodes of hyperinflammation with increased secretion of interleukin 1b (IL-1b).4 MKD is now viewed as a phenotypic continuum based on the degree of enzyme deficiency, with MA being the most severe phenotype and HIDS on the mild end of the spectrum.5 MKD is characterized by autoinflammatory flares with fever, abdominal pain, mucoid and cutaneous lesions, and arthralgias. In addition, more severely affected patients with MA classically have developmental delay, physical dysmorphisms, psychomotor retardation, hepatosplenomegaly, and ocular abnormalities. As MKD remains mysterious in both its precise pathogenesis and full breadth of phenotypic spectrum, we seek to review the most current perspectives in this fascinating disease.
Epidemiology
Although the epidemiology of MKD is largely unknown, it appears to vary greatly by region, with the highest documented prevalence in the Netherlands. Using data from the Eurofever registry, it is estimated that MKD affects at least 300 people worldwide, with a majority representing the HIDS phenotype.6 The distribution and prevalence of MKD in the US are unclear, with the largest case series involving only 20 patients reported as part of an international study group.7 Measuring prevalence is further confounded by a significant number of undocumented and undiagnosed patients. In one study, it took on average 9.9 years for symptomatic patients to reach diagnosis, due to rarity and variability in presentation.7 The frequency of disease in the Dutch population with an estimated 1:200,000 affected nationwide is well documented.7 This is thought to be due to a relatively high carrier frequency of any MVK mutation in the Dutch population, which is estimated as high as 1:65, although notably this would imply a significantly larger number of MKD cases than have been described (as detailed in the next section).8
Genetics and pathophysiology
Molecular genetics
The current genetic understanding of MKD stems in large measure from the parallel discoveries that patients with HIDS carried mutations in MVK, which had been previously linked to MA. Houten et al9 observed an increased urinary excretion of mevalonic acid and diminished mevalonate kinase activity in children with HIDS, prompting the targeted sequencing of the MVK gene. Concurrently, an international study group used linkage analysis in families with HIDS to identify MVK as a candidate gene and ultimately defined mutations.3 Subsequently, >120 sequence variants in this gene have been reported in association with MKD.10 Most reported variants are single-nucleotide polymorphisms leading to missense mutations and are believed to impair mevalonate kinase stability.11 As such, patients with MKD have markedly diminished, to absence of, mevalonate kinase activity.
MKD is considered to be inherited in an autosomal recessive manner. However, although a majority of patients carry biallelic mutations in MVK, some patients have clinical disease with only a single identified mutation.12 There is some degree of genotype–phenotype correlation within MKD; however, this notion is confounded by the variable nature of disease spectrum, and the role of additional genes as modifiers of these phenotypes is an area of active investigation.13 Certain MVK variants are highly associated with the more severe MA phenotype and typically result in very low enzyme activity.14,15 Indeed, variants associated with the MA phenotype largely cluster together in the core of the protein and may have more profound effects on folding and stability.15,16 In contrast, other variants such as the C-terminal V377I substitution typically manifest as the HIDS phenotype and are rarely associated with MA. Interestingly, this variant is the most commonly reported finding in patients with MKD and has a minor allele frequency of up to 0.003 in the healthy Dutch population.8 This relatively high carrier rate would imply a much higher disease incidence than what is typically reported and suggests that many individuals who are homozygous for the V377I variant have a mild phenotype that is not recognized as MKD. Together, these genetic data support a model first proposed by Simon et al in which MKD exists as a phenotypic spectrum dependent on the residual mevalonate kinase activity. HIDS and MA phenotypes are clearly defined entities, but MKD encompasses patients with overlapping features and even those with a minimal or clinically silent course.5
The mevalonate pathway
Mevalonate kinase is a key enzyme in the mevalonate biosynthetic pathway, which leads to the synthesis of both cholesterol and nonsterol isoprenoids (Figure 1). This pathway starts with the action of the highly regulated enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which converts HMG-CoA into mevalonate. Notably, HMG-CoA reductase is a key pharmacological target of the cholesterol-lowering statin agents. Mevalonate kinase subsequently phosphorylates the C5 hydroxyl oxygen of mevalonate to form 5-phosphomevalonic acid.17 Subsequent steps in the pathway convert this to isopentenyl pyrophosphate (IPP), which serves as the substrate for several essential biologic processes (as detailed later in this section). Loss of mevalonate kinase activity leads to both an accumulation of mevalonic acid and deficiency in these downstream compounds. Indeed, while severely affected patients with the MA phenotype have constitutively elevated mevalonic acid levels in plasma and urine,18 those with the more mild HIDS phenotype have MA only during febrile crises.19
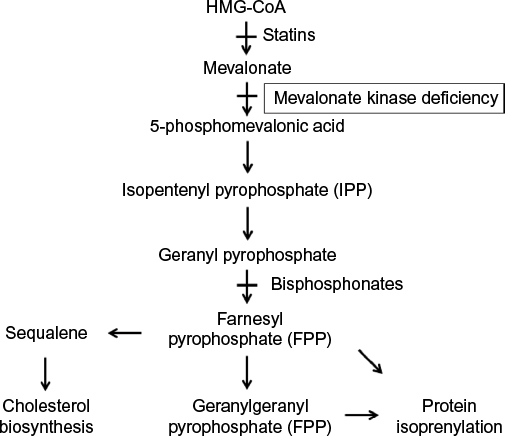 | Figure 1 Overview of the mevalonate pathway. Notes: The pathway starts with conversation of HMG-CoA into mevalonate through the action of HMG-CoA reductase. Mevalonate is then converted into 5-phosphomevalonic acid by mevalonate kinase. In MKD, there is inhibition of the pathway at this step, leading to buildup of mevalonic acid and depletion of downstream products. 5-Phosphomevalonic acid is ultimately converted to geranyl pyrophosphate, which serves as precursor for the isoprenoids farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are the substrates for protein prenylation. This pathway can also be experimentally inhibited using statins, which block HMG-CoA reductase, or bisphosphonates, which block farnesyl pyrophosphate synthase. Abbreviations: HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; MKD, mevalonate kinase deficiency. |
Deficiency in IPP synthesis has several important downstream consequences, including the requirement of IPP itself in modifications of tRNA.20 Its key role, however, is in the synthesis of the isoprenoids farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate, which mediate protein prenylation (Figure 1). These isoprenoids are covalently transferred to the C-terminus of cellular proteins and can serve both to localize proteins as membrane anchors and to regulate protein activity, including the key Ras superfamily of small GTPases.21,22 These G proteins participate in crucial signal transduction events controlling cell cycle and growth, differentiation, and vesicle trafficking. Finally, further downstream roles of some isoprenoids include synthesis of cholesterol, heme, and ubiquinone.20 Accumulating evidence suggests that much of the phenotype and cellular derangements seen in MKD are due to not only accumulation of mevalonic acid but also impaired flux through this isoprenoid pathway (as detailed in the next section).
Autoinflammation in MKD – clues from model systems
The immune manifestations of MKD largely fit the pattern of an autoinflammatory disorder that has been defined by Masters et al23 as seemingly unprovoked bouts of systemic inflammation in the absence of autoreactive lymphocytes or high-titer autoantibodies. The prototypical autoinflammatory disorders include the cryopyrin-associated periodic syndromes and familial Mediterranean fever, where variants in specific inflammasome components directly lead to hyperproduction of cytokines including IL-1b. Patients with MKD similarly experience periodic episodes of systemic inflammation, which are also characterized by excessive secretion of inflammatory mediators. During attacks, patients with MKD have increased levels of acute-phase proteins including C-reactive protein and cytokines such as TNF-a, IL-6, and interferon-g.24,25 In addition, peripheral blood mononuclear cells (PBMCs) from patients with MKD produce increased amounts of IL-1b, IL-6, and TNF-a both at rest and following stimulation with activating ligands.25–27 Crucially, inflammatory hyperresponsiveness in MKD appears to be due to lack of isoprenoid products and not accumulation of mevalonic acid. Blockade of the mevalonate pathway with the HMG-CoA reductase inhibitor lovastatin led to markedly increased IL-1b production from healthy PBMCs, and addition of the isoprenoid farnesyl reversed this, along with decreasing the cytokine hypersecretion seen in MKD. In contrast, addition of mevalonic acid to PBMCs from MKD patients or healthy donors did not promote hyperinflammation.4 This is also supported by the clinical observation that statin therapy in patients with MKD reduces both mevalonic acid levels and residual isoprenoid production and in fact can trigger disease flares.18 Further work has highlighted specifically the need for geranylgeranylation rather than other mevalonate pathway products, such as cholesterol biosynthesis, in mediating the hypersecretion of IL-1b.28 This accumulated evidence has led to the proposal that MKD represents a metabolic autoinflammatory disease, where defective flux through the isoprenoid pathway primes the innate immune system for hyperresponsiveness (Figure 2).20
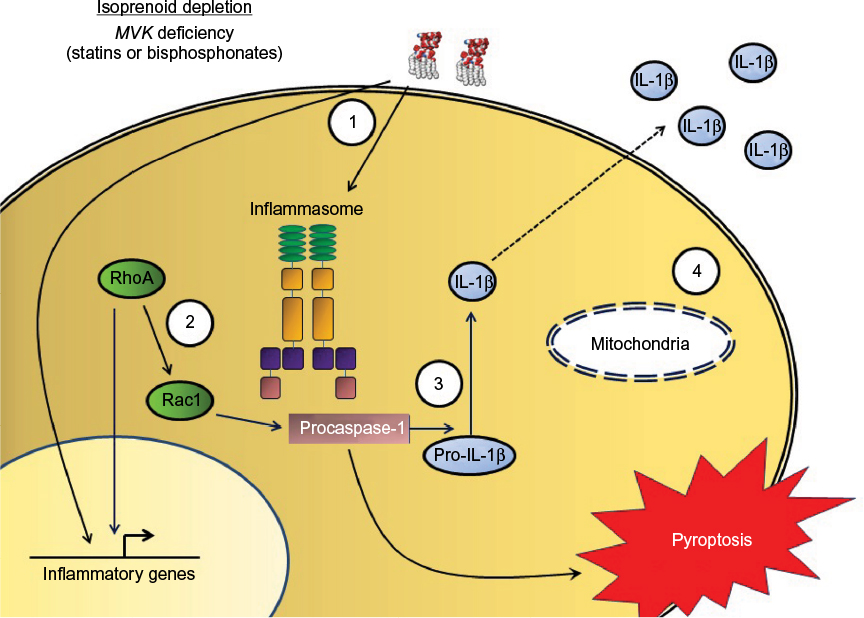 | Figure 2 Proposed pathogenesis of mevalonate kinase deficiency. Notes: Loss of mevalonate kinase activity leads to a cellular isoprenoid deficiency, which can also be mimicked experimentally with statin or bisphosphonate treatment. 1) Activation of monocytes or macrophages, such as by Toll-like receptor ligands, leads to proinflammatory gene expression and priming of the inflammasome. 2) In settings of isoprenoid deficiency, there is increased RhoA activity, leading to further increase in pro-IL-1b gene expression, as well as activation of Rac1, which activates procaspase-1. 3) Inflammasome-mediated caspase-1 activation, augmented by Rac1, leads to processing and release of inflammatory cytokines including IL-1b. It also leads to pyroptosis, a proinflammatory cell death, and possibly induction of apoptosis in bystander cells. 4) Isoprenoid depletion also impairs mitochondrial function and stability, as well as impaired autophagic clearance of damaged mitochondria, further promoting IL-1b hypersecretion. Abbreviation: IL-1b, interleukin 1b. |
A full mechanistic understanding of how MKD leads to autoinflammation has been hampered by the lack of model systems that fully represent the human disease. Mice with heterozygous deletions in MVK have significantly reduced mevalonate kinase activity, but biallelic deletions appear to be nonviable.29 Interestingly, MVK+/- mice do have some features of immune dysfunction, including increased serum IgD and TNF-a levels, as well as increased expression of activation markers on T-lymphocytes and macrophages.29,30 While genetic models of MKD have proven elusive, significant progress has been made using biochemical blockade of the mevalonate pathway with statin drugs (Figure 1). Monocytic cells treated with simvastatin demonstrate increased caspase-1 activation, which mediates IL-1b secretion.31 Further work demonstrated that isoprenoid deficiency specifically leads to caspase-1 activation through the small GTPase Rac1.32 Small GTPases are frequent targets for geranylgeranylation, and several of these including Rac1 and RhoA appear to be sensitive to reduced isoprenylation upon mevalonate pathway blockade.33 Interestingly, recent work has shown that reduced prenylation of RhoA may underlie the excessive IL-1b production seen in MKD. Defective isoprenylation of RhoA augments Rac1 activity and increases IL-1b transcription.34 Relatedly, isoprenoid shortage also compromised mitochondrial stability and led to defective autophagy, further priming monocytes for inflammasome activation and IL-1b production.35 Similar mitochondrial disruption and proinflammatory cell death are also seen in statin-treated neuronal cells cocultured with microglia, suggesting this could contribute to neurologic impairment seen in severely affected MA patients.36
Further insights into MKD pathogenesis have come from a distinct metabolic model of mevalonate pathway dysfunction. Alendronate, an amino bisphosphonate, inhibits the enzyme FPP synthase and thus specifically blocks isoprenoid synthesis distal to mevalonate kinase (Figure 1). When mice are treated with alendronate followed by a Toll-like receptor ligand such as muramyl dipeptide, there is marked systemic inflammation with fever, elevated acute-phase reactants, hepatitis, and IL-1b production.37,38 Indeed, these mice have a proinflammatory cytokine profile that was similar to the MKD patients during attacks.39 In addition, different approaches to augment the cellular isoprenoid pool, including exogenous geraniol and inhibiting farnesyl transferase, had anti-inflammatory effects.37,39,40 This model system has yielded some clues regarding the pathogenesis of MKD. Macrophages treated with alendronate have enhanced inflammasome activation, with increased expression of NALP3, increased production of IL-1b, and increased pyroptosis.41,42 Interestingly, this model may also recapitulate some of the central nervous system pathology seen in severely affected patients, as these mice demonstrated cerebellar microglial activation and inflammasome expression.43 However, this is in contrast to a recent study where MVK knockdown in a murine microglial model did not affect NALP3 expression or pyroptosis.44 Taken together, this metabolic model of MKD may represent a useful system for further mechanistic and therapeutic investigations.
Clinical manifestations
HIDS phenotype
MKD typically presents with recurrent self-limited bouts of multisystem inflammation that are characterized by fever, abdominal pain, adenopathy, rash, and arthralgia. Historically, the milder end of the MKD spectrum has been clinically recognized as HIDS. Typical HIDS episodes can occur in a cyclical fashion (typically every 4–6 weeks) or be provoked by physiologic stress in the form of illness, injury, or iatrogenically following vaccinations. In one series, vaccinations were attributed as the inciting trigger to the first flare in 63% of patients, which could suggest that disease flares are not truly unprovoked but rather initiated by minor immune stimulation.45 Most patients demonstrate their first inflammatory attack prior to 1 year of age.7,46 Total duration of untreated flares last between 3 days and 7 days. The classic course of an episode features a constitutional prodrome, followed by high spiking fevers that can exceed 40°C with associated rigors. After fever, painful cervical adenopathy is the most common finding,7 along with pharyngitis and mucocutaneous ulcerations, making infectious processes difficult to discriminate early in disease course.7,47 Abdominal pain may be the most marked and debilitating feature of systemic inflammation, with severity of presentation prompting surgical evaluation for acute abdomen in some cases.7,47 While arthralgias are commonly seen, erosive arthritis of large joints can also occur.7,48 There is no true pathognomonic rash, with reports ranging from morbilliform to urticarial, appearing exanthems that are largely nonspecific.7,47
MA phenotype
MA represents the most severe end of the MKD spectrum and characteristically presents in the first few months of life, with antenatal presentations linked with a high rate of stillbirth in affected families.49 The phenotypic continuum within MA was specifically first reported by Hoffmann et al,18 who described the distinct presentations of eleven cases. Commonly reported dysmorphisms include frontal bossing, hypertelorism, long eyelashes, and triangular-shaped facies.49 Affected children have significant failure to thrive and varying degrees of developmental delay. Notably, MA also includes autoinflammatory attacks as described in less severe forms of MKD. However, the major delineation between HIDS and MA phenotype is increased involvement in the central nervous system in MA, demonstrated by psychomotor retardation, ataxia, seizures, and myopathies.18,49 Gastrointestinal findings include rare reports of cholestasis and liver dysfunction.50 Ocular findings are also wide ranging including recurrent conjunctivitis, cataracts, and uveitis.7,47 As evidenced by the potential for multisystem involvement, the morbidity and mortality associated with MA are great, with death in infancy being a common occurrence.
The expanding phenotype of MKD
The spectrum and clinical heterogeneity of MKD continues to expand as more novel case presentations are reported in the literature. Interestingly, recent reports have suggested a connection between MA and retinitis pigmentosa, a heritable syndrome of rod photoreceptor degeneration, which induces night blindness and peripheral vision loss. In one cohort of retinitis pigmentosa patients without previously defined gene defects, three patients were noted to have homozygous MVK mutations without other clinical signs of MKD.47,48,51 Similarly, disseminated superficial actinic porokeratosis is a dominantly inherited cause of photosensitive annular skin lesions. In some of these patients, there have been incidental MVK mutations found on exome sequencing but without clinical signs of MKD, indicating a possible genetic overlap relating to impairment in cellular apoptosis.52
Phenotypic relations between the MKD and inflammatory bowel disease (IBD) are also an evolving area of interest as more monogenic syndromes with early-onset autoinflammation including colitis are found to clinically intersect with IBD.53 Bianco et al54 reported six cases of early-onset IBD with both homozygous and heterozygous MVK mutations, along with other known IBD-associated genetic variants, and proposed that these variants could synergistically augment risk of developing bowel inflammation. In line with this notion, two further cases reported in 2013 document severe neonatal onset colitis, responsive to anakinra, as the presenting feature of MVK mutations55
In looking at outcomes of adult patients with MKD, renal angiomyolipoma is documented at rates ranging from 6% to 9% in cohorts, with no clear explanation.47,48 Additionally, the elevated risk for pneumococcal infections and progression to life-threatening sepsis has been documented in some larger cohorts.47,48 It has been speculated that increased plasma mevalonic acid is advantageous for the bacterium’s survival in blood and tissues.47,56
The hematologic complications of MKD are important. Macrophage activation syndrome (MAS) is a potentially life-threatening episode of sustained anomalous inflammation, which is manifested by cytopenias, fevers, organ dysfunction, and hyperferritinemia.57–59 It is most commonly a complication of another autoinflammatory disorder, systemic juvenile idiopathic arthritis, but is increasingly associated with the monogenic periodic fever syndromes.60 MAS was first reported as a complication of MKD in a patient presenting with multiorgan dysfunction and cytopenias who responded to high-dose steroids.61 Additionally, we previously reported a patient with MKD presenting with cholestatic liver failure and recurrent fevers who also developed repeated episodes of MAS in the context of a hypomorphic perforin gene variant.62 MAS can be challenging to diagnose, in part due to lack of standardized diagnostic criteria. However, there is some hint that MAS may be a relatively common complication of MKD. The largest cohort that was specifically screened for MAS included 50 MKD patients with an overall occurrence of 6% noted via retrospective review.47 Nevertheless, this is an entity that deserves clinical forethought and further study as its morbidity is considerable.
Diagnostics laboratory testing
There is significant need for laboratory testing that can accurately identify MKD and discriminate this from other infectious or inflammatory conditions. The HIDS phenotype of MKD acquired its name from its association with elevation in serum IgD.45 Since its nomenclature was assigned, however, it has been noted that elevations in IgD are inconsistent in disease carriers and can be normal in up to 20% of cases.7,63 Additionally, a relationship between severity of phenotype and IgD serum measurements does not appear, although one observational study did postulate that the measurement may be a more adventitious marker of disease in the adult population along with serum IgA.48 During acute febrile attacks, there is a robust acute-phase response with elevated C-reactive protein and erythrocyte sedimentation rate in addition to neutrophil predominant leukocytosis.7 Serum amyloidosis is another long-term sequela of prolonged inflammatory activation, with elevations in serum amyloid A noted in ∼3% of HIDS patients,7,48 although this is lower than rates seen in related hereditary periodic fevers syndromes. The median age at diagnosis of amyloid A amyloidosis in one retrospective study was 25 years, with complications including crescentic and membranous glomerulonephritis and eventual renal transplant requirement.47,48,64 One biologic test that does appear diagnostically valuable is the quantification of urine mevalonic acid excretion. Its utility as a diagnostic tool was studied by Jeyaratnam et al65 who noted that urinary increases of mevalonic acid had a 90% specificity rate, theorizing that this is an adequate and convenient way of screening patients who may need MVK genetic testing.
It is speculated that the root cause of phenotypic variation in MKD lies in the level of residual mevalonate kinase function, with HIDS patients ranging from 1.8% to 28% enzymatic activity compared to <0.5% in MA.9 This increased residual activity may be related to the fact that less severely affected patients are compound heterozygotes for the MVK gene mutations. Indeed, the most prevalent mutation affecting 80% of HIDS patients is V3371, which impacts the folding, maturation, and stability of the protein, whereas mutations associated with MA phenotype have more deleterious effects.3,16,47 However, there are inconsistencies in linking specific genotypes to severity of phenotypic presentation.3,47 Overall, there is a paucity of literature addressing heterozygosity in MVK mutations and symptom development. One study by Barron et al12 documented phenotypic disease in a cohort of patients in which no clinical difference was determined between compound and true heterozygotes. As such, more long-term data specifically assessing genotypic expression variations and their clinical implications are required.
Treatment
As with most rare diseases, evaluation of treatment regimens presents significant challenges. In general, treatment should be tailored to individuals and address symptom and inflammatory control to allow for daily life functioning. The impacts of disease activity are demonstrated by problems with social development, educational delay, unemployment, and mental illness.7,47,48,66 Prior to the advent of biologics, nonsteroidal anti-inflammatory drugs were used supportively to control inflammatory flares. Additionally, the use of corticosteroids was noted to induce a complete response in 24% of patients in one retrospective review.67 Colchicine has performed poorly in MKD in comparison to other heritable periodic fever syndromes.67 Statins were also utilized, with some reports of severe flares in patients with MA phenotype, and that treatment modality has largely been abandoned.6,48
Given the expanding understanding of the role of autoinflammation in MKD, there is increasing interest in the use of IL-1 targeting biologics including anakinra and canakinumab for this disorder. Anakinra, a recombinant human IL-1 receptor antagonist, has been used in both an “on-demand” manner and in daily prophylactic dosing regimens. In a prospective observational study, anakinra was associated with decreased duration and severity of flare when given on-demand.68 Its dosing was most successful if given <24 hours into symptoms, suggesting a finite window of opportunity. In 2015, Rossi-Semerano et al69 retrospectively found that continuous dosing was most beneficial with a 30% full response and 70% partial response. An obvious drawback to daily dosing of anakinra is frequency of painful injections. Canakinumab, a high-affinity human monoclonal anti-IL-1b antibody, has been promising in retrospective studies in dosing ranges from 2 mg/kg to 7 mg/kg every 4 weeks to 8 weeks.67,69,70 There was full response in about half of patients, with less adverse effects and better ease of dosing noted in comparison to those using anakinra.69,70 Other biologics have been reported in MKD, with mixed response. TNF-α blockade has been reviewed in descriptive retrospective studies, with a notable lack of head-to-head comparison with IL-1 blockade.6,71 In one review, including collection of data from the Eurofever Registry, etanercept provided partial response in 65% of patients, with only one complete response noted.67 Given an absence of comparative effectiveness data, TNF-α blocking agents such as etanercept and adalimumab are still considered as a first-line biologics in MKD. Additional consideration should also be given to tocilizumab, a monoclonal antibody targeted against the IL-6 receptor, with published cases of successful reduction in clinical symptoms following treatment failure of anakinra.72 The latest attempt at formalizing therapy guidelines for treatment of autoinflammatory diseases was performed by a European initiative, Single Hub and Access point for paediatric Rheumatology in Europe (SHARE), and their recommendations are summarized in Figure 3.
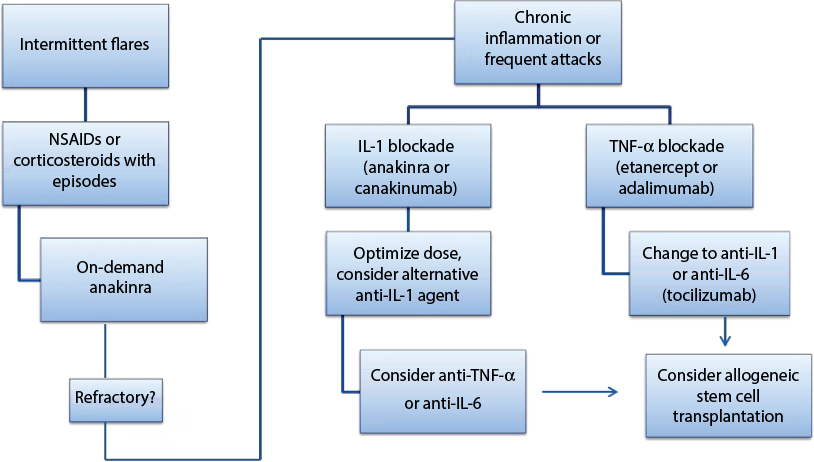 | Figure 3 Treatment algorithm for MKD per the consensus recommendations proposed by the SHARE network. Notes: Patients with intermittent inflammatory flares can be managed with NSAIDs for symptom relief or corticosteroids or anakinra to terminate attacks. Biologic agents, either IL-1 or TNF-α blockade, should be considered for patients who fail this strategy, have more frequent attacks, or have persistent inflammation between attacks. If initial biologic therapy is ineffective, a switch to a different agent or class should be considered. Finally, in patients with unremitting disease and poor quality of life, stem cell transplantation should be considered. Data from Ter Haar et al.6 Abbreviations: MKD, mevalonate kinase deficiency; SHARE, Single Hub and Access point for paediatric Rheumatology in Europe; IL-1b, interleukin 1b; TNF-α, tumor necrosis factor-α; NSAIDs, nonsteroidal anti-inflammatory drugs. |
There have been several attempts to translate the understanding of the mevalonate pathway into potential metabolic therapies. The farnesyltransferase inhibitors (FTIs), tipifarnib and lonafarnib, were studied in 2011 by Marcuzzi et al using cellular models, with the hypothesis that FTIs would help to redistribute mevalonate intermediate compounds to geranylgeranyl pyrophosphate synthesis, thus reducing cytokine upregulation.40,73 Indeed, there was increase in cholesterol biosynthesis and decrease in overall proinflammatory cytokine production in both cellular MKD models and MKD patient isolated monocytes following exposure to FTIs. An alternative metabolic approach using alendronate has also been under study. One patient with MKD, treated with alendronate for his steroid-induced osteoporosis achieved complete remission while on this particular therapy.74 Paradoxically, as described earlier alendronate inhibits FPP synthase (Figure 1) and leads to biochemical defects that mimic MKD in cell culture and mouse models. Indeed, a more recent study found less compelling anti-inflammatory properties when this compound was tested in vitro.75
For refractory cases of MA phenotype, the last consideration for therapy includes liver transplantation or hematopoietic stem cell transplantation (HSCT). The first documented HSCT for the treatment of severe neonatal MKD was in 2007.76 Since that time, there have been several case reports, including one in which HSCT was performed following liver transplantation.77 Interestingly, there was stabilization of MKD-related white matter abnormalities on brain MRI following liver transplantation, perhaps hinting that this may correct cholesterol biosynthetic defects, which is protective for brain development. In the same patient, however, HSCT was required for correction of autoinflammatory attacks. This case suggests that liver transplantation may be more efficacious for neurologic manifestations and HSCT may provide more corrective benefits for immune regulation.77,78
Conclusions and unanswered questions
MKD represents a broad range of disease phenotypes, with HIDS and MA located on either end of the severity spectrum. These entities lack homogeneity in both genotypic and phenotypic expression, and there remains opportunity for overlap in presentation based upon residual mevalonate kinase activity. Further dedicated study, focusing on the particular MVK variants and their pathogenicity, is required in order to better anticipate disease penetrance. Mutation in the mevalonate pathway leads to interruption in cholesterol and nonsterol isoprenoid biosynthesis and also hyperproduction of IL-1b, leading to its classification as a metabolic autoinflammatory disorder. There still persists a lack of full understanding of disease pathogenesis, with a notable absence of representative animal models. Treatment is largely tailored to individual patients and includes biologic blockade to halt autoinflammation, with more intensive measures including liver and HSCT for severe disease phenotype. Further understanding of metabolic therapy modalities may provide an additional avenue of disease control, as well as the hope to reverse the more severe MA clinical features. In assessing this disease spectrum as a whole, it is clear that its heterogeneous presentation poses challenges for both diagnosis and treatment, making multicenter collaboration in studying this disease imperative.
Acknowledgments
Doctor Schulert is supported by K12 HL119986 from the National Institutes of Health and a Scientist Development Award from the Rheumatology Research Foundation.
Disclosure
Doctor Schulert has received consulting fees from Novartis. The authors report no other conflicts of interest in this work.
References
Berger R, Smit GP, Schierbeek H, Bijsterveld K, le Coultre R. Mevalonic aciduria: an inborn error of cholesterol biosynthesis? Clin Chim Acta. 1985;152(1–2):219–222. | ||
van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. 1984;1(8386):1087–1090. | ||
Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22(2):178–181. | ||
Frenkel J, Rijkers GT, Mandey SH, et al. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. 2002;46(10):2794–2803. | ||
Simon A, Kremer HP, Wevers RA, et al. Mevalonate kinase deficiency: evidence for a phenotypic continuum. Neurology. 2004;62(6):994–997. | ||
Ter Haar NM, Oswald M, Jeyaratnam J, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636–1644. | ||
van der Hilst JC, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore). 2008;87(6):301–310. | ||
Houten SM, van Woerden CS, Wijburg FA, Wanders RJA, Waterham HR. Carrier frequency of the V377I (1129G>A) MVK mutation, associated with hyper-IgD and periodic fever syndrome, in the Netherlands. Eur J Hum Genet. 2003;11(2):196–200. | ||
Houten SM, Kuis W, Duran M, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22(2):175–177. | ||
Touitou I [webpage on the Internet]. Infevers: an online database for autoinflammatory mutations; 2016. Available from: http://fmf.igh.cnrs.fr/ISSAID/infevers/. Accessed May 28, 2016. | ||
Mandey SH, Schneiders MS, Koster J, Waterham HR. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Hum Mutat. 2006;27(8):796–802. | ||
Barron KS, Ombrello AK, Goldsmith DP, Aksentijevich I, Jones A, Kastner DL. Single MVK mutation and recurrent fevers. Arthritis Rheum. 2013;65:S926. | ||
Moura R, Tricarico PM, Campos Coelho AV, Crovella S. GRID2 a novel gene possibly associated with mevalonate kinase deficiency. Rheumatol Int. 2015;35(4):657–659. | ||
Houten SM, Koster J, Romeijn GJ, et al. Organization of the mevalonate kinase (MVK) gene and identification of novel mutations causing mevalonic aciduria and hyperimmunoglobulinaemia D and periodic fever syndrome. Eur J Hum Genet. 2001;9(4):253–259. | ||
Cuisset L, Drenth JP, Simon A, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet. 2001;9(4):260–266. | ||
Ter Haar NM, Jeyaratnam J, Lachmann HJ, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the Eurofever Registry. Arthritis Rheum. Epub 2016, May 23. | ||
Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys. 2011;505(2):131–143. | ||
Hoffmann GF, Charpentier C, Mayatepek E, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91(5):915–921. | ||
Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis. 2000;23(4):363–366. | ||
van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol. 2013;147(3):197–206. | ||
Ando S, Kaibuchi K, Sasaki T, et al. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem. 1992;267(36):25709–25713. | ||
McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63(3):255–267. | ||
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–668. | ||
Drenth JP, Powell RJ, Brown NS, Van der Meer JW. Interferon-gamma and urine neopterin in attacks of the hyperimmunoglobulinaemia D and periodic fever syndrome. Eur J Clin Invest. 1995;25(9):683–686. | ||
Drenth JP, van Deuren M, van der Ven-Jongekrijg J, Schalkwijk CG, van der Meer JW. Cytokine activation during attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Blood. 1995;85(12):3586–3593. | ||
Drenth JP, van der Meer JW, Kushner I. Unstimulated peripheral blood mononuclear cells from patients with the hyper-IgD syndrome produce cytokines capable of potent induction of C-reactive protein and serum amyloid A in Hep3B cells. J Immunol. 1996;157(1):400–404. | ||
Stoffels M, Jongekrijg J, Remijn T, Kok N, van der Meer JWM, Simon A. TLR2/TLR4-dependent exaggerated cytokine production in hyperimmunoglobulinaemia D and periodic fever syndrome. Rheumatology (Oxford). 2015;54(2):363–368. | ||
Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum. 2006;54(11):3690–3695. | ||
Hager EJ, Tse HM, Piganelli JD, et al. Deletion of a single mevalonate kinase (Mvk) allele yields a murine model of hyper-IgD syndrome. J Inherit Metab Dis. 2007;30(6):888–895. | ||
Hager EJ, Piganelli JD, Tse HM, Gibson KM. Aberrant expression of costimulatory molecules in splenocytes of the mevalonate kinase-deficient mouse model of human hyper-IgD syndrome (HIDS). J Inherit Metab Dis. 2012;35(1):159–168. | ||
Kuijk LM, Mandey SH, Schellens I, et al. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol Immunol. 2008;45(8):2158–2165. | ||
Kuijk LM, Beekman JM, Koster J, Waterham HR, Frenkel J, Coffer PJ. HMG-CoA reductase inhibition induces IL-1beta release through Rac1/PI3K/PKB-dependent caspase-1 activation. Blood. 2008;112(9):3563–3573. | ||
Henneman L, Schneiders MS, Turkenburg M, Waterham HR. Compromised geranylgeranylation of RhoA and Rac1 in mevalonate kinase deficiency. J Inherit Metab Dis. 2010;33(5):625–632. | ||
van der Burgh R, Pervolaraki K, Turkenburg M, Waterham HR, Frenkel J, Boes M. Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J Biol Chem. 2014;289(40):27757–27765. | ||
van der Burgh R, Nijhuis L, Pervolaraki K, et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J Biol Chem. 2014;289(8):5000–5012. | ||
Tricarico PM, Piscianz E, Monasta L, Kleiner G, Crovella S, Marcuzzi A. Microglia activation and interaction with neuronal cells in a biochemical model of mevalonate kinase deficiency. Apoptosis. 2015;20(8):1048–1055. | ||
Marcuzzi A, Pontillo A, De Leo L, et al. Natural isoprenoids are able to reduce inflammation in a mouse model of mevalonate kinase deficiency. Pediatr Res. 2008;64(2):177–182. | ||
Marcuzzi A, Decorti G, Pontillo A, Ventura A, Tommasini A. Decreased cholesterol levels reflect a consumption of anti-inflammatory isoprenoids associated with an impaired control of inflammation in a mouse model of mevalonate kinase deficiency. Inflamm Res. 2010;59(5):335–338. | ||
Marcuzzi A, Zanin V, Kleiner G, Monasta L, Crovella S. Mouse model of mevalonate kinase deficiency: comparison of cytokine and chemokine profile with that of human patients. Pediatr Res. 2013;74(3):266–271. | ||
De Leo L, Marcuzzi A, Decorti G, Tommasini A, Crovella S, Pontillo A. Targeting farnesyl-transferase as a novel therapeutic strategy for mevalonate kinase deficiency: in vitro and in vivo approaches. Pharmacol Res. 2010;61(6):506–510. | ||
Pontillo A, Paoluzzi E, Crovella S. The inhibition of mevalonate pathway induces upregulation of NALP3 expression: new insight in the pathogenesis of mevalonate kinase deficiency. Eur J Hum Genet. 2010;18(7):844–847. | ||
Marcuzzi A, Piscianz E, Girardelli M, Crovella S, Pontillo A. Defect in mevalonate pathway induces pyroptosis in Raw 264.7 murine monocytes. Apoptosis. 2011;16(9):882–888. | ||
Kleiner G, Celsi F, Tricarico PM, Zacchigna S, Crovella S, Marcuzzi A. Systemic and neuronal inflammatory markers in a mouse model of mevalonate kinase deficiency: a strain-comparative study. In Vivo. 2013;27(6):715–722. | ||
Celsi F, Piscianz E, Romano M, Crovella S. Knockdown of MVK does not lead to changes in NALP3 expression or activation. J Inflamm (Lond). 2015;12:7. | ||
Drenth JPH, Haagsma CJ, van der Meer JWM, et al. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Baltimore). 1994;73(3):133–144. | ||
Van Der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep. 2010;12(2):101–107. | ||
Bader-Meunier B, Florkin B, Sibilia J, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. 2011;128(1):e152–e159. | ||
Durel C-A, Aouba A, Bienvenu B, et al. Observational study of a French and Belgian multicenter cohort of 23 patients diagnosed in adulthood with mevalonate kinase deficiency medicine ® OBSERVATIONAL STUDY. Medicine (Baltimore). 2016;95(11):1–7. | ||
Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006;1:13. | ||
Hinson DD, Rogers ZR, Hoffmann GF, et al. Hematological abnormalities and cholestatic liver disease in two patients with mevalonate kinase deficiency. Am J Med Genet. 1998;78(5):408–412. | ||
Siemiatkowska AM, Van Den Born LI, Van Hagen PM, et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120(12):2697–2705. | ||
Zhang S-Q, Jiang T, Li M, et al. Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet. 2012;44(10):1156–1160. | ||
Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62(12):1795–1805. | ||
Bianco AM, Girardelli M, Vozzi D, Crovella S, Kleiner G, Marcuzzi A. Mevalonate kinase deficiency and IBD: shared genetic background. Gut. 2014;63(8):1367–1368. | ||
Levy M, Arion A, Berrebi D, et al. Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics. 2013;132(3):e779–e783. | ||
Wilding EI, Brown JR, Bryant AP, et al. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol. 2000;182(15):4319–4327. | ||
Ravelli A. Macrophage activation syndrome. Medscape. 2002;14:548–552. | ||
Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol. 2010;22(5):561–566. | ||
Stabile A, Bertoni B, Ansuini V, La Torraca I, Sallì A, Rigante D. The clinical spectrum and treatment options of macrophage activation syndrome in the pediatric age. Eur Rev Med Pharmacol Sci. 2006;10(2):53–59. | ||
Rigante D, Emmi G, Fastiggi M, Silvestri E, Cantarini L. Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clin Rheumatol. 2015;34(8):1333–1339. | ||
Rigante D, Capoluongo E, Bertoni B, et al. First report of macrophage activation syndrome in hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheum. 2007;56(2):658–661. | ||
Schulert GS, Bove K, McMasters R, Campbell K, Leslie N, Grom AA. Mevalonate kinase deficiency associated with recurrent liver dysfunction, macrophage activation syndrome and perforin gene polymorphism. Arthritis Care Res (Hoboken). 2015;67(8):1173–9. | ||
Ammouri W, Cuisset L, Rouaghe S, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology(Oxford). 2007;46(10):1597–1600. | ||
Lane T, Loeffler JM, Rowczenio DM, et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013;65(4):1116–1121. | ||
Jeyaratnam J, Ter Haar NM, de Sain-van der Velden MGM, Waterham HR, van Gijn ME, Frenkel J. Diagnostic value of urinary mevalonic acid excretion in patients with a clinical suspicion of mevalonate kinase deficiency (MKD). JIMD Rep. 2016;27:33–38. | ||
Berody S, Galeotti C, Koné-Paut I, Piram M. A retrospective survey of patient’s journey before the diagnosis of mevalonate kinase deficiency. Joint Bone Spine. 2015;82(4):240–244. | ||
Ter Haar N, Lachmann H, Ozen S, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013;72(5):678–685. | ||
Bodar EJ, Kuijk LM, Drenth JPH, van der Meer JWM, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70(12):2155–2158. | ||
Rossi-Semerano L, Fautrel B, Wendling D, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10:19. | ||
Galeotti C, Meinzer U, Quartier P, et al. Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatol (Oxford). 2012;51(10):1855–1859. | ||
Bodar EJ, van der Hilst JCH, Drenth JPH, van der Meer JWM, Simon A. Effect of etanercept and anakinra on inflammatory attacks in the hyper-IgD syndrome: Introducing a vaccination provocation model. Neth J Med. 2005;63(7):260–264. | ||
Shendi HM, Devlin LA, Edgar JD. Interleukin 6 blockade for hyperimmunoglobulin D and periodic fever syndrome. J Clin Rheumatol. 2014;20(2):103–105. | ||
Marcuzzi A, De Leo L, Decorti G, Crovella S, Tommasini A, Pontillo A. The farnesyltransferase inhibitors Tipifarnib and Lonafarnib inhibit cytokines secretion in a cellular model of mevalonate kinase deficiency. Pediatr Res. 2011;70(1):78–82. | ||
Cantarini L, Vitale A, Magnotti F, et al. Weekly oral alendronate in mevalonate kinase deficiency. Orphanet J Rare Dis. 2013;8:196. | ||
Tricarico PM, Girardelli M, Kleiner G, et al. Alendronate, a double-edged sword acting in the mevalonate pathway. Mol Med Rep. 2015;12(3):4238–4242. | ||
Neven B, Valayannopoulos V, Quartier P, et al. Allogeneic bone marrow transplantation in mevalonic aciduria. N Engl J Med. 2007;356(26):2700–2703. | ||
Chaudhury S, Hormaza L, Mohammad S, et al. Liver transplantation followed by allogeneic hematopoietic stem cell transplantation for atypical mevalonic aciduria. Am J Transplant. 2012;12(6):1627–1631. | ||
Giardino S, Lanino E, Morreale G, et al. Long-term outcome of a successful cord blood stem cell transplant in mevalonate kinase deficiency. Pediatrics. 2015;135(1):e211–e215. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.