Back to Journals » Drug Design, Development and Therapy » Volume 17
Metastasis Related Epithelial-Mesenchymal Transition Signature Predicts Prognosis and Response to Chemotherapy in Acute Myeloid Leukemia
Authors Qu S, Huang X, Guo X, Zheng Z, Wei T, Chen B
Received 3 April 2023
Accepted for publication 2 June 2023
Published 6 June 2023 Volume 2023:17 Pages 1651—1663
DOI https://doi.org/10.2147/DDDT.S415521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Shuang Qu,1,* Xiaoli Huang,2,* Xiaoling Guo,3 Zhihai Zheng,1 Tiannan Wei,1 Biyun Chen1
1Department of Hematology, Shengli Clinical Medical College of Fujian Medical University, Fujian Provincial Hospital, Fuzhou, People’s Republic of China; 2Department of Clinical Laboratory Medicine, Shengli Clinical Medical College of Fujian Medical University, Fujian Provincial Hospital, Fuzhou, People’s Republic of China; 3Translational Medicine Centre, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuang Qu, Email [email protected]
Background: Acute myeloid leukemia (AML) is a highly heterogenous disease with varying clinical outcomes among patients. Epithelial-mesenchymal transition (EMT) is an important mechanism underlying cancer metastasis and chemotherapy resistance. However, few EMT-based signatures have been established to predict AML prognosis and treatment efficacy.
Methods: By conducting comparative RNA-seq analysis, we discovered the differential expression of EMT genes between AML patients with relapse and those without relapse. Based on the prognostic analysis of the differentially expressed EMT genes, a metastasis-related EMT signature (MEMTs) was constructed. An analysis was conducted on both TARGET and TCGA cohorts to explore the possible association between MEMTs and prognosis in AML. Three separate chemotherapy treatment cohorts were utilized to assess the predictive efficacy of MEMTs for chemotherapy response. In addition, the potential correlation between MEMTs and the tumor microenvironment was also investigated. Finally, random forest analysis and functional experiments were conducted to verify the key MEMTs gene associated with AML metastasis.
Results: Based on expression and prognostic analysis, we constructed MEMTs that include three EMT genes (CDH2, LOX, and COL3A1). Our findings suggested that the MEMTs could act as a prognostic factor for AML patients, and furthermore, it proved to be a predictor of their response to chemotherapy. Specifically, high MEMTs was associated with worse prognosis and poor response to chemotherapy, while low MEMTs was linked to better prognosis and higher response rates. Random forest and functional experiments demonstrate that CDH2 is a key gene promoting leukemia cell metastasis among the three MEMTs genes.
Conclusion: The identification of MEMTs could potentially act as a predictor for the prognosis and the response to chemotherapy in AML patients. Individual tumor evaluation based on MEMTs could provide personalized treatment options for AML patients in the future.
Keywords: acute myeloid leukemia, prognosis, metastasis, epithelial-mesenchymal transition, chemotherapy
Introduction
Acute Myeloid Leukemia (AML) is an intricate illness with diverse genetic alterations and heterogeneous clinical outcomes.1–3 It is a highly aggressive malignancy that is associated with high relapse rates and poor overall survival outcomes.4 Chemotherapy is an effective treatment for AML, but it is heterogeneous in its effectiveness.5 While many patients with AML respond well to chemotherapy and achieve remission, others may have a poorer response or experience relapse despite treatment. This heterogeneity is likely due to the genetic and clinical diversity of AML, as well as the varying responses of individual patients to chemotherapeutic agents.6–8 Therefore, there is a pressing need to create novel molecular markers that can precisely classify subtypes of AML patients who may derive greater benefits from specific treatment strategies.
Epithelial-mesenchymal transition (EMT) is a biological phenomenon in which epithelial cells undergo a transformation and acquire mesenchymal characteristics, resulting in weakened cell-cell adhesion, enhanced motility, and invasiveness.9–11 The importance of EMT in tumorigenesis and tumor advancement lies in its ability to facilitate cancer cell invasion into adjacent tissues and spread to distant locations.12,13 Specifically, EMT facilitates the detachment of tumor cells from their primary site, as well as their migration into the circulatory system for eventual colonization in secondary organs. Moreover, EMT has been linked to drug resistance, immune evasion, and stem cell-like properties in cancer cells, further underscoring its impact on cancer pathogenesis.14–20 Currently, there is a growing interest in developing prognostic models for cancer using EMT features. Recent studies have shown that EMT-related gene expression signatures can predict patient survival and clinical outcomes across various cancer types, including breast, lung, and colon cancers.21–24 Additionally, a number of bioinformatic tools and algorithms have been developed to analyze EMT-related features and build predictive models using machine learning and other techniques. The potential of EMT-based prognostic models to improve cancer patient stratification and treatment decision-making continues to generate interest and investment in the research community.
In this study, our study involved establishing a MEMTs signature linked to AML metastasis and scrutinizing the transcriptomic, genomic, and tumor microenvironment facets of various MEMTs subtypes, alongside their reaction to chemotherapy treatment. We concluded that MEMTs is an effective prognostic biomarker that can reliably predict the response of AML to chemotherapy treatment.
Methods
Clinical Data Acquisition
We acquired mRNA-seq expression profiles and clinical data of patients with Acute myeloid leukemia from the TARGET database (https://ocg.cancer.gov/programs/target/data-matrix), The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga/), and Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GEO database includes the GSE10358, GSE107465, and GSE213056 cohort.
Identification of Differentially Expressed EMT Genes
To distinguish the expression patterns of EMT genes among relapsed and non-relapsed AML cases in the TARGET-AML dataset, we employed the limma package for differential gene expression analysis. The log2(fold change) > 1.5 and False-discovery rate (FDR) < 0.05 were filtered as the statistically significant.
Survival Prognosis Analysis
To evaluate and compare the rates of disease-free survival (DFS) and overall survival (OS) in AML patients, we employed Kaplan-Meier curves supplemented by the log rank test.
Least Absolute Shrinkage and Selection Operator (LASSO) Analysis
LASSO is a widely used method for regression analysis of high-dimensional data. It can identify gene pairs that are related to prognosis of AML by shrinking regression coefficients. We employed a grid search method in a 10-fold cross-validation process to determine the optimal penalty weight for the Lasso-Cox model. The LASSO method allowed us to reduce the coefficients of most genes to zero, while highlighting a small number of genes with non-zero coefficients, which were found to be significantly associated with AML prognosis. These genes were used to construct a MEMTs model. The MEMTs score was calculated as follows: 0.056×expression level of CDH2 + 0.029 × expression level of LOX + 0.026 × expression level of COL3A1. LASSO regression analysis allowed us to divide AML patients into MEMTs-low and MEMTs-high subtypes based on the median MEMTs score.
Drug Sensitivity Analysis
To identify molecular compounds that could potentially be used for targeted therapy, we conducted a drug sensitivity analysis on MEMTs genes utilizing the Gene Set Cancer Analysis (GSCA) website (http://bioinfo.life.hust.edu.cn/web/GSCALite/)25 as a tool in our study.
Single-Cell Level Analysis
The Tumor Immune Single-cell Hub 2 (TISCH2)26 is a dedicated platform that focuses on the tumor microenvironment (TME), leveraging scRNA-seq database (http://tisch.comp-genomics.org/home/) to provide a rich, detailed and comprehensive set of cell-type annotations at the single-cell level. This platform enables us to investigate the intricacies of the TME in various types of cancer.
Cell Culture
HL-60 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and were cultured in RPMI1640 medium containing 10% FBS. The cells were seeded in 6-well plates at 5.0×105 cells/well. The culture medium was changed every two days, and the cells were subcultured using 0.25% trypsin/EDTA solution when they attained about 80% confluency.
Cell Transfection
Transfection of CDH2-shRNA plasmids and control plasmids was performed according to the Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) manual. HL-60 cells with stable transfection of CDH2 shRNA were selected by 700 μg/mL G418 (Sigma, Louis, MI, USA) for 1 week and maintained with 100 μg/mL G418. The target sequence of CDH2-shRNAs is described as follows:CDH2-shRNA1:5’-CACCGCTGGGCAACAATTCTCCTTTTCAAGAGAAGGAGAATTGTTGCCCAGCTTTTG-3’;CDH2-shRNA2:5’-CACCGCGCAGTATAATTTTCTCCATACTCGAGTATGGAGAAATTATACTGCGCTTTTTG-3’.
Real-Time Quantitative PCR
The total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNA template was obtained by TaKaRa reverse transcription reagents (TaKaRa, Dalian, China). Real-time quantitative PCR was carried out using SYBR Green PCR kit (Takara, Dalian, China). The reaction mixture (20 µL) contained 2 µL cDNA, 10 µL RT-qPCR-Mix, 0.5 µL forward primer, 0.5 µL reverse primer, and 7 µL ddH2O. The reaction conditions involved an initial denaturation at 95°C for 10 minutes followed by 40 cycles of 95°C for 1 minute and 60°C for 30 seconds. GAPDH was utilized as an internal control. Primer sequences were as follows: CDH2 forward 5’-GGCTTAATGGTGATTTTGCTCAG-3’; CDH2 reverse 5’-TCCATACCACAAACATCAGCAC-3’. GAPDH forward 5’-CCTGCACC ACCAACTGCTTAG-3’; GAPDH reverse:5’-GTGGATGCAGGGATGATGTTC-3’.
Cell Counting Kit-8 (CCK8) Assay
To evaluate the impact of CDH2 knockdown on cell proliferation, we conducted CCK8 assays. In brief, HL-60 cells were seeded at a density of 1.0×104 cells per well in 96-well plates supplemented with 100 μL of complete medium. After 24, 48, and 72 hours of incubation, 10 μL of CCK8 reagent was introduced to every well and left to incubate for 2 hours at 37°C. At that point, a microplate reader was employed to determine absorbance levels at 450 nm.
Transwell Assay
Transwell assay was used to detect the invasiveness of cells. Transwell chambers were placed in 24-well plates. The upper chamber was coated with culture matrix and 1×105 cells were added to the upper chamber in 200 μL of medium. The lower chamber was filled with 500 μL of complete medium. After incubation at 37°C and 5% CO2 for 24 hours, the migrated cells on the lower surface of the filters were quantified by crystal violet staining.
Statistical Analysis
Statistical analysis was carried out using GraphPad Prism software (San Diego, USA). The data are expressed as mean ± standard error of the mean (SEM), and p-values <0.05 were deemed statistically significant.
Results
Identification of EMT Genes Associated with AML Metastasis
By analyzing the TARGET-AML cohort, we investigated the differential expression of EMT genes between metastasis and non-metastasis AML. Thirteen EMT genes (BGN, CDH2, COL1A1, COL1A2, COL3A1, CXCL12, LOX, LOXL1, MAGEE1, MXRA5, SFRP1, TNFRSF11B, VCAM1) were found to be significantly upregulated in metastasis AML and identified as potential genes associated with disease metastasis (Figure 1A). Subsequent analysis focused on these genes. Chromosomal distribution analysis revealed that the genes, except for BGN, MAGEE1, and MXRA5 which are located on sex chromosomes, are located on autosomes (Figure 1B). Through Spearman correlation analysis, we found that the majority of EMT genes exhibited significant correlations with other genes (Figure 1C). Our functional analysis demonstrated that these genes are capable of activating the EMT pathway while simultaneously inhibiting cell growth (Figure 1D). These findings suggest that these genes may hold substantial influence over cancer progression and metastasis. Given the crucial role of gene mutations in cancer progression, we investigated the mutation patterns of 13 EMT genes in AML patients. Our analysis identified MXRA5 as the gene with the highest mutation frequency (2.1%), followed by COL1A1 (1.4%), COL3A1 (0.7%), and VCAM1 (0.7%). The remaining genes had the lowest mutation frequency of 0% (Figure 1E). In addition, our overall survival and disease-free survival analyses revealed that only CDH2, LOX, and COL3A1 were significantly associated with poor prognosis in AML patients among the 13 genes examined (Figure 1F and G). These findings suggest that CDH2, LOX, and COL3A1 may serve as potential prognostic biomarkers for AML, while the other genes may have a less significant impact on patient outcomes.
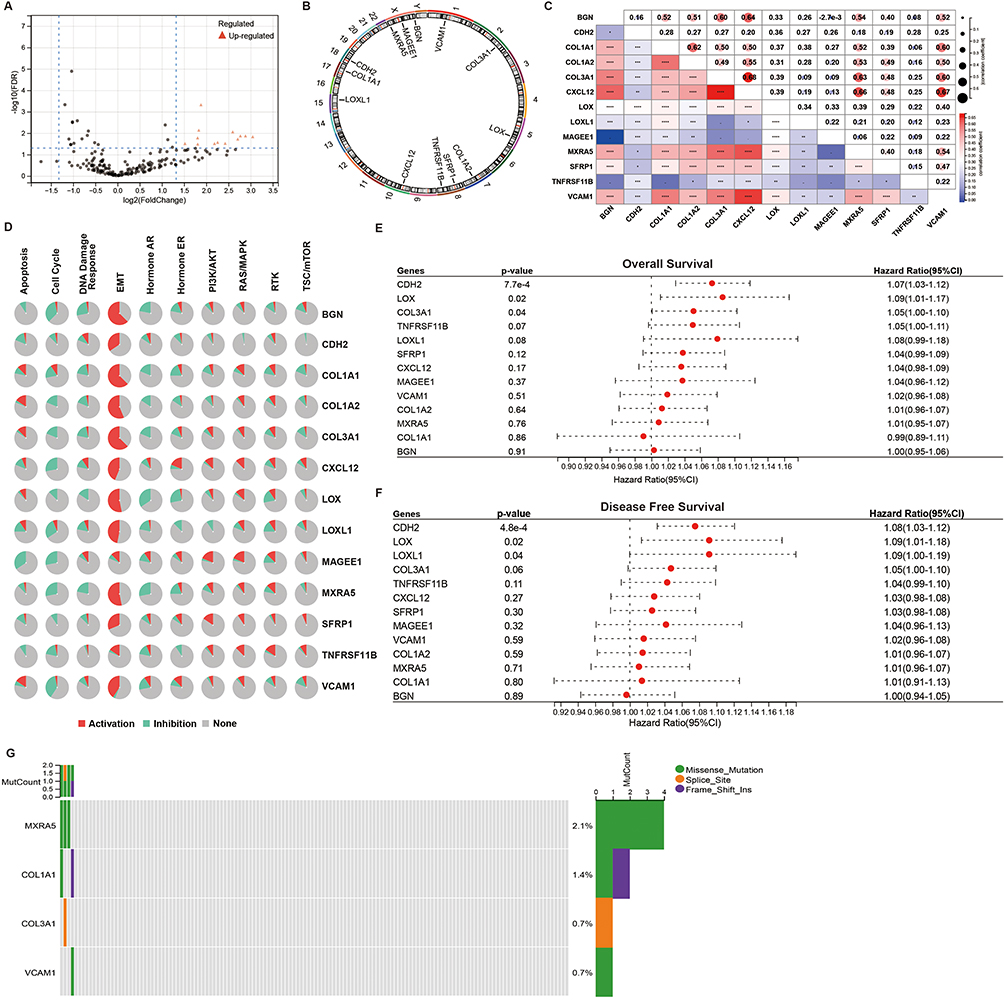 |
Figure 1 Analysis of genetic variations and correlations of EMT genes in AML. (A) The volcano plot shows the DEG between non-recurrent AML and recurrent AML. (B) Distribution of 13 EMT genes on chromosomes. (C) Spearman correlation analysis of the 13 genes. (D) Relationship between 13 genes and pathways. (E) OS survival analysis of the 13 genes. (F) DFS survival analysis of the 13 genes. (G) Oncoplots show the mutation status of the 13 genes in AML patients from the TCGA database. *p <0.05; **p <0.01; ***p <0.001; ****p <0.0001. |
Construction of a MEMTs-Based Prognostic Model
To establish a prognostic model, we employed LASSO regression analysis using the expression levels of CDH2, LOX, and COL3A1 genes. Figures 2A and B depict the process of building the metastasis related epithelial-mesenchymal transition-related signature (MEMTs) model. The MEMTs score was calculated as follows: 0.056×expression level of CDH2 + 0.029 × expression level of LOX + 0.026 × expression level of COL3A1. LASSO regression analysis allowed us to divide AML patients into MEMTs-low and MEMTs-high subtypes based on the median MEMTs score. Figure 2C displays the expression patterns of the three genes in both subtypes, along with their survival status and survival time. The results of our survival analysis indicated a significant difference in overall survival between patients with MEMTs-high subtype and MEMTs-low subtype, with the former exhibiting poorer outcomes compared to the latter. (Figure 2D). To validate the robustness of the prognostic model, we further confirmed our findings in an independent cohort (TCGA-AML) by categorizing AML patients into MEMTs-low and MEMTs-high subtypes using the median MEMTs score. Consistent with the results from the TARGET cohort, we found that MEMTs-high patients exhibited significantly worse overall survival than MEMTs-low patients (Figure 2E), supporting the notion that MEMTs can serve as a reliable predictor of AML prognosis.
 |
Figure 2 Construction of a MEMTs-Based prognostic model. (A and B) By utilizing the LASSO-Cox regression model, coefficients were calculated which enabled the derivation of MEMTs. (C) Through analysis and visualization of the distribution of risk scores, survival status, and corresponding heatmap of gene expression levels for each patient. (D and E) Kaplan-Meier analysis was employed to determine the prognostic significance of MEMTs with respect to overall survival in two distinct cohorts: the (D) TARGET-AML cohort and the (E) TCGA-AML cohort. |
Characterization of Molecular Signatures of MEMTs Low and High Subtypes in AML
Subsequently, we conducted a genomic analysis to compare the alterations found in MEMTs-low and MEMTs-high subtypes in AML patients. The top 20 mutated genes are generally similar between the two subtypes in terms of frequency (Figure 3A and B). We analyzed the impact of these genetic mutations on the prognosis of AML patients, and the results showed that patients with mutations in the DMNT3A, FTL3, TP53, and RUNX1 genes had significantly worse prognoses than those without mutations (Supplementary Figure 1). Additionally, we found that the mutation rates of these genes were significantly higher in the MEMTs-high subtype of patients compared to the MEMTs-low subtype. KRAS (Kirsten rat sarcoma viral oncogene homolog) gene mutation and IDH1 gene mutation are common genetic mutations that occur in AML. KRAS gene mutation can promote the proliferation of hematopoietic cells, leading to the development of AML. IDH1 gene mutation can cause metabolic pathway disorders and exacerbate the development of AML. Both genetic mutations are crucial in both the progression and management of leukemia, so they are highly valued in the research and clinical treatment of AML. Therefore, we analyzed the differences in KRAS and IDH1 mutations between the two subtypes. Our findings revealed that the occurrence of KRAS (8%) and IDH1 (23%) mutations were higher in the MEMTs-high subtype as opposed to the MEMTs-low subtype, where rates of these mutations were relatively lower (KRAS: 2.5%, IDH1: 10%) (Figure 3C and D). Tumor mutation burden (TMB) has emerged as a promising indicator for immune therapy, owing to the production of immunogenic neoantigens. Our findings suggest that there is a discrepancy in TMB between the MEMTs-low and MEMTs-high subtypes, with the former exhibiting a higher level of TMB (Figure 3E).
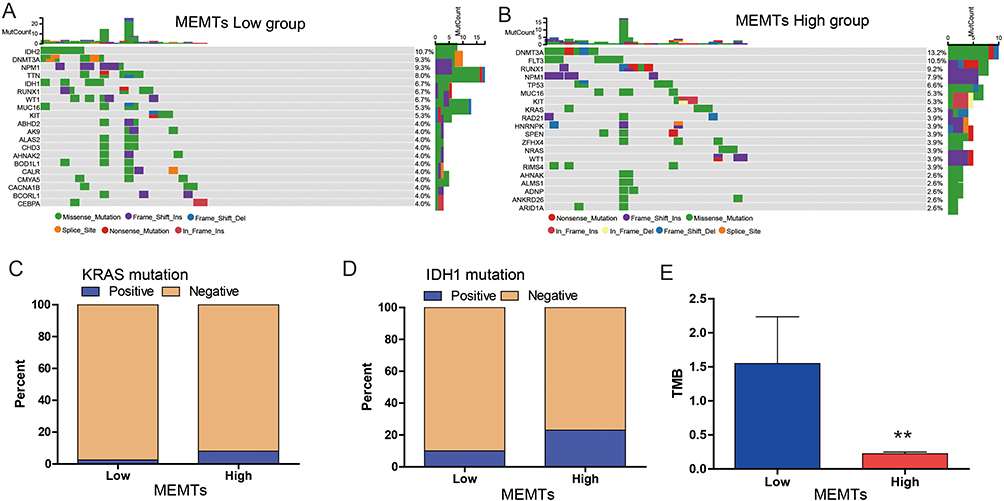 |
Figure 3 Characterization of molecular features of the MEMTs low and high subtypes. (A and B) Oncoplots show the genomic landscape of (A) MEMTs low subtype and (B) MEMTs high subtype. (C and D) The mutation status of (C) KRAS gene and (D) IDH1 gene between MEMTs low and MEMTs high subtypes. (E) Difference in tumor mutation burden (TMB) between MEMTs low and high subtypes. **p <0.01. |
MEMTs Predicts AML Response to Chemotherapy Treatment
Various clinical studies have demonstrated the efficacy of adjuvant chemotherapy in improving the prognosis of AML patients. However, resistance triggered by EMT poses a significant challenge to chemotherapy response. To further explore this relationship, we evaluated the correlation between drug sensitivity and MEMTs gene expression using the CTRP database. Our analyses revealed a positive association between MEMTs gene expression and IC50 values of anti-cancer drugs (Figure 4A). These findings can be utilized to guide the design of novel and improved chemotherapy protocols.
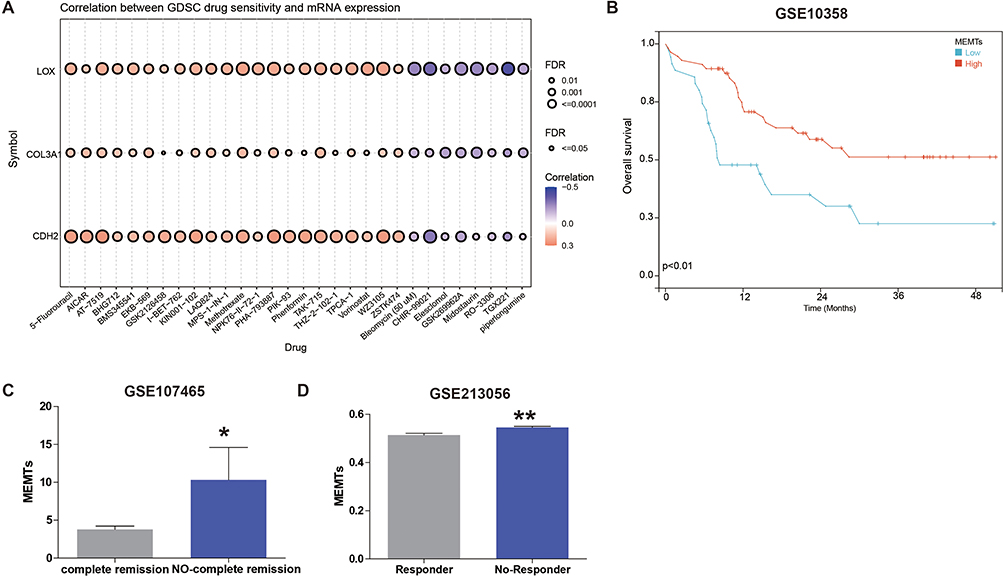 |
Figure 4 Correlation of three MEMTs genes to chemotherapy drugs. (A) The correlation between drug sensitivity in GDSC and the expression of three MEMT genes. (B) Kaplan-Meier plot shows the overall survival (OS) of AML patients (GSE10358 cohort) who received chemotherapy treatment, stratified by low and high subgroups based on MEMTs. (C) Differential analysis of MEMTs between AML patients with complete remission and those without complete remission after chemotherapy treatment (GSE107465 cohort). (D) Differential analysis of MEMTs between AML patients who responded to chemotherapy treatment and those who did not respond (GSE213056 cohort). *p <0.05; **p <0.01. |
Next, we conducted an analysis of the correlation between MEMTs and AML patient prognosis, as well as complete remission in the context of chemotherapy treatment, using three different datasets: GSE10358, GSE107465, and GSE213056. While GSE10358 did not have chemotherapy CR data available, we focused our analysis on the relationship between MEMTs and overall survival in this dataset. In contrast, GSE107465 and GSE213056 had chemotherapy response data available, so we analyzed the correlation between MEMTs and complete remission in these dataset.
Firstly, we examined the relationship between MEMTs expression and overall survival in a cohort of 91 patients who received chemotherapy treatment in the GSE10358 dataset. Notably, our findings indicated that patients classified as MEMTs-low subtype exhibited significantly improved overall survival rates compared to those classified as MEMTs-high subtype (Figure 4B). Furthermore, in the GSE107465 cohort, 30 AML patients who underwent chemotherapy were divided into two subgroups: those with complete remission after treatment (n=15) and those without complete remission after treatment (n=15). We analyzed the MEMTs in both subgroups and found that the subgroup with complete remission had significantly lower MEMTs compared to the subgroup without complete remission (Figure 4C). Similarly, in the GSE213056 cohort, which included 11 patients with AML who received chemotherapy treatment, we analyzed the MEMTs in subgroups of patients with/without response to treatment. Our data revealed a distinct difference in MEMTs value between responders and non-responders to treatment, with significantly lower MEMTs value observed in the responsive subgroup compared to the non-responsive subgroup (Figure 4D).
Taken together, these findings suggest that MEMTs may serve as a potential predictive biomarker for chemotherapy response in AML patients. It is important to note, however, that caution must be exercised when interpreting these results due to the limited sample size. Nevertheless, our results underscore the possibility of using MEMTs expression levels to identify AML patients who are likely to benefit from chemotherapy treatment.
The Relation Between MEMTs and Tumor Microenvironment in AML
To explore the relationship between MEMTs subtypes and immune microenvironment in AML, we investigated the distribution of 22 infiltrating immune cells across MEMTs-low and MEMTs-high subtypes. Our analysis revealed that the MEMTs-high subtype exhibited a statistically significant increase in immunosuppressive cell infiltration, including T cells CD4 memory resting, T cells CD4 memory activated, T cells gamma delta, and Neutrophils (as depicted in Figure 5A). Furthermore, we observed that the MEMTs-high subtype had higher infiltration of cancer-associated fibroblasts (CAFs) compared to the MEMTs-low subtype (shown in Figure 5B). It is important to note that these findings highlight the potential role of MEMTs in shaping the immune microenvironment, which could influence AML tumor progression and treatment response. To assess the function of MEMTs, we investigated their association with established molecular characteristics (Figure 5C). We observed positive correlations between MEMTs and several features including EMT, Mitotic spindle, G2M checkpoint, Androgen response, Protein secretion, Unfolded protein response, Mtorc1 signaling, E2F targets, and MYC targets V1. Further analysis using ssGSEA revealed enriched carcinogenic pathways, such as WNT beta-catenin, KRAS signaling, and EMT, in the MEMTs-high subtype (Figure 5D). The results of our study indicate a link between MEMTs and TME, which sheds light on the potential predictive role of MEMTs in response to AML adjuvant chemotherapy.
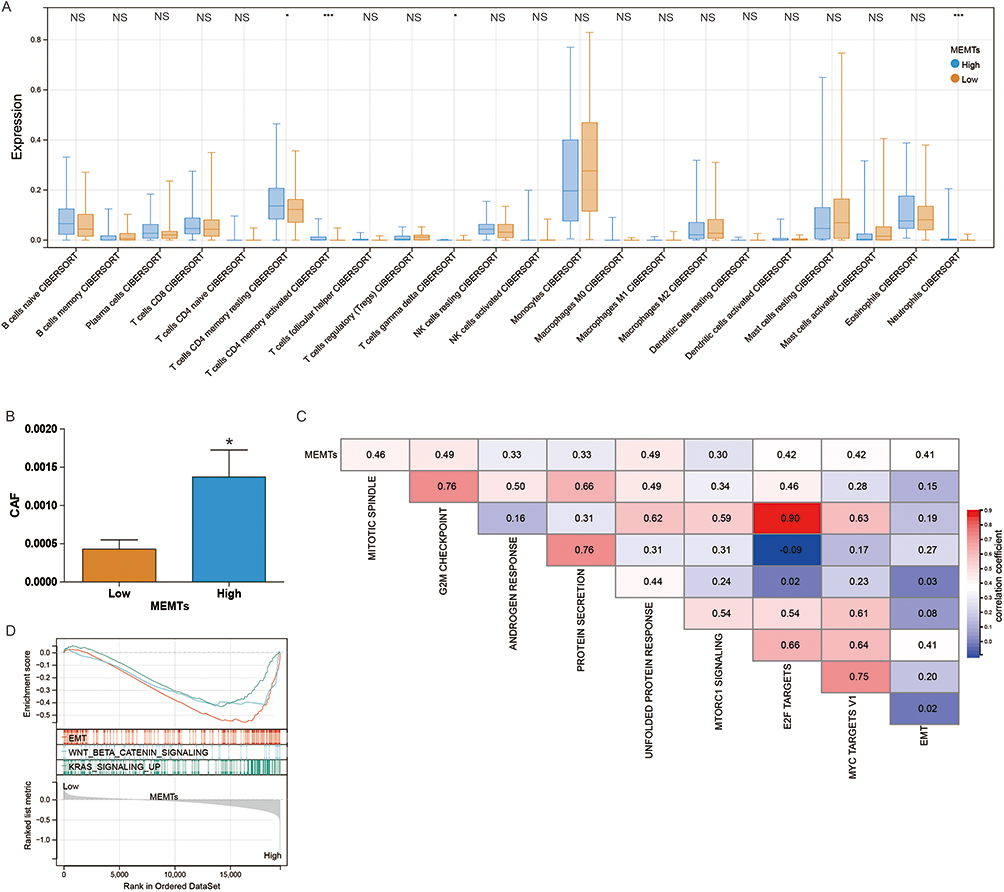 |
Figure 5 The correlation between MEMTS and the AML microenvironment. (A) Box plots showing the relationship between MEMTS subtypes and the infiltration of 22 immune cells. (B) Differential analysis of cancer-associated fibroblasts (CAF) between low and high MEMTS subtypes. (C) Spearman analysis showing the correlation between MEMTS and known gene characteristics. (D) Gene enrichment results for different MEMTS subtypes. NS, p >0.05; *p <0.05. ***p <0.001. |
The emergence of single-cell RNA sequencing (scRNA-seq) has presented a potent approach to explore the molecular features of individual cells and attain improved insights into the TME, thus reducing redundancy in research. To clarify the function of MEMTs within the TME, we conducted an analysis of the GSE16256 cohort. This cohort was derived from scRNA-seq experimentation utilizing Smart-seq2 on 21 AML patients. Using the UMAP algorithm, the filtered 38,348 cells were divided into 13 cell clusters (Figure 6A). Subsequently, we investigated the expression of three MEMTs genes. CDH2 and LOX were highly expressed in plasma cells, progenitor cells and Hematopoietic Stem Cells (HSC) (Figure 6B and C), while COL3A1 was highly expressed in Proliferating T cells (Figure 6D).
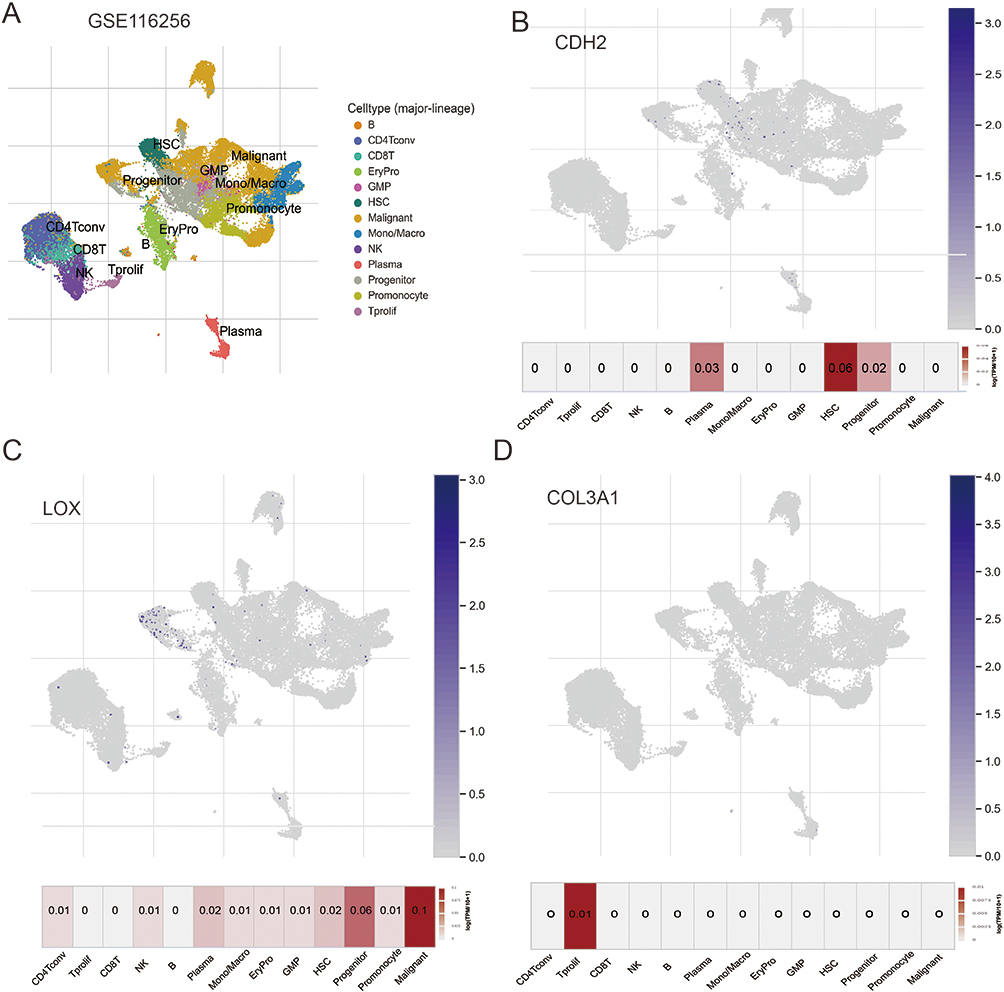 |
Figure 6 The distribution of MEMTs in the tumor microenvironment. (A) UMAP plot showing 13 cell types from 38,348 cells. (B) Expression of CDH2 gene in the 13 cell types. (C) Expression of LOX gene in the 13 cell types. (D) Expression of COL3A1 gene in the 13 cell types. |
Random Forest Diagnostics and Functional Experiments to Identify Significant Genes
Based on random forest analysis of the three MEMTs genes, we identified important genes related to AML relapse. Our analysis revealed that CDH2 was the most significant gene among them (Figure 7A). We then proceeded to knock down CDH2 expression in HL-60 cells (Figure 7B). Results from CCK-8 assays indicated that downregulating CDH2 expression did not affect cell proliferation (Figure 7C); however, invasion assays demonstrated that downregulation of CDH2 significantly suppressed cell invasion abilities (Figure 7D). These findings highlight the critical role of CDH2 in AML progression.
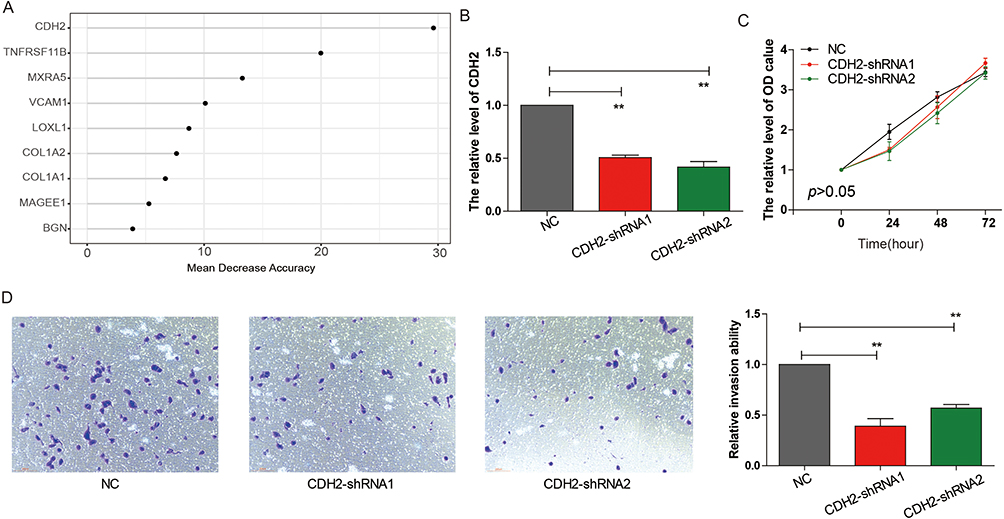 |
Figure 7 CDH2 knockdown inhibits HL-60 cells proliferation and invasion capability. (A) Random Forest feature importance ranking for the 13 EMT genes. (B) Detection of CDH2 expression in HL-60 cells by RT-PCR. (C) CCK8 experiment to assess the effect of CDH2 on HL-60 cells proliferation. (D) Trans-well experiment to evaluate the impact of CDH2 on HL-60 cells invasion capability. **p <0.01. |
Discussion
AML is a hematological malignancy that impacts the bone marrow and blood cells.27 The disease progresses rapidly and has a serious impact on human health. While there have been improvements in the treatment of AML, metastasis remain major threats for AML patients.28 One important process involved in tumor metastasis is EMT, which enhances the tumor’s ability to spread. In this study, we analyzed the differential expression and prognostic value of EMT-related genes between leukemia patients with or without metastasis. Subsequently, we identified a gene set consisting of 3 genes (CDH2, LOX, COL3A1) and constructed MEMTs to predict the survival rate of AML and its response to chemotherapy.
CDH2, also known as N-cadherin, is a transmembrane glycoprotein involved in cell adhesion and motility. Many studies have shown that CDH2 expression is dysregulated in different tumors. Specifically, CDH2 expression is upregulated in breast, prostate, colon, lung, and ovarian cancers,29–32 while it is downregulated in gliomas.24 CDH2 overexpression has been implicated in promoting tumor invasion and metastasis by enhancing the migratory and invasive ability of cancer cells through activation of multiple signaling pathways. On the other hand, CDH2 downregulation or inhibition has been shown to decrease cancer cell migration and invasion. LOX is a copper-dependent enzyme involved in the cross-linking of collagen and elastin fibers in the extracellular matrix (ECM). Recent research has shown that LOX is integral to cancer advancement and metastasis, as it exerts influence over the structure and composition of the ECM. Elevated levels of LOX have been noted in diverse cancer cell types, such as breast, pancreatic, and colorectal cancer.33–36 Overexpression of LOX facilitates the invasion, migration, and metastasis of tumor cells. COL3A1 is a crucial constituent of the extracellular matrix (ECM). Current research has demonstrated that COL3A1 plays a pivotal function in tumor advancement, and metastasis by influencing tumor cell invasion and migration. COL3A1 overexpression has been discovered in multiple cancer cell types, including breast, ovarian, and colorectal cancer.37,38
More and more studies have revealed the important relationship between genetic mutations and tumor metastasis. Mutations in KRAS genes play a critical role in the pathogenesis of various hematologic malignancies, including AML. These mutations lead to constitutive activation of the KRAS pathway, which promotes cell proliferation, survival, and differentiation, thereby contributing to AML initiation and progression. A commonly observed genetic mutation in AML is the IDH1 gene mutation, which exerts a significant influence on its development and progression. For example, recent research shows that IDH1 mutations are closely associated with AML incidence and prognosis, with additional clinical value in older adults, giving guidance to patient treatment strategies. Our analysis results indicate that the MEMTs-high subgroup has a higher mutation rate of RAS and IDH1 genes compared to the MEMTs-low subgroup. TMB has emerged as a crucial factor in augmenting the effectiveness of immunotherapy in cancer treatment due to the heightened presence of tumor neoantigens. Our results also indicate that MEMTs-high subgroup exhibit lower TMB, whereas MEMTs-low subgroup show the opposite trend.
AML is usually treated with chemotherapy as one of the primary treatment options. Due to the continuous proliferation of AML cells and the presence of many gene abnormalities, chemotherapy drugs can be used to kill or inhibit the growth of these cells, thereby achieving the goal of controlling and treating AML. However, not all AML patients respond effectively to chemotherapy. Some patients do not respond to chemotherapy drugs while others tend to relapse after treatment. Our research has found that compared to the MEMTs-high subgroup, patients in the MEMTs-Low subgroup have a better prognosis after chemotherapy treatment. In addition, we found that the MEMTs values of AML patients who responded to chemotherapy treatment were lower than those of AML patients who did not respond to chemotherapy treatment. All of the above results indicate that MEMTs is a useful predictive tool for chemotherapy treatment in AML.
However, we acknowledge the limitations of our study, including the small sample size and conventional chemotherapy drugs of AML were less in Figure 4A, which may limit the generalizability of our findings. While our results only hint at a trend, they nonetheless provide a promising avenue for future research in this area.
Based on random forest analysis of 13 differential expression of EMT genes, we identified CDH2 as the most significant gene related to AML relapse. The cell functional experiments indicate that knocking down CDH2 does not affect the proliferation ability of AML cells, but it does suppress the invasion ability of the cells. A study has found that the expression of CDH2 can enhance the resistance of AML cells to chemotherapy drugs. These studies suggest that by inhibiting the expression of CDH2, we may be able to reduce the metastatic potential and drug resistance of AML cells, which can ultimately improve clinical outcomes. However, more research is needed to better understand the underlying mechanisms involved in CDH2-mediated tumor metastasis in AML. Further investigations may also help identify potential therapeutic targets and strategies for the treatment of AML.
Conclusion
In conclusion, we have developed an MEMTs score for the purpose of categorizing AML patients across various cohorts. Through this approach, we have delved into the underlying mechanisms that give rise to distinct characteristics between samples with high and low MEMTs. This work has contributed to our comprehension of EMT advancement in AML. Our findings indicate that MEMTs may be utilized as a means of stratifying patients and identifying those who are inclined to benefit most from chemotherapy treatments.
Data Sharing Statement
The corresponding author can provide the datasets used and/or analyzed during the current study upon reasonable request.
Funding
This work was supported by grants from Youth and Middle-aged Backbone Talents Training Project of Fujian Health Commission (2016-ZQN-11), Natural Science Foundation of Fujian Province of China (2022J011007).
Disclosure
The authors have declared that they do not have any significant affiliations or financial interests in any commercial entity related to the subject matter discussed in this article.
References
1. Pelcovits A, Niroula R. Acute myeloid leukemia: a review. Rhode Island Med J. 2020;103(3):38–40.
2. De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J. 2016;6(7):e441.
3. Infante MS, Piris M, Hernández-Rivas J. Molecular alterations in acute myeloid leukemia and their clinical and therapeutical implications. Med Clin. 2018;151(9):362–367.
4. Rubnitz JE, Gibson B, Smith FO. Acute myeloid leukemia. Hematol Oncol Clin North Am. 2010;24(1):35–63.
5. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474.
6. Roloff GW, Odenike O, Bajel A, Wei AH, Foley N, Uy GL. Contemporary Approach to Acute Myeloid Leukemia Therapy in 2022. Am Soc Clin Oncol Educ Book Am Soc Clin Oncol Annu Meeting. 2022;42:1–16.
7. Floren M, Gillette JM. Acute myeloid leukemia: therapy resistance and a potential role for tetraspanin membrane scaffolds. Int J Biochem Cell Biol. 2021;137:106029.
8. Sami SA, Darwish NHE, Barile ANM, Mousa SA. Current and future molecular targets for acute myeloid leukemia therapy. Curr Treat Options Oncol. 2020;21(1):3.
9. Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412.
10. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
11. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344):re8.
12. Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules. 2016;21(7):564.
13. Taki M, Abiko K, Ukita M, et al. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin Cancer Res. 2021;27(17):4669–4679.
14. Sommers CL, Heckford SE, Skerker JM, et al. Loss of epithelial markers and acquisition of vimentin expression in Adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992;52(19):5190–5197.
15. Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69(14):5820–5828.
16. McConkey DJ, Choi W, Marquis L, et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28(3–4):335–344.
17. Huang J, Li H, Ren G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int J Oncol. 2015;47(3):840–848.
18. Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010;30(7):2513–2517.
19. Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 2014;4(5):411–435.
20. Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–950.
21. Grasset EM, Dunworth M, Sharma G, et al. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Sci Transl Med. 2022;14(656):eabn7571.
22. Lee HW, Jose CC, Cuddapah S. Epithelial-mesenchymal transition: insights into nickel-induced lung diseases. Semin Cancer Biol. 2021;76:99–109.
23. Stehr AM, Wang G, Demmler R, et al. Neutrophil extracellular traps drive epithelial-mesenchymal transition of human colon cancer. J Pathol. 2022;256(4):455–467.
24. Camand E, Peglion F, Osmani N, Sanson M, Etienne-Manneville S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J Cell Sci. 2012;125(Pt 4):844–857.
25. Liu CJ, Hu FF, Xie GY, et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 2023;24(1):76.
26. Sun D, Wang J, Han Y, et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021;49(D1):D1420–d1430.
27. Nie Y, Su L, Li W, Gao S. Novel insights of acute myeloid leukemia with CEBPA deregulation: heterogeneity dissection and re-stratification. Crit Rev Oncol Hematol. 2021;163:103379.
28. Nagata A, Kanemasa Y, Kikuchi M, et al. Bone marrow metastasis of glioblastoma multiforme mimicking acute myeloid leukemia. Oxford Medical Case Reports. 2020;2020(6):omaa040.
29. Xu M, Liu C, Pu L, et al. Systemic analysis of the expression levels and prognosis of breast cancer-related cadherins. Exp Biol Med. 2021;246(15):1706–1720.
30. Zeng H, Huang Y, Liu Q, et al. MiR-145 suppresses the motility of prostate cancer cells by targeting cadherin-2. Mol Cell Biochem. 2021;476(10):3635–3646.
31. Liu T, Xia R, Li C, Chen X, Cai X, Li W. mRNA expression level of CDH2, LEP, POSTN, TIMP1 and VEGFC modulates 5-fluorouracil resistance in colon cancer cells. Exp Ther Med. 2021;22(3):1023.
32. Wang L, Peng Q, Sai B, et al. Ligand-independent EphB1 signaling mediates TGF-β-activated CDH2 and promotes lung cancer cell invasion and migration. J Cancer. 2020;11(14):4123–4131.
33. Mao F, Wang M, Wang J, Xu WR. The role of 15-LOX-1 in colitis and colitis-associated colorectal cancer. Inflammation Res. 2015;64(9):661–669.
34. Vishnupriya P, Aparna A, Viswanadha VP. Lipoxygenase (LOX) Pathway: a Promising Target to Combat Cancer. Curr Pharm Des. 2021;27(31):3349–3369.
35. Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12(8):540–552.
36. Leo C, Cotic C, Pomp V, Fink D, Varga Z. Overexpression of Lox in triple-negative breast cancer. Ann Diagn Pathol. 2018;34:98–102.
37. Wang L, Sun Y, Guo Z, Liu H. COL3A1 overexpression associates with poor prognosis and cisplatin resistance in lung cancer. Balkan Med J. 2022;39(6):393–400.
38. Wu Y, Xu Y. Integrated bioinformatics analysis of expression and gene regulation network of COL12A1 in colorectal cancer. Cancer Med. 2020;9(13):4743–4755.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.