")
Back to Journals » OncoTargets and Therapy » Volume 13
Metformin Increases the Chemosensitivity of Pancreatic Cancer Cells to Gemcitabine by Reversing EMT Through Regulation DNA Methylation of miR-663
Authors Gu Y , Zhang B, Gu G, Yang X, Qian Z
Received 15 May 2020
Accepted for publication 6 September 2020
Published 14 October 2020 Volume 2020:13 Pages 10417—10429
DOI https://doi.org/10.2147/OTT.S261570
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Yuqing Gu, Bin Zhang, Guangliang Gu, Xiaojun Yang, Zhuyin Qian
Pancreas Center, The Second Affiliated Hospital to Nanjing Medical University, Nanjing 210003, People’s Republic of China
Correspondence: Zhuyin Qian Email [email protected]
Background: Pancreatic cancer is a devastating malignancy with poor prognosis. Metformin, a classic anti-diabetes drug, seems to improve survival of pancreatic cancer patients in some studies.
Methods: Cell counting kit-8 assay was used to detect the BxPC-3 and MIAPaCa-2 cell viability after treatment with gemcitabine only or with different concentrations of metformin. The methylation state and expression level of miR-663 were detected by methylation analysis and RT-PCR. Dual-luciferase reporter gene analysis, Western blot and RT-PCR were used to confirm the target of miR-663. Moreover, xenograft experiment was also performed to validate the role of metformin in chemosensitivity in vivo.
Results: We found that metformin increased the chemosensitivity of pancreatic cancer cells to gemcitabine, and epithelial–mesenchymal transition (EMT) progress caused by gemcitabine was suppressed by metformin. We further explored the possible molecular mechanisms and it was demonstrated that CpG islands of miR-663 were hypomethylated and relative expression level of miR-663 was up-regulated after treatment of metformin. miR-663, an important cancer suppressor miRNA, was confirmed to increase the chemosensitivity of pancreatic cancer cells by reversing EMT directly targeted TGF-β 1. Moreover, we identified that metformin increased the chemosensitivity through up-regulating expression of miR-663.
Conclusion: We demonstrated that metformin increased the chemosensitivity of pancreatic cancer cells to gemcitabine by reversing EMT through regulation DNA methylation of miR-663.
Keywords: metformin, pancreatic cancer, epithelial-mesenchymal transition; EMT, DNA methylation, miR-663
Introduction
Pancreatic cancer is one of the devastating malignancies of the digestive system. The incidence and mortality of pancreatic cancer have increased rapidly in recent years.1 Patients, whether they received radical resection or were diagnosed at an advanced stage, are all recommended to receive comprehensive treatment including chemotherapy to improve prognosis.2 However, the effect of chemotherapy in pancreatic cancer is still not satisfactory.3 The overall 5-year survival rate of pancreatic cancer is about 8%.1,4 Increasing the chemosensitivity of pancreatic cancer is one of the hotspots in research field of pancreatic cancer. In recent years, some evidence shows that metformin, a classic drug for diabetes mellitus, seems to improve survival of pancreatic cancer patients.5,6 The combination of metformin and gemcitabine has synergistic effect, and metformin can be used as an adjuvant drug to improve the clinical efficacy of gemcitabine of pancreatic cancer patients.7–9
It was found that epithelial-mesenchymal transition (EMT) can lead to chemotherapeutic resistance to gemcitabine in pancreatic cancer.10 When EMT process occurs in cancer cells, cell division is partly blocked, which reduces the efficiency of gemcitabine, a cell division dependent drug. Therefore, inhibiting EMT process can increase the sensitivity to gemcitabine, which provides new clues and ideas for reversing drug resistance in pancreatic cancer.11,12 We observed that metformin could reverse the gemcitabine-induced EMT process in pancreatic cancer cells. Thus, it is speculated that the reversal of the EMT process may be the mechanism of metformin to increase the sensitivity to gemcitabine. We further studied the molecular mechanism caused by metformin in pancreatic cancer cells.
Metformin can alter the DNA methylation status of cancer cells in a variety of ways.13,14 Our previous study found that the promoter region of miR-663 was abnormally hypermethylated in pancreatic cancer cells, which inhibit the expression of miR-663.15 After treatment with metformin in pancreatic cancer cells, the methylation status of miR-663 and its expression level was detected. We took miR-663 into further research, and then focused on its role in the chemosensitivity of pancreatic cancer cells. Our results indicate that metformin can increase the chemosensitivity to gemcitabine in pancreatic cancer cells by reversing EMT through regulation DNA methylation of miR-663.
Materials and Methods
In vitro Cell Culture
Pancreatic cancer cell lines BXPC-3 and MIAPaCa-2 were purchased from Shanghai Cell Bank and cultured in DMEM (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Sigma, St Louis, MO, USA) in a 5% CO2 incubator at 37°C. The gemcitabine/metformin was dissolved in PBS with different concentrations.
Quantitative Real‐Time Polymerase Chain Reaction
Pancreatic cancer cells were treated with TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Inc) to extract RNA according to the manufacturer’s protocol. The expression level of miRNA was detected using All-in-One Customized Human qPCR Array (GeneCopoeia Inc, Rockville, Md, USA). The TGF-β1 mRNA level was quantified using SYBR-Green PCR Master Mix kit (Takara, Dalian, China) normalized to β-actin. The primer sequences used were as follows. TGF-β1, forward, 5ʹ-GGACACCAACTATTGCTTCAG-3ʹ, TGF-β1 reverse, 5ʹ-TCCAGGCTCCAAATGTAG-3ʹ, β-actin, forward, 5ʹ-AGAAAATCTGGCACCACACC-3ʹ, β-actin, reverse, 5ʹ-TAGCACAGCCTGGATAGCAA-3ʹ.
Western Blot
Total protein was extracted using RIPA lysis buffer. Protein samples were subjected to separation on SDS-PAGE gels and then transferred onto polyvinylidene difluoride membranes, which were blocked using 5% non-fat milk. The membranes were incubated with antibodies against TGF-β1 (1:1000 dilution, Abcam, Cambridge, MA), E-cadherin (1:500 dilution, Santa Cruz, Dallas, TX, USA), Vimentin (1:1000 dilution, Abcam, Cambridge, MA), and β-actin (1:1000 dilution, Beyotime, Nantong, China) at 4°C overnight. The membranes were then incubated with secondary antibody at room temperature for 2 h. The protein brands were visualized using an ECL chemiluminescent kit (Pierce, Rockford, IL, USA). Quantification of Western blots was conducted using Index of Density.
Cell Transfection
miR-663 mimic or inhibitor and negative control was purchased from RiboBio (Guangzhou, Guangdong, China). Pancreatic cancer cell lines BxPC-3 and MIAPaCa-2 cells were cultured in 6-well plates for 24 h to 50–60% confluence and transfected with 30nmol/L miRNA or negative control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Cell Counting Kit-8 (CCK-8) Assay
The BxPC-3 and MIAPaCa-2 were seeded into 96-well plates. After being treated with gemcitabine, or gemcitabine+metformin for 48 h, CCK-8 reagent was added to each well and further cultured for 2 h. Absorbance values were measured at 550 nm wavelength with a microplate reader (Tecan, Shanghai, China).
Wound Healing Assay
Cells were cultured in 6-well plates for 24 h to 70–80% confluence. The artificial wound was created using a 10 μL sterile pipette tip. After washed using PBS, cells were incubated in FBS-free DMEM for 24 h. The migrated cells were imaged at 0, 12 and 24 h. The cell migration ability was calculated by the proportion of the wound.
Cell Invasion Assay
Cell invasion assay was performed with a Transwell assay kit and invasion chambers (Corning, Corning, NY, USA). The upper chamber of 8 μm pore transwells were overlaid with 1mg/mL Matrigel-coated membrane matrix. 0.25 × 105 cells per well (BxPC-3) or 0.5×105 cells per well (MIAPaCa-2) were plated in the upper chamber with serum-free medium. The lower chamber contained DMEM supplemented with 10% fetal bovine serum. After 24 h, cells invaded through the membrane were stained with 0.1% crystal violet for 30 min at 37 °C. Stained cells were soaked in 33% ice-cold acetic acid and absorbance values were measured at 550 nm wavelength with a microplate reader.
Dual-Luciferase Reporter Gene Analysis
miRanda, TargetScan, and TarBase were used to predict the miRNA targets. The mutant or wild-type 3ʹ-UTR of TGF-β1 sequence was cloned into the pGL3-control vector (Promega, Madison, WI, USA). After co-transfected with vector and miRNA, the luciferase activity was detected using the Dual-Luciferase Reporter Assay System (Promega).
DNA Isolation and Methylation Analysis
Genomic DNA was extracted from pancreatic cancer cell lines using the DNeasy kit (Qiagen, Hilden, Germany). Bisulfite modification procedure was according to the manufacturer’s instructions of Epitect Bisulfite kit (Qiagen).
The PCR primers were designed as follows: miR-615 forward, 5′-AGTTAAGAGTAGTGGGGAGATTAA-3′, reverse, 5′-AAATTTTTTTTCTTTATTTACCCC-3′, miR-663 forward, 5′-GTTGAGGAGGGTTTTTTTGTA-3′, reverse, 5′-CCTCGATAAAAAAACCTTCTCT-3′, miR-663b forward, 5′-TATTTTATTAAGGGGGAAGTGG-3′, reverse, 5′-CCTCGATAAAAAAACCTTCTCT-3′, miR-675 forward, 5′-GGGTTTTGGAGGTTAGTGT-3′, reverse, 5′-CTTAACAAACAATACAACATCCA-3′. The PCR products of sodium bisulfite-treated DNA were cloned into pTA2 vector using the TA cloning kit (Toyobo, Osaka, Japan), and single clones of six samples were sent for sequencing.
Xenograft Experiment
Six-week-old BALB/c nude mice were obtained from the Shanghai Experimental Animal Center. There were two groups, gemcitabine group and gemcitabine + metformin group. Five mice were assigned per group. A total of 1 × 107 MIAPaCa-2 cells were injected subcutaneously into the neck area of each mouse. After cell injection, the mice were treated with drugs, and the scheme was as following, gemcitabine only group (100 mg/kg of gemcitabine injected intraperitoneally twice weekly), combination gemcitabine with metformin group (gemcitabine 100 mg/kg injected intraperitoneally twice weekly and 800 mg/L metformin in drinking water each day). The tumor width, length, and height were measured by external caliper every 5 days and calculated as follows: Volume (mm3) = width × length × height/2. The mice were sacrificed by cervical dislocation following 25 days of treatment and the tumors were collected, fixed, and embedded. The animal experiments were approved by the Animal Care and Use Committee of The Second Affiliated Hospital of Nanjing Medical University, approval number 2018-KY-027, and in accordance with the Guide for the Care and Use of Laboratory Animals (eighth edition, 2011, National Institutes of Health, USA).
Immunohistochemistry
Immunohistochemistry was used to examine TGF-β1 expression in tumor xenografts. After fixed in formalin, embedded in paraffin blocks, tumor tissues were incubated with primary antibody against TGF-β1 (1:500 dilution, Abcam, Cambridge, MA) overnight at 4 °C. Goat anti-rabbit IgG antibody conjugated to HRP (1:200 dilution, Santa Cruz, Dallas, TX, USA) were incubated for 2 h at room temperature. Subsequently, the sections were stained with DAB and counterstained with hematoxylin.
Statistical Analysis
All experimental results were shown as the means ± standard deviation of three separate experiments. The statistical significance of differences was performed using the Student t tests when only two groups were compared. The difference among multiple groups was compared using one-way ANOVA using Dunnett’s multiple comparison test. P<0.05 was considered to indicate a significant.
Results
Metformin Increased the Chemosensitivity of Pancreatic Cancer Cells to Gemcitabine by Reversing EMT in vitro
To study the role of metformin in the chemosensitivity of pancreatic cancer cells to gemcitabine, the CCK-8 assay was used to detect the BxPC-3 and MIAPaCa-2 cell viability after treatment with gemcitabine only or with different concentrations of metformin. It showed that the survival of pancreatic cancer cells was inhibited by gemcitabine, and when combined with different concentrations of metformin, cell viability was further inhibited which exhibited synergistic effect (Figure 1A). The difference was statistically significant. We used Calcusyn software to evaluate the synergistic effect of gemcitabine and metformin, and found that the best CI value was 0.549 when the concentration of gemcitabine and metformin was 143.2nmol/L and 6.8mmol/L for MIAPaCa-2, and the best CI value was 0.645 when the concentration of gemcitabine and metformin was 633.9nmol/L and 4.8mmol/L for BxPC-3.
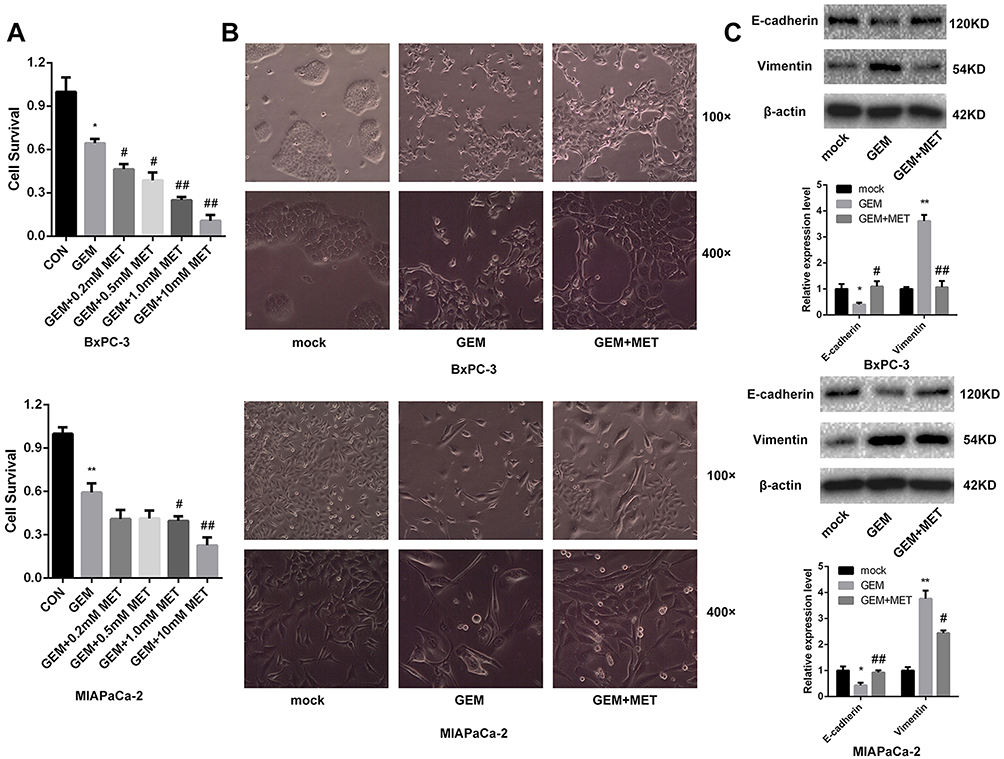 |
Figure 1 Metformin increased the chemosensitivity of pancreatic cancer cells by reversing EMT. (A) Cell viability was determined by CCK-8 assay after treated with gemcitabine only or with different concentrations of metformin. *PCON vs. GEM < 0.05, **PCON vs. GEM < 0.01, #PGEM vs. GEM+MET < 0.05, ##PGEM vs. GEM+MET < 0.01. (B) Morphology changes of BxPC-3 and MIAPaCa-2 cells were taken before and after treated with gemcitabine only or with metformin. (C) The protein levels of E-cadherin and Vimentin in BxPC-3 and MIAPaCa-2 cells were determined by Western blot analyses. *PCON vs. GEM < 0.05, **PCON vs. GEM < 0.01, #PGEM vs. GEM+MET < 0.05, ##PGEM vs. GEM+MET < 0.01. |
Before and after treatment of gemcitabine only or with metformin, morphological changes were taken by microscope of BxPC-3 and MIAPaCa-2. BxPC-3 and MIAPaCa-2 cells clustered together, and after treatment of gemcitabine, cells gradually transitted from epithelial-like cells to mesenchymal-like cells, which were elongated, and grew individually. While it was treated with gemcitabine and metformin, EMT progress was suppressed and epithelial-like cells reappeared. (Figure 1B).
EMT-related protein E-cadherin and Vimentin were detected in pancreatic cancer cells using Western blot.
The lower expression level of E-cadherin and higher expression level of Vimentin was found after gemcitabine treatment in both BxPC-3 and MIAPaCa-2 cells, while it was treated with gemcitabine and metformin, E-cadherin was increased and Vimentin was decreased comparing with metformin only group (Figure 1C). The difference was statistically significant.
CpG Islands of miR-663 Were Hypomethylated and Relative Expression of miR-663 Was Up-Regulated After Treatment of Metformin in Pancreatic Cancer Cells
As we described before,15 CpG islands of miR-615, miR-663, miR-663b and miR-675 were confirmed to be hypermethylated. It had been reported that metformin could alter the DNA methylation status of cancer cells, so after treated of metformin for BxPC-3, the percentage of methylated CpG sites of the four miRNAs was re-examined and miR-663 was shown to be hypomethylated (80.3±5.5% vs. 30.3±13.8%, the difference was statistically significant), while there was no significant change of miR-615, miR-663b and miR-675 (Figure 2A). Furthermore, the expression of miR-663 in BxPC-3 and MIAPaCa-2 cells was up-regulated by 5.7±0.3 and 5.1±0.6 fold after treatment of metformin (Figure 2B), and the difference was statistically significant.
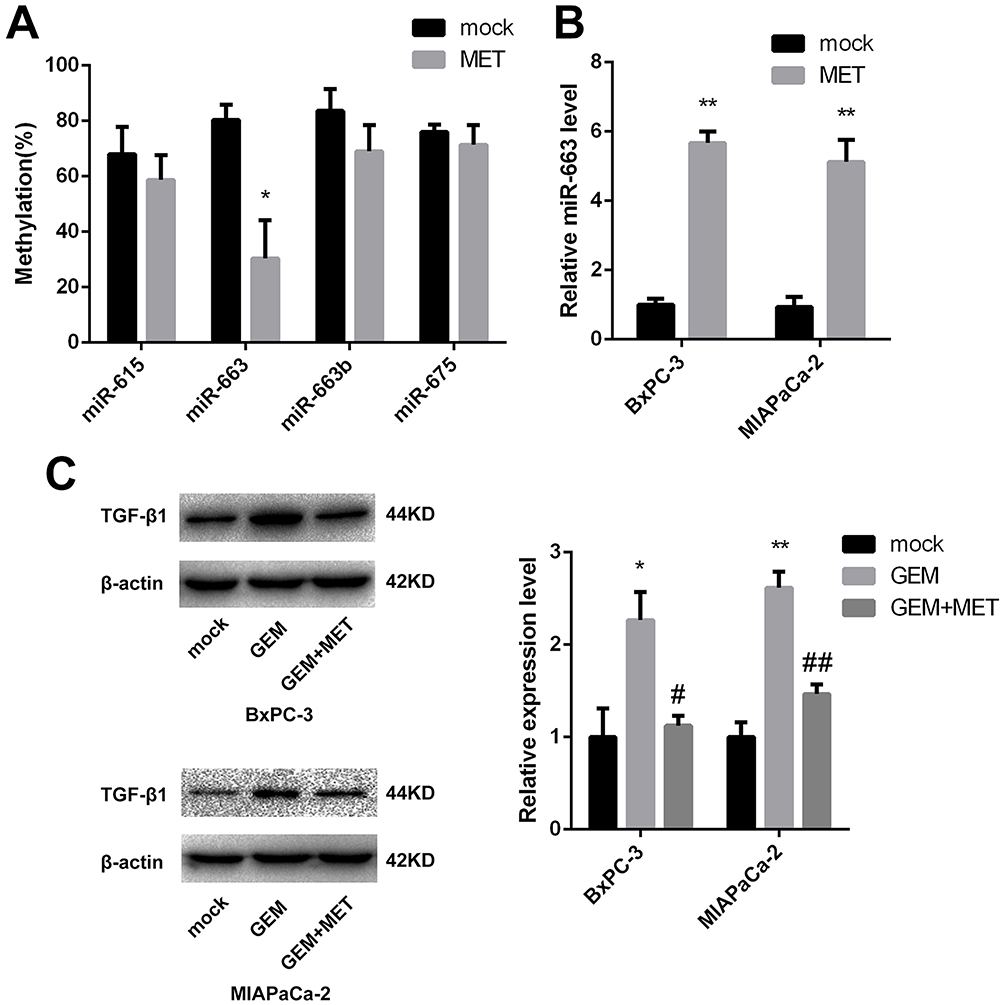 |
Figure 2 DNA methylation status and relative expression of miR-663 was altered after treatment of metformin. (A) The percentage of methylated CpG sites of miR-615, miR-663, miR-663b and miR-675 was shown for BxPC-3 before and after treatment of metformin. *P < 0.05. (B) Relative expression of miR-663 was shown for BxPC-3 and MIAPaCa-2 before and after treatment of metformin. **P < 0.01. (C) The protein level of TGF-β1 in BxPC-3 and MIAPaCa-2 cells were determined by Western blot analyses. *PCON vs. GEM < 0.05,**PCON vs. GEM < 0.01, #PGEM vs. GEM+MET < 0.05, ##PGEM vs. GEM+MET < 0.01. |
miR-663 Increased the Chemosensitivity of Pancreatic Cancer Cells to Gemcitabine by Reversing EMT in vitro
Because miR-663 was up-regulated after treatment of metformin, we further evaluated the role of miR-663 in chemosensitivity of pancreatic cancer cells to gemcitabine. CCK-8 assay revealed that up-regulation of miR-663 significantly increased the chemosensitivity to gemcitabine of BxPC-3 and MIAPaCa-2 cells (Figure 3C). The difference was statistically significant.
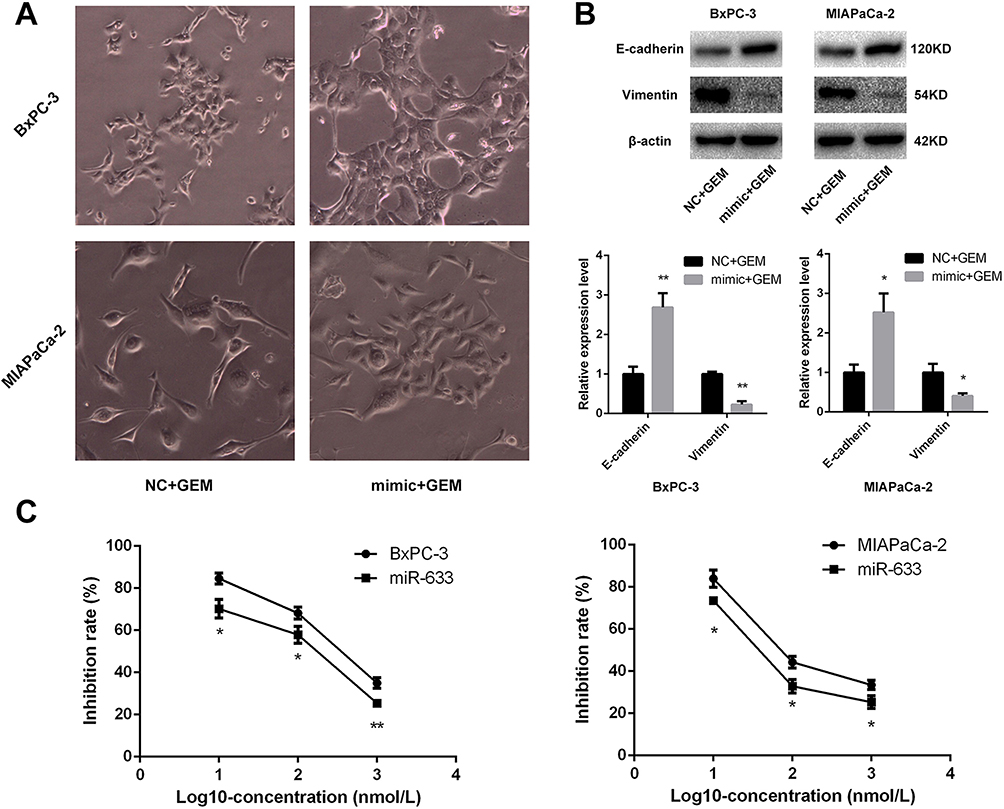 |
Figure 3 miR-663 increased the chemosensitivity of pancreatic cancer cells by reversing EMT. (A) Morphology changes of BxPC-3 and MIAPaCa-2 cells were taken treating with gemcitabine after transfected with miR-663 mimic or negative control (NC). Magnification: × 400. (B) The protein levels of E-cadherin and Vimentin in BxPC-3 and MIAPaCa-2 cells were determined by Western blot analyses. *P < 0.05, **P < 0.01. (C) Cell inhibition rate was determined by CCK-8 assay treated with different concentrations of gemcitabine after transfected with miR-663 mimic or NC. *P < 0.05, **P < 0.01. |
Morphological changes was also taken after treatment of gemcitabine on the miR-663-overexpressed BxPC-3 and MIAPaCa-2 cells, and the EMT progress caused by gemcitabine was suppressed by miR-663 (Figure 3A). The expression levels of EMT-related protein were detected, and it was found that E-cadherin was increased and Vimentin was decreased after overexpression of miR-663 (Figure 3B). The difference was statistically significant.
It was reported that EMT was also related to invasion and metastasis of pancreatic cancer. The migratory ability of BxPC-3 and MIAPaCa-2 cells measured by the wound healing assay demonstrated that miR-663 significantly inhibited the cell migration (Figure 4A), and miR-663 reduced the cell invasive ability using a Matrigel invasion assay (Figure 4B). The difference was statistically significant.
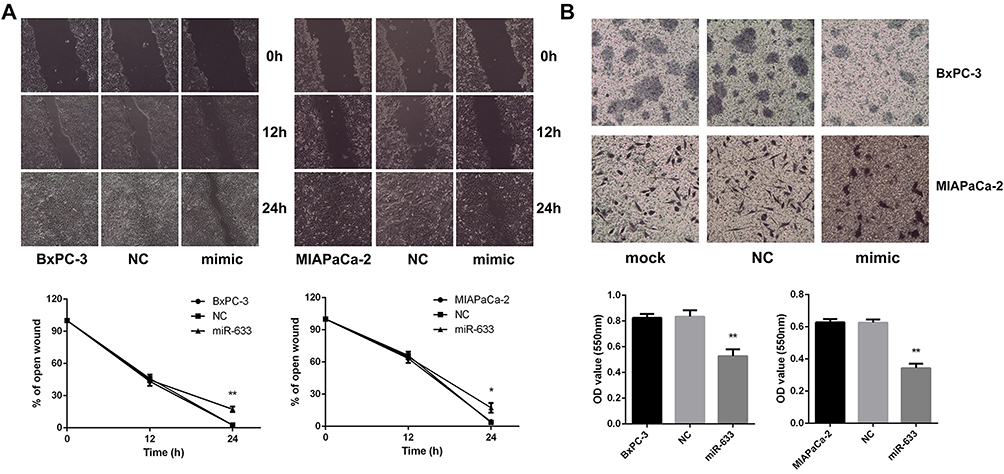 |
Figure 4 miR-663 inhibited cell migration and invasion of BxPC-3 and MIAPaCa-2 in vitro. (A) Wound healing assay was performed on BxPC-3 and MIAPaCa-2 cells after transfected with miR-663 mimic or NC. Magnification: × 200. *P < 0.05, **P < 0.01. (B) Transwell invasion assay was performed on BxPC-3 and MIAPaCa-2 cells after transfected with miR-663 mimic or NC. Magnification: × 200. **P < 0.01. |
miR-663 Directly Targeted TGF-β1
In order to investigate the role of miR-663 in pancreatic cancer, we used Targetscan to explore the potential targets of miR-663, and EMT-related gene TGF-β1 was selected for further analysis (Figure 5A). Two groups of luciferase reporter constructs were used to confirm the prediction. It was shown that the luciferase activity was significantly reduced to 65.3±4.9% of BxPC-3 and 26.0±5.0% of MIAPaCa-2 in miR-663 mimic + TGF-β1_WT co-transfected group comparing with WT+NC group (Figure 5B).
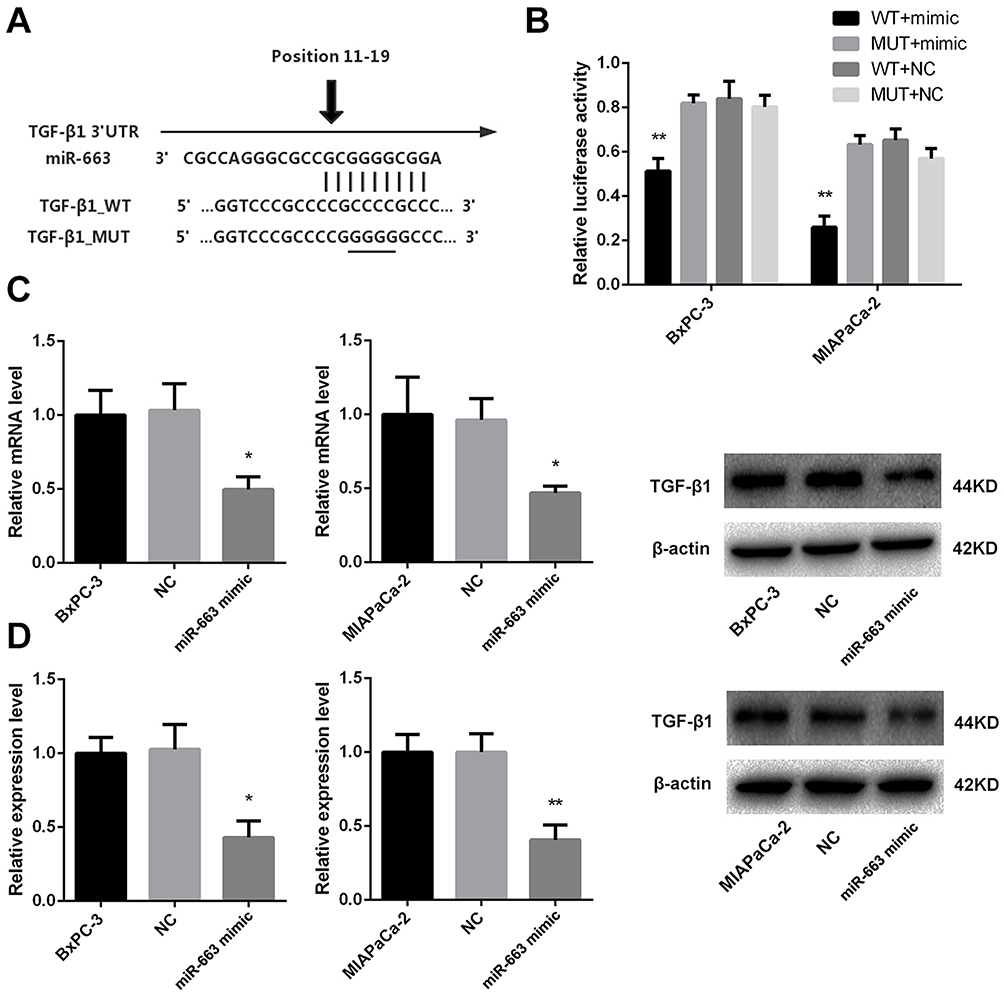 |
Figure 5 miR-663 inhibited expression of TGF-β1 by targeting 3ʹ-UTR of TGF-β1 in pancreatic cancer cells. (A) Sequence of the miR-663 binding site with TGF-β1 3ʹ-UTR (TGF-β1_WT) and the mutated TGF-β1 3ʹ-UTR sequence (TGF-β1_MUT). (B) BxPC-3 and MIAPaCa-2 cells were co-transfected with miR-663 mimic or NC and luciferase reporter vectors containing theTGF-β1_WT or TGF-β1_MUT sequences of the miR-663 binding sites. Luciferase activities were measured 48 h after transfection. **P < 0.01. (C) The relative expression of TGF-β1 mRNA was measured by real-time PCR in BxPC-3 and MIAPaCa-2 cells. *P < 0.05. (D) TGF-β1 protein in BxPC-3 and MIAPaCa-2 cells was detected by Western blot. *P < 0.05, **P < 0.01. |
We further examined the regulation of miR-663 on TGF-β1 expression in pancreatic cancer cells. The results indicated that miR-663 significantly inhibited the expression level of TGF-β1 mRNA (Figure 5C) and protein (Figure 5D) in BxPC-3 and MIAPaCa-2 cells. These data indicated that TGF-β1 was the direct target of miR-663. The difference was statistically significant.
Metformin Increased the Chemosensitivity of Pancreatic Cancer Cells Through Up-Regulating Expression of miR-663
In order to test our hypothesis that the effect of metformin resulted from up-regulating the expression of miR-663, BxPC-3, and MIAPaCa-2 cells were co-treated with miR-663 inhibitor and metformin. CCK-8 assay revealed that down-regulation of miR-663 significantly decreased the chemosensitivity to gemcitabine of BxPC-3 and MIAPaCa-2 cells. When co-treated with metformin and miR-663 inhibitor, there were no statistical differences between control group and miR-663 inhibitor + metformin group (Figure 6A).
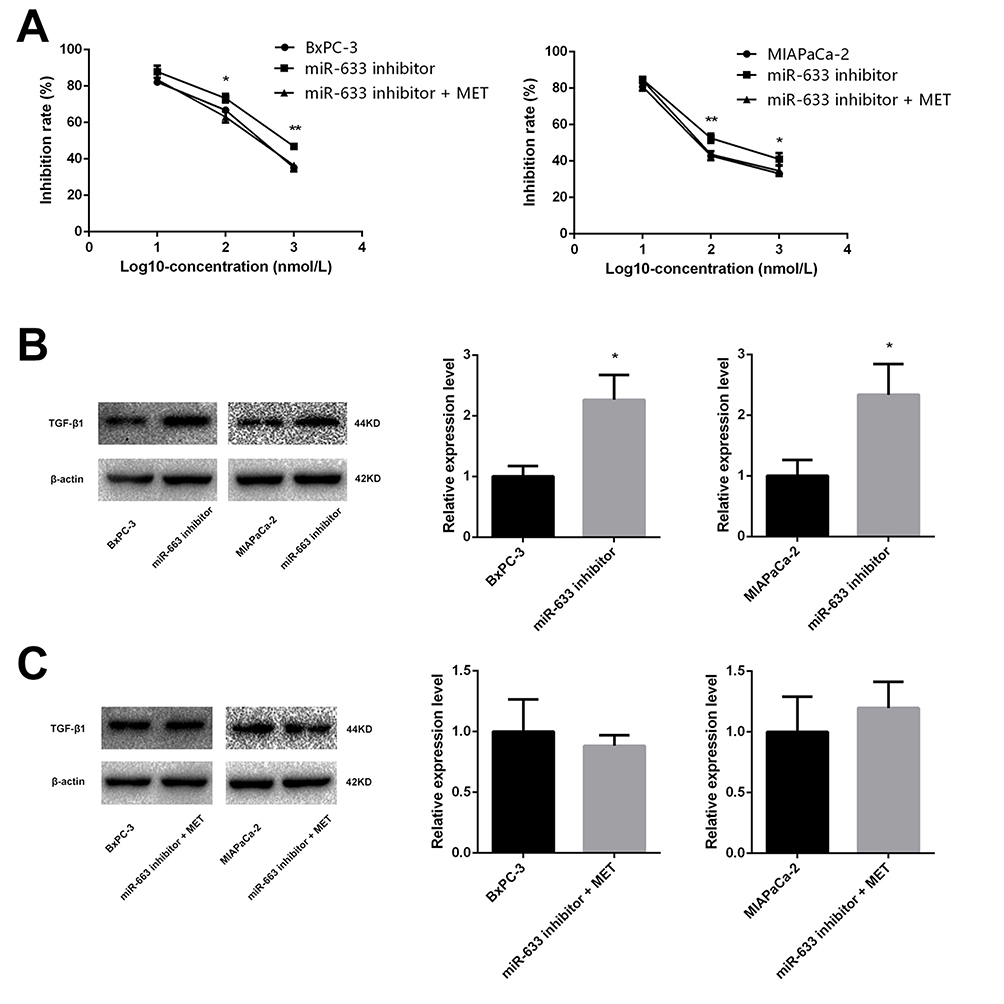 |
Figure 6 Metformin increased the chemosensitivity through up-regulating expression of miR-663. (A) Cell inhibition rate was determined by CCK-8 assay treated with different concentrations of gemcitabine after transfected with miR-663 inhibitor only or with metformin. *PCON vs. inhibitor < 0.05, **PCON vs. inhibitor < 0.01. (B and C) The protein level of TGF-β1 in BxPC-3 and MIAPaCa-2 cells were determined by Western blot analyses. *P < 0.05. |
The expression level of TGF-β1 protein was detected in pancreatic cancer cells using Western blot. Compared with control group, TGF-β1 protein was increased in gemcitabine group. When treated with gemcitabine and metformin, the increased TGF-β1 protein trend disappeared (Figure 2C). We also detected the TGF-β1 protein in miR-663 inhibitor group and miR-663 inhibitor + metformin group of BxPC-3 and MIAPaCa-2 cells, the TGF-β1 protein was down-regulated in the miR-663 inhibitor group compared with the control group (Figure 6B). The difference was statistically significant. When co-treated with metformin and miR-663 inhibitor, there were no statistical differences of TGF-β1 protein between the control group and miR-663 inhibitor + metformin group (Figure 6C).
Metformin Increased the Chemosensitivity of Pancreatic Cancer Cells to Gemcitabine in vivo
To investigate the role of metformin in chemosensitivity of pancreatic cancer cells to gemcitabine in vivo, xenograft experiments were conducted. The tumor volume was measured every 5 days, and tumor growth was slower in the gemcitabine group than in the gemcitabine + metformin group (Figure 7A, 7B). The difference was statistically significant. Mice were sacrificed at the 25th day and the weights of the tumors were measured. The average weight of tumors from the gemcitabine + metformin group was significantly less than that from the gemcitabine group (360.0±85.4 vs. 180.0±60.0 mg, the difference was statistically significant) (Figure 7C). Furthermore, the protein expression levels of TGF-β1 in tumor tissues were examined by immunohistochemistry and it was markedly down-regulated in the gemcitabine + metformin group than in the gemcitabine group (83.3±8.6% vs. 45.3±6.7%, the difference was statistically significant) (Figure 7D,7E).
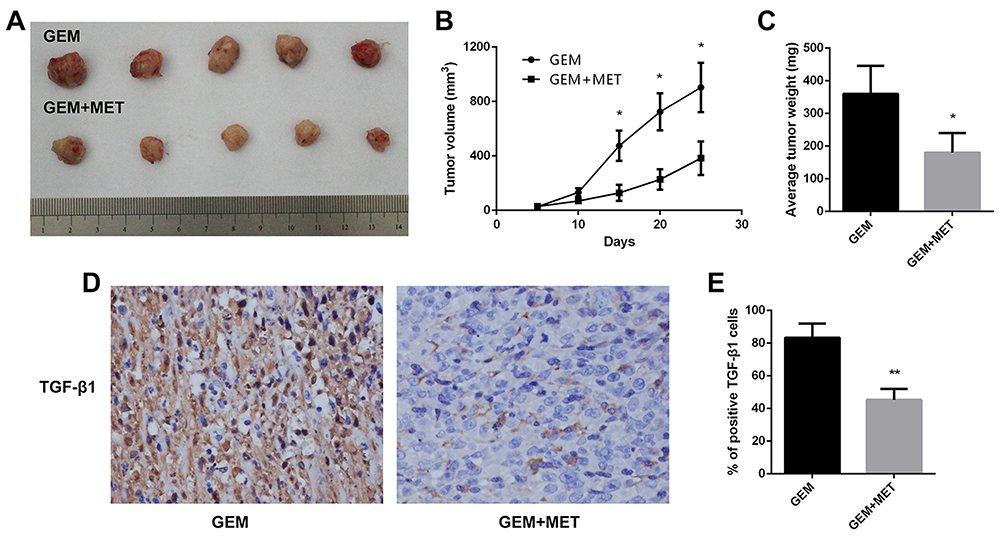 |
Figure 7 Metformin increased the chemosensitivity of pancreatic cancer in vivo. (A) Tumors were harvested from gemcitabine group and gemcitabine+metformin group of nude mice. (B) The tumor volume growth curve in vivo was shown. *P < 0.05. (C) The tumor weight of each group were calculated. *P < 0.05. (D) TGF-β1 protein levels were measured by immunohistochemistry in the tumor tissues isolated from the mice. Magnification: × 40. (E) The percentage of TGF-β1 positive cells was calculated. **P < 0.01. |
Discussion
Chemotherapy is widely used in all stages of pancreatic cancer patients, which is an important part of comprehensive treatment of pancreatic cancer. However, the effect of chemotherapy in pancreatic cancer is still not satisfactory, and drug resistance is the main reason for the failure of chemotherapy. EMT process appeared in the process of gemcitabine treatment of pancreatic cancer cells.16 The volume of epithelial cells reduced gradually, and the intercellular junction decreased, which turned to the characteristics of mesenchymal cells. The expression level of epithelial marker E-cadherin was down-regulated and mesenchymal marker Vimentin was up-regulated, which resulted in the loss of cell polarity in epithelial cells. EMT process can lead to resistance of gemcitabine in pancreatic cancer.17
Tumor cells show mesenchymal transition in the process of acquired drug resistance, and EMT process has gradually been considered as an important mechanism of drug resistance of tumor cells.10 More and more studies have shown that there are common regulatory signal pathways between EMT process and drug resistance of tumor cells.18,19 The inactivation and imbalance of EMT process-related signaling pathway play an important role in drug resistance. Therefore, reversal of the process of EMT provides a new clue to solve the problem of drug resistance.
Metformin has been proven to increase chemosensitivity to gemcitabine in pancreatic cancer, while its mechanism is not yet clear. Zhong et al13 found that metformin affected the expression of DNA methylase 3B by regulating the activity of S-adenosylhomocysteine (SAHH), thus altering the percentage of genomic DNA methylation. Cuyas et al14 found that metformin could increase the S-adenosylmethionine (SAM)/SAHH ratio and regulate the percentage of DNA methylation. In our previous study, we used methylation chip to detect the DNA methylation status of pancreatic cancer cells. The results showed that the promoter regions of 11 microRNAs were abnormally methylated in pancreatic cancer cells compared with normal pancreatic tissues. Pyrosequencing and TA clone sequencing was then used to confirm the presence of aberrant methylation in the promoter regions of miR-615, 663, 663b and 675.15 We explored whether metformin could alter the methylation status and the expression of the above microRNAs. After treatment with metformin in pancreatic cancer cells, the percentage of methylation of miR-663 was down-regulated and its expression level was up-regulated.
miR-663 is an important miRNA in tumorigenesis. The role of miR-663 in gastric cancer,20 breast cancer,21 liver cancer,22 lung cancer,23 bladder cancer,24 nasopharyngeal cancer,25 and other malignant tumors has been found, and its main functions focus on regulation of tumor proliferation, invasion, metastasis. and apoptosis. The expression level of miR-663 is varied in different types of tumors, and we noticed that it is regulated by DNA methylation26 and drugs (retinoic acid,27 fluoxetine)28 in some reported research. At the same time, the abnormal expression of miR-663 also has been found in drug-resistant breast cancer cells21 and castrated refractory prostate cancer.29 In pancreatic cancer, Zhang et al30 reported that the expression of miR-663 was lower than that of normal tissues, and miR-663 inhibited the proliferation and invasion of cancer cells. However, there are few reports about the function of miR-663 in chemotherapy of cancers. Our previous study found that abnormal DNA methylation inhibits the expression of miR-663 in pancreatic cancer cells. We over-expressed miR-663 using miRNA mimic in pancreatic cancer cell lines, and found that miR-663 significantly increased the chemosensitivity to gemcitabine.
The reported target genes of miR-663 in cancer cells include JUND, JUNB, PI3K, EEF1A2, which are closely related to multiple cellar functions.30–32 TGF-β1, a recognized gene that induces EMT process,33 is also the target gene of miR-663. TGF-β1 can activate Smad and bind with related nuclear transcription factors through the classical signaling pathway, then regulates the expression of downstream genes. Thereby, the signaling pathway adjusts the junctional structure between epithelial cells, and makes epithelial cells lose polarity and turn into mesenchymal cells.34 Bioinformatics prediction software showed that the 18–24 base of 3ʹ-UTR region of TGF-β1 paired with miR-663, and when it was over-expressed of miR-663 in pancreatic cancer cells, the expression of TGF-β1 was down-regulated at both the level of mRNA and protein. Luciferase reporter gene experiment further confirmed the targeting pairing relationship between miR-663 and the 3ʹ-UTR region of TGF-β1. Therefore, we speculated that the expression of TGF-β1 was regulated by miR-663, and then inhibited the process of EMT in pancreatic cancer cells.
It has been shown that another metformin-like drug AICAR is effective in combination with mTOR inhibitor-Rapamycin as anti-cancer agent.35 Interestingly, metformin activates AMPK while AMPK regulates the mTOR pathway in a feedback loop manner.36 In this study, we found that whether gemcitabine or gemcitabine + metformin treatment would affect the protein level of TGF-β, which is known to activate Akt through PI3K, that in turn activates the mTOR complex 1 (mTORC1).37 It will be interesting to see how this gemcitabine and metformin combination treatment affects mTOR signaling pathways. This may be meaningful in possible future studies.
The antitumor effect of metformin in pancreatic cancer patients is still controversial. Although the meta-analysis suggests that the usage of metformin is associated with a significantly lower mortality rate in pancreatic cancer patients,6 different results also existed that metformin use was not statistically significantly associated with improved survival.38 Therefore, we suppose that some selected pancreatic cancer patients can benefit from the usage of metformin, and further studies are needed.
In conclusion, the present study demonstrated that inhibition of EMT process accompanied with up-regulation of miR-663 was found in pancreatic cancer cells when treated with metformin. Over-expression of miR-663 increased the sensitivity of pancreatic cancer cells to gemcitabine and inhibited the EMT process. The analysis of the mechanism showed that TGF-β1 was the target gene of miR-663. Our results provide a new theoretical basis of metformin for improving chemosensitivity to gemcitabine in pancreatic cancer.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15(6):333–348. doi:10.1038/s41575-018-0005-x
3. Deplanque G, Demartines N. Pancreatic cancer: are more chemotherapy and surgery needed? Lancet. 2017;389(10073):985–986. doi:10.1016/S0140-6736(17)30126-5
4. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
5. Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract. 2018;143:409–419. doi:10.1016/j.diabres.2018.05.023
6. Wirunsawanya K, Jaruvongvanich V, Upala S. Survival Benefits From Metformin Use in Pancreatic Cancer: A Systemic Review and Meta-analysis. Pancreas. 2018;47(3):e11–e14. doi:10.1097/MPA.0000000000001000
7. Zhou DC, Gong H, Tan CQ, Luo JQ. Prognostic significance of anti-diabetic medications in pancreatic cancer: A meta-analysis. Oncotarget. 2017;8(37):62349–62357. doi:10.18632/oncotarget.17728
8. Li M, Li X, Zhang H, Lu Y. Molecular Mechanisms of Metformin for Diabetes and Cancer Treatment. Front Physiol. 2018;9:1039.
9. Chai X, Chu H, Yang X, Meng Y, Shi P, Gou S. Metformin Increases Sensitivity of Pancreatic Cancer Cells to Gemcitabine by Reducing CD133+ Cell Populations and Suppressing ERK/P70S6K Signaling. Sci Rep. 2015;5(1):14404. doi:10.1038/srep14404
10. Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. doi:10.1038/nature16064
11. Kuwada K, Kagawa S, Yoshida R, et al. The epithelial-to-mesenchymal transition induced by tumor-associated macrophages confers chemoresistance in peritoneally disseminated pancreatic cancer. J Exp Clin Cancer Res. 2018;37(1):307. doi:10.1186/s13046-018-0981-2
12. Hiramoto H, Muramatsu T, Ichikawa D, et al. miR-509-5p and miR-1243 increase the sensitivity to gemcitabine by inhibiting epithelial-mesenchymal transition in pancreatic cancer. Sci Rep. 2017;7(1):4002. doi:10.1038/s41598-017-04191-w
13. Zhong T, Men Y, Lu L, et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene. 2017;36(17):2345–2354. doi:10.1038/onc.2016.391
14. Cuyas E, Fernandez-Arroyo S, Verdura S, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. 2018;37(7):963–970. doi:10.1038/onc.2017.367
15. Gao W, Gu Y, Li Z, et al. miR-615-5p is epigenetically inactivated and functions as a tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene. 2015;34(13):1629–1640. doi:10.1038/onc.2014.101
16. Bulle A, Dekervel J, Libbrecht L, et al. Gemcitabine induces Epithelial-to-Mesenchymal Transition in patient-derived pancreatic ductal adenocarcinoma xenografts. Am J Transl Res. 2019;11:765–779.
17. Gaianigo N, Melisi D, Carbone C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers. 2017;9.
18. Wang H, Wang HS, Zhou BH, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS One. 2013;8(2):e56664. doi:10.1371/journal.pone.0056664
19. Gungor C, Zander H, Effenberger KE, et al. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011;71(14):5009–5019. doi:10.1158/0008-5472.CAN-11-0036
20. Pan J, Hu H, Zhou Z, et al. Tumor-suppressive mir-663 gene induces mitotic catastrophe growth arrest in human gastric cancer cells. Oncol Rep. 2010;24:105–112.
21. Hu H, Li S, Cui X, et al. The overexpression of hypomethylated miR-663 induces chemotherapy resistance in human breast cancer cells by targeting heparin sulfate proteoglycan 2 (HSPG2). J Biol Chem. 2013;288(16):10973–10985. doi:10.1074/jbc.M112.434340
22. Huang Y, Liu J, Fan L, et al. miR-663 overexpression induced by endoplasmic reticulum stress modulates hepatocellular carcinoma cell apoptosis via transforming growth factor beta 1. Onco Targets Ther. 2016;9:1623–1633. doi:10.2147/OTT.S96902
23. Fiori ME, Villanova L, Barbini C, De Angelis ML, De Maria R. miR-663 sustains NSCLC by inhibiting mitochondrial outer membrane permeabilization (MOMP) through PUMA/BBC3 and BTG2. Cell Death Dis. 2018;9(2):49. doi:10.1038/s41419-017-0080-x
24. Wang S, Liu J, Li C, Yang X. Research of the effect of miR-663 on the proliferation of prostate cancer cells, and the correlations of miR-663 with pathological grade and clinical stage. J BUON. 2017;22:1011–1016.
25. Yi C, Wang Q, Wang L, et al. MiR-663, a microRNA targeting p21(WAF1/CIP1), promotes the proliferation and tumorigenesis of nasopharyngeal carcinoma. Oncogene. 2012;31(41):4421–4433. doi:10.1038/onc.2011.629
26. Yang Y, Wang LL, Wang HX, et al. The epigenetically-regulated miR-663 targets H-ras in K-562 cells. FEBS J. 2013;280(20):5109–5117. doi:10.1111/febs.12485
27. Jian P, Li ZW, Fang TY, et al. Retinoic acid induces HL-60 cell differentiation via the upregulation of miR-663. J Hematol Oncol. 2011;4(1):20. doi:10.1186/1756-8722-4-20
28. Mundalil VM, Anitha A, Takahashi T, et al. Fluoxetine Increases the Expression of miR-572 and miR-663a in Human Neuroblastoma Cell Lines. PLoS One. 2016;11:e164425.
29. Jiao L, Deng Z, Xu C, et al. miR-663 induces castration-resistant prostate cancer transformation and predicts clinical recurrence. J Cell Physiol. 2014;229(7):834–844. doi:10.1002/jcp.24510
30. Zang W, Wang Y, Wang T, et al. miR-663 attenuates tumor growth and invasiveness by targeting eEF1A2 in pancreatic cancer. Mol Cancer. 2015;14(1):37. doi:10.1186/s12943-015-0315-3
31. Tili E, Michaille JJ, Adair B, et al. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31(9):1561–1566. doi:10.1093/carcin/bgq143
32. Shi Y, Chen C, Zhang X, et al. Primate-specific miR-663 functions as a tumor suppressor by targeting PIK3CD and predicts the prognosis of human glioblastoma. Clin Cancer Res. 2014;20(7):1803–1813. doi:10.1158/1078-0432.CCR-13-2284
33. Ooshima A, Park J, Kim SJ. Phosphorylation status at Smad3 linker region modulates transforming growth factor-beta-induced epithelial-mesenchymal transition and cancer progression. Cancer Sci. 2019;110(2):481–488. doi:10.1111/cas.13922
34. Moustakas A, Heldin CH. Mechanisms of TGFbeta-Induced Epithelial-Mesenchymal Transition. J Clin Med. 2016;5.
35. Mukhopadhyay S, Chatterjee A, Kogan D, Patel D, Foster DA. 5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle. 2015;14(20):3331–3339. doi:10.1080/15384101.2015.1087623
36. Mukhopadhyay S, Saqcena M, Chatterjee A, Garcia A, Frias MA, Foster DA. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem. 2015;290(11):6986–6993. doi:10.1074/jbc.M114.622571
37. Mukhopadhyay S, Frias MA, Chatterjee A, Yellen P, Foster DA. The Enigma of Rapamycin Dosage. Mol Cancer Ther. 2016;15(3):347–353. doi:10.1158/1535-7163.MCT-15-0720
38. Chaiteerakij R, Petersen GM, Bamlet WR, et al. Metformin Use and Survival of Patients With Pancreatic Cancer: A Cautionary Lesson. J Clin Oncol. 2016;34(16):1898–1904. doi:10.1200/JCO.2015.63.3511
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.